

Abstract
Fluorinated molecules are core to contemporary drug discovery programs and critical for advancing innovation in numerous fields. In merging these important chemical themes, fluorinated Diels-Alder cycloaddition products are a particularly attractive subset of compounds with significant utility. Herein, an in-depth computational and experimental study of fluorine substitution effects on dienophile partners in Diels-Alder reactions is reported. Of particular focus to this study is understanding the origin of reaction rate deceleration as a consequence of employing fluorinated dienophiles and the factors controlling endo- vs. exo-selectivity. To unlock insight into this unique reactivity, density function theory calculations, distortion/interaction-activation strain models, energy decomposition analysis and natural bond orbital analysis, among other computational methods, were applied. In addition, the influence of oriented external-electric-field-effects (OEEFs) and local electric field effects were explored. To further probe this effect, experimental studies of charge-enhanced Diels-Alder reactivity with fluorinated dienophiles were conducted. Collectively, this work offers novel mechanistic understanding pertinent to Diels-Alder reactions of fluorinated dienophiles providing valuable fluorinated scaffolds.
Keywords: fluorine, density functional theory, charge-enhanced, Diels-Alder, smart reagents
Graphical Abstract

An in-depth computational and experimental study of the effects of fluorinated dienophiles in Diels-Alder cycloadditions is disclosed, along with insights into the origin of reaction rate deceleration and the factors controlling endo- vs. exo-selectivity. In addition, the design and synthesis of a Brønsted acid activated fluorinated dienophile facilitating charge-enhanced Diels-Alder reactivity is reported.
Introduction
Fluorinated compounds are coveted chemical building blocks and strategies for their direct synthesis remain a top priority in materials science, agrochemicals, and pharmaceuticals.[1,2] Prompting this progress is the ability of fluorine to impart stability, improve solubility and permeability, dramatically influence molecular conformation, as well as modulate bioavailability and protein binding,[3] having gained this “small atom with a big ego” (van der Waals radius of 1.47 Å and high Pauling electronegativity of 3.98)[4] a sizable reputation. This is amply evidenced in drug discovery by numerous breakthrough therapeutics[5] and recent estimates that 20% of prescribed or clinically administered pharmaceuticals contain at least one fluorine atom. Moreover, in general, 30–50% of the most profitable drugs (depending on the sales period) contain fluorine.[6]
Over the past years, many elegant strategies for the construction of organofluorine compounds have emerged,[7] including electrophilic and nucleophilic fluorination approaches or mild C–H and C–C bond “radical” fluorination tactics enabling the late-stage, site-selective monofluorination of complex molecules.[2,8–9] Notably, the latter approach has increasingly found use for the derivatization of potential drug analogues of interest.[10] One less explored route is the use of the Diels-Alder reaction for preparing organofluorine compounds where a combination of steric and electronic effects contribute to rate and stereoselection, with or without a catalyst.
In this context, several limitations to employing Diels-Alder reactions include the lower reactivity of fluoroalkene dienophiles and unpredictable stereochemical trends.[11] For example, Haufe et al. reported a 30-fold rate decrease in the reaction of fluorinated dienophile, p-fluoro-α-fluorostyrene with 1,3-diphenylisobenzofuran (1) relative to that with p-fluorostyrene.[12] Additionally, in the reaction of (1) with α-fluorostyrene (2), endo product (3a) was favored. Conversely, an inversion in stereoselectivity was reported upon reaction of (1) with β-fluorostyrene (4), favoring the formation of exo product (5b) (Scheme 1a). Similar is the observation that the Diels-Alder reaction of cyclopentadiene (6) with benzylacrylate dienophile (7) was selective towards the endo product (8a). Cycloaddition of its fluorinated counterpart α-fluorobenzyl acrylate (9) was 10.7 times slower and displayed a flip in stereoselectivity, favoring exo product (10b) as seen in Scheme 1b.[13] The origin of these effects has previously been studied;[14] one such example is a computational investigation by Chermette et al. exploring the fluorine substituent effect on Diels-Alder reactions of butenone with cyclopentadiene as well as Lewis acid activating effects on this reactivity. Stereoselectivity of fluorinated dienophiles was largely attributed to strain interactions along with hyperconjugation. Despite the observed electron-donating effects of fluorine that lowered reactivity, fluorinated substrates proved to be more electrophilic than non-fluorinated substrates.[15] In addition to this work, a limited number of studies have also attempted to outline diastereoselection associated with fluoroalkenes in Diels-Alder reactions, though many details remain undefined. These include: the degree to which the resonance-donating and inductively-withdrawing effects of fluorine attenuate reactivity, and the role of electrostatic effects in modulating reactivity. Accordingly, deeper insight into the many facets of this reactivity is warranted, e. g., the significance of deformation and strain energies, Pauli repulsive interactions, and diminished orbital and electrostatic interactions. Further, the poorly understood role of fluorine lone pair donation in this setting or the impact of electrostatic effects remains to be clarified. Against this backdrop, electrostatic catalysis and external electric fields or local electric fields with their capacity to stabilize polar transition states are well-known to catalyze chemical processes. This is abundantly shown and exploited in enzymes, while in organic synthesis, the use of electrostatic effects for catalysis is hampered by the problems of generating a sufficiently large field and orienting it in a solution-phase environment.[16] In addition, there is the frequent requirement to pre-install charged and/or polar functional groups. Even so, we speculated it might be possible to harness positive electrostatic interactions to temper fluorine’s innate tendency to suppress the rates of Diels-Alder reactions of fluorinated dienophiles, thus providing enhanced reaction rates.
Scheme 1.

Examples of previous experimental work probing the role of fluorine in Diels-Alder reactions. [4+2] cycloaddition reaction schemes of (a) 1,3-diphenylisobenzofuran (1) with α-fluorostyrene (Rα=F, Rβ=H) (2) and β-fluorostyrene (Rα=H, Rβ=F) (4),[12] (b) cyclopentadiene (6) with benzyl acrylate, R=H (7) and α-fluorobenzyl acrylate, R=F (9)[13]
In the present work using both computational and experimental tools, we disclose an in-depth study offering novel insights into the origin of reduced reaction rates of charged and non-charged fluorinated dienophiles and the factors controlling endo- vs. exo-selectivity in Diels-Alder reactions (Scheme 1). To arrive at this understanding, we employ density functional theory calculations, distortion/interaction-activation strain models, energy decomposition analysis, non-covalent interaction analysis and the unprecedented use of natural bond orbital analysis as an effective tool to gauge fluorine lone pair delocalization upon fluoroalkene reactivity as a dienophile. Beyond this important understanding, to expand the field of electrostatic catalysis, we explore the influence of oriented external-electric-field-effects (OEEFs) and local electric field effects. From this insight as a proof-of-concept, we experimentally document rate enhancement in Diels–Alder reactions of fluorinated dienophiles via electrostatic effects achieved through Brønsted acid activation. Critical to this electrostatic catalysis is the role of local internal oriented electric fields and attenuating fluorine lone pair donation. Collectively, this work offers more detailed mechanistic understanding pertinent to Diels-Alder reactions of fluorinated dienophiles providing valuable guidance for the synthesis of complex fluorinated scaffolds.
Results and Discussion
At the outset of our theoretical study exploring Diels-Alder reactions of fluorinated dienophiles, our focus turned to investigating the reactivity of 1,3-diphenylisobenzofuran (1) with α- and β-F-substituted styrene (2), (4) and styrene (11). Our selection of these fluorinated and non-fluorinated dienophiles derived from the intriguing variability in exo/endo-selectivity and relative reaction rates as reported by Haufe et al.[12] (Scheme 1a). Moreover, furan is a less common diene, and it adds the possibility of repulsive interactions with the oxygen that is absent in carbon analogs such as cyclopentadiene. From this analysis, we located transition states endo/exo-TS1, endo/exo-TS2 and endo/exo-TS3 with computed Gibbs free activation energies (ΔG‡) of 24–28 kcal/mol.
Evident as well were reduced reaction rates consistent with experiments and a slight 1.3 kcal/mol and 1.2 kcal/mol preference for endo product formation (12a and 3a) from the cycloaddition of styrene (11) and α-fluorostyrene (2), while exo selectivity was preferred for β-fluorostyrene by 0.5 kcal/mol (Scheme 2a,b). Defining these structures were asynchronous C–C bond forming distances of 2.06 Å to 2.10 Å at the alkene β-carbon. Repulsive fluorine and furan oxygen lone pair-lone pair interactions, favorable π-π-stacking interactions minimizing charge separation, and sterics also were contributing factors steering endo/exo selectivity. For example, destabilizing fluorine and furan oxygen lone pair-lone pair repulsion was absent from exo-TS2 owing to an anti-alignment between the electro-negative furan oxygen and alkene fluorine, while π-π-stacking was evident in both exo-TS2 and endo-TS2. Conversely, oxygen and fluorine lone pair-lone pair repulsion was present in endo-TS2 as seen from a short contact of 2.82 Å, placing the two atomic nuclei well inside the sum of their van der Waals radii (distance=2.99 Å).
Scheme 2.
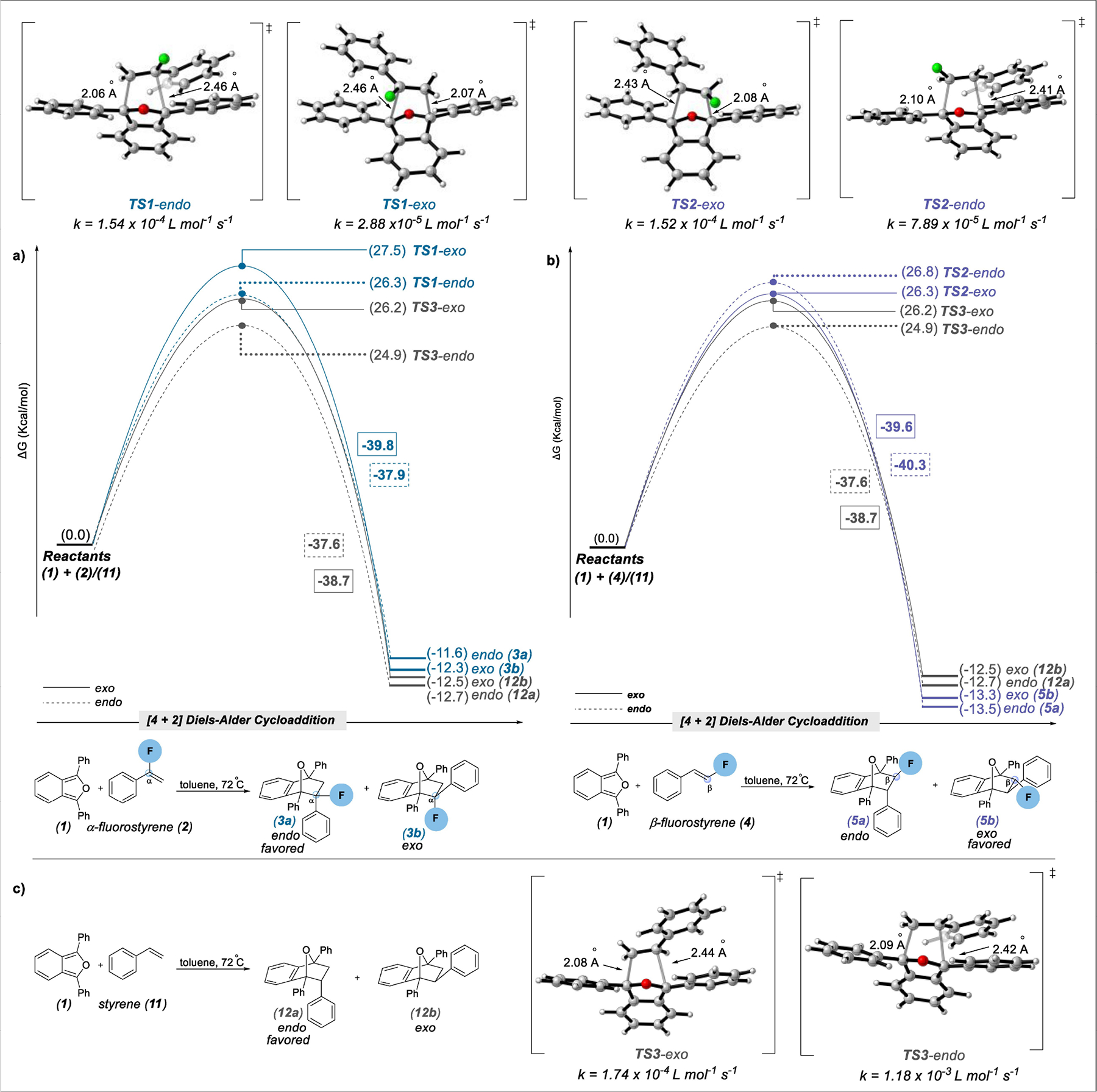
Reaction coordinate diagrams and schemes for Diels-Alder reactions of (a) 1,3-diphenylisobenzofuran (1) with α-fluorostyrene (2) and styrene (11), (b) 1,3-diphenylisobenzofuran (1) with β-fluorostyrene (4) and styrene (11). (c) Diels-Alder reaction scheme of 1,3-diphenylisobenzofuran (1) with styrene (11). All energies (kcal/mol) were computed at the ωB97X–D/6–311+G(d,p) (IEFPCM=Toluene)// ωB97X–D/6–31+G(d,p) level of theory.[17a,b]
Our attention next turned to investigating cyclopentadiene (6) with benzyl acrylate (7) and α-fluorobenzyl acrylate (9) to reveal asynchronous concerted transition states endo/exo-TS4 and endo/exo-TS5 with activation barriers of 31.8–34.3 kcal/mol (Scheme 3). Governing endo/exo-selectivity, much like in the previous cases, were sterics and a unique element of hydrogen bonding. Further, reaction rates were strongly correlated and dependent upon the presence or absence of fluorine and its ability to donate lone pair electron density into the dienophile alkene double bond. Absent from these structures, however, were repulsive fluorine and furan oxygen lone pair-lone pair interactions. Overall, these trends corroborate fluorine’s role in Diels-Alder reaction rate deceleration, increased activation energy barriers, and exo/endo stereoinversion.
Scheme 3.

Reaction coordinate diagrams for Diels-Alder reactions of cyclopentadiene (6) with benzyl acrylate (7) and α-fluorobenzyl acrylate (9). All energies (kcal/mol) were computed at the ωB97X–D/6–311+G(d,p) (IEFPCM=Toluene)// ωB97X–D/6–31+G(d,p) level of theory.[17a]
To further probe this reactivity, we conducted a non-covalent interaction (NCI) analysis[18] to reveal the presence of greater repulsive interactions in unfavorable exo-TS1 vs. preferred exo-TS2, while present in both structures was H-bonding interactions and π-π stacking (see SI). Moreover, crucial to these transition state structures, was fluorine lone pair donation to the adjacent dienophile alkene. To clarify this role, we applied second-order perturbation theory natural bond orbital (SOPT-NBO) donor-acceptor analysis. Inspection of this interaction for the various cases showed a large degree of fluorine lone pair donation to the dienophile π-systems (Flp→π*) amounting to −27.2 kcal/mol and −26.4 kcal/mol for α-fluorostyrene (2) and β-fluorostyrene (4). This donation by fluorine was also seen through fluorine lone pair orbital overlap with adjacent C5–C6 orbitals for both fluorinated dienophiles (Figure 1a). Further still, NBO donor-acceptor interactions of α-fluorostyrene cycloaddition transition states endo/exo-TS1 displayed similar fluorine lone pair donation into the dienophile π-systems (C5-C6), with −0.2 kcal/mol greater donation observed in the unfavored exo-TS1 relative to endo-TS1 (Figure 1b). Interestingly, in both transition states endo/exo-TS1 as well was significant fluorine lone pair donation into antibonding alkene π-orbitals (C2–C5), contributing −19.8 kcal/mol and −18.4 kcal/mol. Likewise, as seen in Figure 1c, analysis of β-fluorostyrene transition states endo/exo-TS2, show similar fluorine lone pair donation with a −0.1 kcal/mol greater donation into the dienophile π-systems (C5–C6) of favored exo-TS2 relative to endo-TS2. Furthermore, analysis of endo/exo-TS2, also indicated significant fluorine lone pair donation into antibonding orbitals (C3–C6) of −17.0 kcal/mol for exo-TS2 and −15.0 kcal/mol for endo-TS2. Collectively these NBO results offer the first quantitative insights into the significance of fluorine lone pair donation into the alkene π-bond of styrene dienophiles as a deactivating effect in Diels-Alder reactions.
Figure 1.
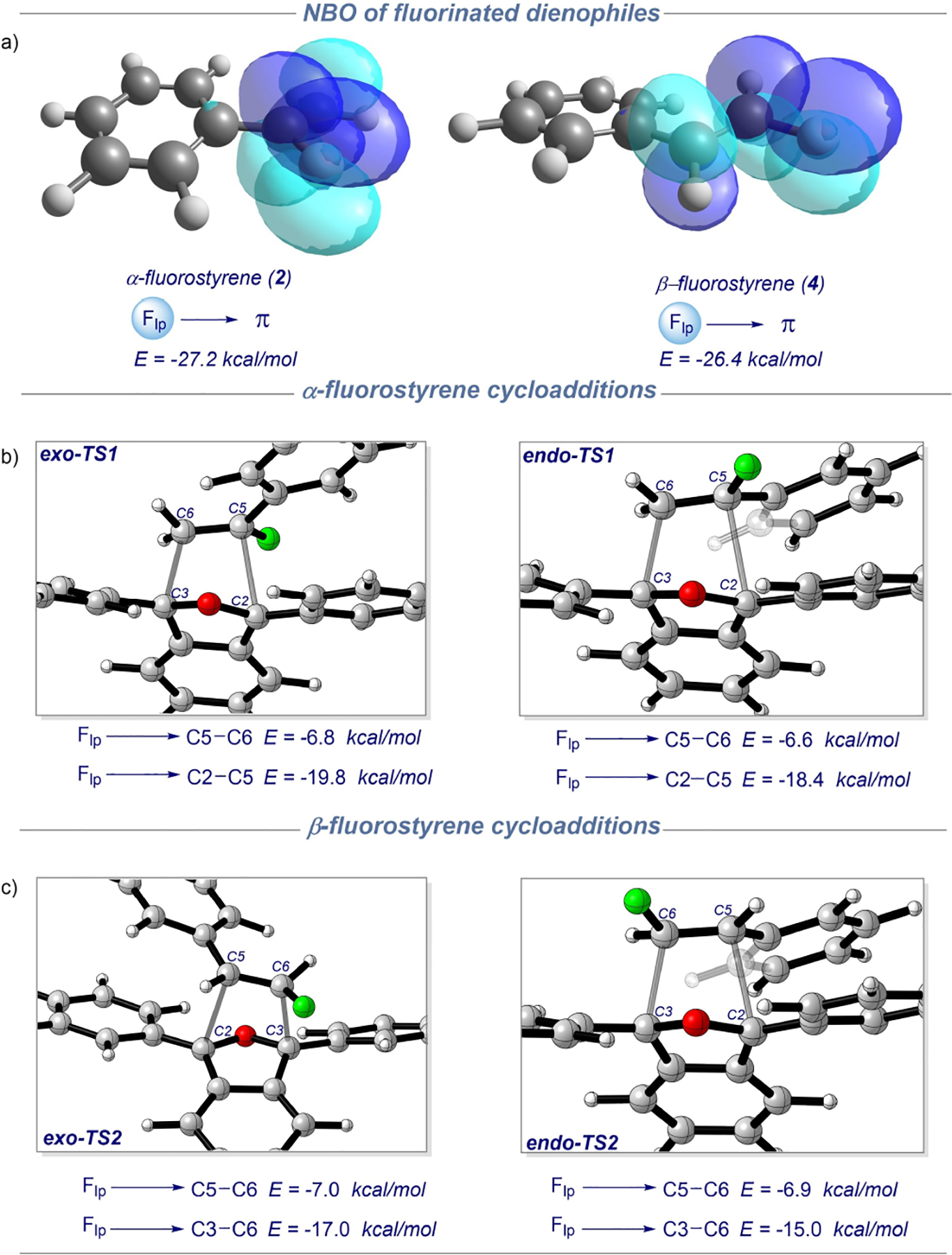
Second-order perturbation theory natural bond orbital (SOPT-NBO) donor-acceptor analysis of (a) α-fluorostyrene (2) and β-fluorostyrene (4) dienophiles. (b) α-fluorostyrene cycloaddition transition states exo/endo-TS1 (c) β-fluorostyrene cycloaddition transition states exo/endo-TS2.
These [4+2] cycloadditions of 1,3-diphenylisobenzofuran (1) and cyclopentadiene (6) with (fluoro)styrenes (2, 4, 11) and (fluoro)benzyacrylates, (7, 9) were next analyzed with the Distortion/Interaction–Activation Strain Model (D/I-ASM).[19] This analysis deconstructs the electronic activation energies into distortion and interaction energies along the intrinsic reaction coordinate (IRC) C…C bond forming distance. Distortion energy is the energy required to geometrically deform the ground state geometries of the reactants into transition state geometries. Meanwhile, the interaction energy is the energy exchange that results from the interaction of the reactants during bond formation. Shown in Figure 2a, are the distortion/interaction analyses for the Diels-Alder reactions of 1,3-diphenylisobenzofuran (1) with (fluoro)styrene (2, 4, 11). The distortion energy, ΔEstrain (ζ) curves for transition states exo/endo-TS1 and exo/endo-TS3 show little variation between endo/exo orientations. This is while the interaction energy ΔEint (ζ) curves show greater stabilization for endo-TS1 relative to exo-TS1, with a −6.1 kcal/mol difference at the critical bond forming points. Endo-TS3 also displays a slightly stabilized ΔEint (ζ) compared to exo-TS3. For β-fluorostyrene (4) cycloadditions, ΔEstrain (ζ) energy curves display marginally larger strain associated with unfavored endo-TS2 relative to favored exo-TS2. Simultaneously, ΔEint (ζ) slightly stabilizes the exo-TS2 orientation by −1.3 kcal/mol at the critical bond forming points, thus governing exo stereoselectivity of this Diels–Alder reaction. Notably, β-fluorostyrene (4) cycloadditions exo/endo-TS2 display overall greater ΔEstrain (ζ) contribution compared to non-fluorinated exo/endo-TS3 (Figure 2a). With respect to the cycloadditions of α-fluorobenzyl acrylate (9) with cyclopentadiene (6), similar ΔEint (ζ) curves were found for exo/endo-TS5 conformations. However, marginally larger destabilizing strain energy ΔEstrain(ζ) is observed in the unfavored endo-TS5 structure compared to the exo-TS5 approach. By comparison, benzyl acrylate (7) cycloadditions display parallel trends, with the favored endo-TS4 pathway having reduced deformation energy ΔEstrain (ζ) relative to exo-TS4 and analogous stabilization of ΔEint(ζ) for exo/endo-TS4. Additionally, fluorinated exo/endo-TS5 cycloadditions display greater ΔEstrain(ζ) in relation to their non-fluorinated counterparts (exo/endo-TS5) (Figure 2b). Notably, similar behavior has been observed in many other pericyclic reactions.[20] Overall, with the aim of revealing the origin of Diels-Alder rate deceleration of fluorinated dienophiles, our D/I-ASM models shed light on the ability of fluorine to impart greater geometric deformation of reagents to achieve respective transition state conformations.
Figure 2.

Computed D/I-ASM energies ΔE for [4+2] cycloadditions of (fluoro)styrenes (2, 4, 11) with diphenylisobenzofuran (1) and cycloadditions of benzyl acrylate (7) and α-fluorobenzyl acrylate (9) with cyclopentadiene (6). All energies (kcal/mol) were computed at ωB97X–D/6–31+G(d,p) level of theory.[17a]
Next, we implemented an energy decomposition analysis (EDA) method.[20] Employing this analysis, the instantaneous interaction energies (ΔEint (ζ)) were mapped out along the intrinsic reaction coordinate (IRC) plots from reactants to transition states TS1-TS5. ΔEint (ζ) was partitioned into four critical components: (1) Pauli repulsion (ΔEPauli), (2) electrostatic interactions (ΔVelstat), (3) orbital interactions (ΔEoi), and (4) dispersion energy(ΔEdisper).[21,22] With an initial focus on the exo- and endo-cycloadditions of cyclopentadiene (6) and α-fluorobenzyl acrylate (9) via exo/endo-TS5, similar dispersion energies and the rapid increase in repulsive Pauli interactions with a progressive enhancement in attractive electrostatic and orbital interactions were observed (Figure S55). Markedly, the latter components, i. e., attractive, electrostatic, and orbital interactions, had a role in shifting reactivity towards exo-product formation, while cyclopentadiene (6) and benzyl acrylate (7) selectivity was reversed in favor of endo-product formation. Despite greater, electrostatic and orbital attractive interactions present in exo-TS4, repulsive Pauli interactions were slightly reduced for endo cycloaddition by endo-TS4 (Figure S54). Similarly, the Diels-Alder reactions of diphenylisobenzofuran (1) with α-fluorostyrene (2) β-fluorostyrene (4) and styrene (11) revealed little variation in dispersion interactions (Figure S51–S53). In contrast, Pauli repulsion was marginally greater for α-fluorostyrene (2) cycloaddition by endo-TS1 and flipped for β-fluoroethylene (4) transition state exo-TS2. Meanwhile, α-fluorostyrene cycloaddition structure endo-TS1 revealed slightly greater orbital and electrostatic stabilizing interactions relative to unfavored exo-TS1. For β-fluorostyrene cycloadditions, the favored transition state exo-TS2 displayed similar trends whereby more extensive electrostatic stabilizing interactions were observed relative to endo-TS2. Finally, analysis of styrene (11) cycloaddition transition states exo/endo-TS3 showed marginally more stabilizing attractive orbital and electrostatic interactions for endo-TS3, with dispersion interactions and Pauli repulsion relatively equivalent for both exo/endo.
Notably, these computed trends align with previous experimental findings and show a clear inversion in endo- vs. exo-selectivity along with rate deceleration for fluorine-substituted alkenes. The basis for this intriguing reactivity pattern is attributed to increased alkene π-electron density brought about by fluorine lone pair donation, leading to greater Pauli repulsion manifesting in decelerated rates of [4+2] cycloaddition. Of course, this reactivity trend contrasts the electron-withdrawing character of fluorine and conventional logic, suggesting that a LUMO-lowering effect might arise, providing accelerated reaction rates.[23]
To further explore elements of electrostatic catalysis affecting these Diels-Alder reactions, our focus turned to applying electric field conditions. In terms of oriented external electric fields (OEEFs) and local electric fields in organic synthesis, the seminal studies by Wilcox et al. in the 1990s, investigating ion pair effects on reaction rates and selectivity demonstrated how electrostatic fields could influence the rates of reactions with polar transition states,[24] including cycloadditions.[24c,g] In more recent times, Shaik and co-workers, as well as others through theoretical studies, have shown the potential of electrostatic catalysis to enhance reaction rates and selectivities,[25] including the use of ion pairs to alter gold-catalyzed cyclizations[25g,h] and the application of oriented electric fields on Diels–Alder and other reactions.[26] Additionally, Maruoka and co-workers, in a recent report, disclosed the application of tetraalkylammonium and trialkylsulfonium salts as catalysts for the aza-Diels–Alder reactions between imines and Danishefsky’s diene, which was computationally studied by Houk et al.[27,28] Opposed to using ionic catalysts to generate intense local oriented fields to promote reactivity are several excellent substrate-based examples,[24,29] including an eloquent report by Coote et al. of a computational approach showing dienophiles furnished with non-π-conjugated charged functional groups dramatically altering the rate, regioselectivity, and diastereoselectivity of a Diels–Alder reaction.
Accordingly, electric fields were oriented along the reaction axis of Diels-Alder reactions in both the positive (direction of electron flow) and negative (against electron flow) directions and with varying field magnitudes (Fz= ±0.01 au, ±0.005 au and ±0.008 au). We initially evaluated the effects of OEEF on the reactivity of 1,3-diphenylisobenzofuran (1) with α-fluorostyrene (2), β-fluorostyrene (4), and styrene (11). Intriguingly, both increasing and decreasing the OEEF field strength (Fz) displayed no change in endo/exo stereoselectivity; however, it appeared to have an inhibitory role on α-fluorostyrene (2) and styrene (11) as seen in the increase in activation energies of exo/endo-TS1 and endo/exo-TS3 (Table S15 and S17). Conversely, a positive OEEF of +0.005 au appeared to have a catalytic role on the cycloaddition of β-fluorostyrene (4), evident by a decrease in activation energy of endo/exo-TS2 while displaying a minimal reversal in endo/exo stereoselectivity (Scheme 4).
Scheme 4.

Reaction coordinate diagram of Diels-Alder reactions of 1,3-diphenylisoenzofuran (1) and β-fluorostyrene (4) under Oriented External Electric Field (OEEF) conditions and non-OEEF conditions. All structures are calculated at the ωB97X–D/6–31+G(d,p) level of theory.[17]
In mapping out the reactivity of cyclopentadiene (6) with benzyl acrylate (7) and α-fluorobenzyl acrylate (9), catalytic and inhibitory trends were also observed. Notably, it was found that increasing the positive OEEF strength (0.005 au, 0.008 au and 0.010 au) decreased the activation energy barriers of endo/exo stereoisomers for both fluorinated and nonfluorinated cycloadditions (endo/exo-TS4 and endo/exo-TS5) as noted in Table S18 and S19. Upon increasing OEEF magnitude, the endo and exo selective transition states for benzyl acrylate cycloadditions endo/exo-TS4 exhibited a reduction in activation free energy up to 6.2 kcal/mol and 7.0 kcal/mol compared to non-OEEF models. Interestingly, α-fluorobenzyl acrylate cycloadditions endo/exo-TS5 displayed a larger reduction in activation free energy up to 6.6 kcal/mol and 7.7 kcal/mol, indicating a greater catalytic activity of OEEFs in the presence of fluorine (Table S18 and S19). In undertaking this computational investigation, we have arrived at a means for selectively modulating Diels-Alder reactivity with fluorinated dienophiles and in doing so, offer direction for advancing “smart reagents” - reagents that are responsive to electric field stimuli. Crucial to this reagent and well-known is charge with its ability to induce local internal electric fields.[30]
Informed by our computational studies, we finally sought to develop a fluorinated dienophile with unconventional reactivity on par or superior to its non-fluorinated counterpart. Guiding this enterprise were several criteria exhumed from our studies, including (1) attenuation of fluorine lone pair donation to the dienophile alkene, (2) generation of local internal oriented electronic fields linked to favorable LUMO lowering effects, and (3) activation by electrostatic interactions and/or potential Brønsted acid catalysis for “switching-on” reactivity. In fulfilling these conditions, charged dienophile (13) was envisioned as the easily obtained conjugate acid of (14). Innate to this analog, as well, was the possibility of engaging in intramolecular fluorine hydrogen bonding (Scheme 5), thus adding to the growing entries of recent research revealing fluorine’s ability to participate in hydrogen bonding, as shown for amide N–H groups and acidic side chains in proteins.[31] Further were prospects for C–F interactions (e. g., F···π and/or hydrophobic arrangements offering favorable dispersion forces) and cationic charge as factors contributing to rate acceleration.[32]
Scheme 5.

(a-d) Computationally predicted reactions of cyclopentadiene (6) with (a) 2-vinyl pyridine (15), (b) vinyl pyridinum (16), (c) α-fluorovinyl pyridine (14) and (d) α-fluorovinyl pyridinium (13). (e) NBO analysis of α-fluorovinyl pyridinium (13), (f) NCI analysis of α-fluorovinyl pyridinium cycloadditions endo/exo-TS6, (g) QTAIM analysis of compound (13) depicting electron density at the bond critical point (ρbcp). All energies (kcal/mol) were computed at the (SMD=Acetonitrile) ωB97X–D/6–311+G(d,p)// ωB97X–D/6–31+G(d,p) level of theory.[17a,c] (h-k) Experimental reactions of (6) with dienophiles (14 and 15) under thermal and catalytic conditions.
Indeed, calculations probing the [4+2] cycloadditions of dienophiles (α-fluoro)-2-vinyl pyridine (14 and 15) and (α-fluoro)-2-vinyl pyridinium (13 and 16) with cyclopentadiene (6), revealed enhanced reactivity exhibited using charged substrates (13 and 16). From our analysis, evident is a Brønsted acid activation effect shown by the 13.4 kcal/mol and 12.7 kcal/mol decrease in activation energy barriers of endo/exo-TS7 upon protonation of dienophile (15) to (16) (Scheme 5a, b). Further, consistent with our predictions a 13.9 kcal/mol and 13.0 kcal/mol decrease in activation energy was observed for endo/exo-TS9 cycloadditions with α-fluoro-2-vinyl pyridinium (13) relative to α-fluoro-2-vinyl pyridine (14) along with an inversion in stereoselectivity (Scheme 5c, d). Interestingly, neutral substrates displayed an anti-orientation with fluorine directed away from pyridine nitrogen to alleviate lone pair-lone pair repulsion, while charged substrates preferred an inverted syn-orientation with fluorine nearby the pyridinium N–H bond. SOPT-NBO analysis was conducted to shed light on the latter syn-orientation to reveal a donation of fluorine lone pair into the σ*-orbital of the N–H bond (Flp→σ*N-H) providing −3.7 kcal/mol of stabilization (Scheme 5e). Furthermore, NCI analysis of α-fluoro-2-vinyl pyridinium cycloadditions (endo/exo-TS9) revealed the presence of weakly attractive interactions, including F···H contacts as seen by the green surface regions (Scheme 5f). Next, we turned to quantum theory of atoms in molecules (QTAIM) to determine the electron density at the bond critical point (BCP), or the ρbcp value of these F···H interactions of (13),[33] which indicated the presence of a closed shell, moderately strong halogen bond (ρbcp= 0.0206 au) as seen in Scheme 5g. Overall, the transformations of charged dienophile substrates results in greater stabilization of cycloaddition transition states.
To corroborate these computational results, we next turned to experimental methods that investigated the thermal reactions of 2-vinyl pyridines, expecting to recapitulate the deactivating effect of fluorine in cycloadditions with cyclopentadiene. The reaction of 2-vinyl pyridine (15) with cyclopentadiene required elevated temperature (150°C, microwave) but led to cycloadducts (17) with 3:1 endo:exo selectivity in 70% yield. By comparison, α-fluoro-2-vinyl pyridine (14) produced (18) in only 23% yield with the standard 1 hour reaction time, and a reversed selectivity of 1:3.5 endo/exo-18. A brief study of Lewis and Brønsted acids led to the finding that triflimidic acid (Tf2NH) provided substantial rate acceleration for both substrates. 2-Vinyl pyridinium (16) was generated in situ from 2-vinyl pyridine (15), using 50 mol% catalyst loading and reacted with cyclopentadiene at room temperature to afford bicycle (19) in 80% yield with 30:1 endo/exo selectivity, and with notable improvement relative to its thermal counterpart. α-Fluoro-2-vinyl pyridinium (13) was generated from α-fluoro-2-vinyl pyridine (14) in a similar fashion and exhibited accelerated reactivity, producing (20) in 55% yield, but again slower overall relative to its non-fluorinated counterpart (16). Interestingly, selectivity inverted to 2:1 endo/exo-20 with the use of Brønsted acid to accelerate the reaction. In aligning with our calculated predictions, experimental results display stereoinversion and phenomenal rate enhancement of Diels-Alder reactivity through charge-enhanced fluorinated dienophiles. As such, these findings confirm the capacity of Brønsted acid activation to counteract the limitations brought about by fluorine.
Conclusions
In closure, we have reported an in-depth study exploring the role of fluorinated dienophiles within Diels-Alder reactivity. Our results elucidate that rate and stereochemical selectivity is indeed modified in the presence of fluorine leading to reaction rate deceleration, increased activation energy barriers, and endo/exo stereochemical inversion, collectively, aligning with the earlier observation of Haufe.[12,13] In uncovering the origin of this effect, we found fluorine substitution results in larger geometric deformation of starting reagent and increased Pauli repulsive interactions within transition states. Our analysis additionally revealed significant fluorine lone pair donation into the dienophile alkene π-bond contributed to deactivation of the dienophile. Further, electric field analyses predicted modulation of Diels-Alder reactivity in the presence of an oriented external electric field. Building upon these findings, we designed a fluorinated ‘smart reagent’ dienophile with the capacity to undergo Brønsted acid activation resulting in charge enhanced reactivity leading to exquisite rate enhancement and stereoselective inversion. Overall, these results provide a means of overcoming the conventional rate decelerating effects of fluorine and controlling stereoselectivity within Diels-Alder reactivity, to better access fluorinated molecular scaffolds.
Supplementary Material
Supporting information for this article is available on the WWW under https://doi.org/10.1002/ejoc.202401203
Acknowledgements
T.D. thanks the Natural Sciences and Engineering Research Council of Canada for Discovery Grants (RGPIN-2019-04205). Computations were carried out using facilities at Digital Research Alliance of Canada; this research was enabled in part by support provided by SHARCNET (Shared Hierarchical Academic Research Computing Network) and Compute/Calcul Canada and the Digital Research Alliance of Canada. J.N.J. is grateful to the National Institute of General Medical Sciences (NIH GM084333, GM063557) for financial support, and the VUSRP program for summer undergraduate research.
Footnotes
Conflict of Interests
The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- [1].a) Li W-B, Cheng Y-ZD-H, Yang, Liu Y–W, Han B-H, Macromol. Rapid Commun 2023, 44, 2200778; [DOI] [PubMed] [Google Scholar]; b) Kirsch P, Lenges M, Kühne D, Wanczek KP, Eur. J. Org. Chem 2005, 2005, 797–802; [Google Scholar]; c) Wang Q, Song H, Wang Q, Chin. Chem. Lett 2022, 33, 626–642; [Google Scholar]; d) Meanwell NA, J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- [2].Dehnen S, Schafer LL, Lectka T, Togni A, Inorg. Chem 2021, 60, 17419–17425. [DOI] [PubMed] [Google Scholar]
- [3].a) Shah P, Westwell AD. J Enzyme Inhib. Med. Chem 2007, 22, 527–540; [DOI] [PubMed] [Google Scholar]; b) Vara BA, Mayasundari A, Tellis CJ, Danneman MW, Arredondo V, Davis TA, Min J, Finch K, Guy RK, Johnston RJN, J. Org. Chem 2014, 79, 6913–6938; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Davis TA, Johnston JN, Chem. Sci 2011, 2, 1076–1079; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Vara BA, Johnston JN, J. Am. Chem. Soc 2016, 138, 13794–13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shannon RD, Acta Cryst. 1976, A32, 751–767. [Google Scholar]
- [5].a) Dombrowski AW, Gesmundo NJ, Aguirre AL, Sarris KA, Young JM, Bogdan AR, Martin MC, Gedeon S, Wang Y, ACS Med. Chem. Lett 2020, 11, 597–604; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blakemore DC, Castro L, Churcher I, Rees DC, Thomas AW, Wilson DM, Wood A, Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- [6].a) Mei H, Remete AM, Zou Y, Moriwaki H, Fustero S, Kiss L, Soloshonok VA, Han J, Chin. Chem. Lett 2020, 31, 2401–2413; [Google Scholar]; b) O’Hagan D, J. Fluor. Chem 2010, 131, 1071–1081. [Google Scholar]
- [7].a) Deng Z, Padalino MA, Jan JEL, Park S, Danneman MW, Johnston JN, J. Am. Chem. Soc 2024, 146, 1269–1275; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bing JA, Johnston JN, Org. Lett 2023, 25, 950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Britton R, Gouverneur V, Lin JH, Meanwell M, Ni C, Pupo G, Xiao JC, Hi J, Nat Rev Methods Primers. 2021, 1, 47; [Google Scholar]; b) Rajabalinia S, Hoford S, Dudding T, ACS Omega 2024, 9, 21152–21163; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang M, Rowshanpour R, Guan L, Ruskin J, Nguyen PM, Wang Y, Zhang QA, Liu R, Ling B, Woltornist R, Stephens AM, Prasad A, Dudding T, Lectka T, Pitts CR, J. Am. Chem. Soc 2023. 145, 22442–22455; [DOI] [PubMed] [Google Scholar]; d) Holt E, Ruskin J, Garrison NG, Vemulapalli S, Lam W, Kiame N, Henriquez N, Borukhova F, Williams J, Dudding T, Lectka T, J . Org. Chem 2023, 88, 17538–17543; [DOI] [PubMed] [Google Scholar]; e) Capilato JN, Siegler MA, Rowshanpour R, Dudding T, Lectka T, J. Org. Chem 2021, 86, 1300–1307; [DOI] [PubMed] [Google Scholar]; f) Dilman AD Levin VV, Mendeleev Commun. 2015, 25, 239–244; [Google Scholar]; g) Rozatian N, Hodgson DRW, Chem. Comm 2021, 57, 683–712. [DOI] [PubMed] [Google Scholar]
- [9].Crocker MS, Foy H, Tokumaru K, Dudding T, Pink M, Johnston JN, Chem. 2019, 5, 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Szpera R, Moseley DFJ, Smith LB, Sterling AJ, Gouverneur V, Angew. Chem. Int. Ed 2019, 58, 14824–14848; [DOI] [PubMed] [Google Scholar]; b) Yan H, Zhu C, Sci. China Chem 2017, 60, 214–222; [Google Scholar]; c) Nobile E, Castanheiro T, Besset T, Angew. Chem 2021, 133, 12278–12299. [DOI] [PubMed] [Google Scholar]
- [11].For studies of similar diastereodivergence, see:; a) Struble TJ, Smajlagic TI, Foy H, Dudding T, Johnston JN, J. Org. Chem 2021, 86, 15606–15617; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sprague DJ, Singh A, Johnston JN, Chem. Sci 2018, 9, 2336–2339; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Review: Krautwald S, Carreira EM, J. Am. Chem. Soc 2017, 139, 5627–5639. [DOI] [PubMed] [Google Scholar]
- [12].Ernet E, Haufe G, Tetrahedron Lett. 1996, 37, 7251–7252. [Google Scholar]
- [13].Essers M, Mück-Lichtenfeld C, Haufe G, J. Org. Chem 2002, 67, 4715–4721. [DOI] [PubMed] [Google Scholar]
- [14].a) Dyan OT, Andreev RV, Zaikin PA, J. Fluor. Chem 2021, 250, 109859; [Google Scholar]; b) Ernet T, Maulitz AH, Würthwein E-U, Haufe G, J. Chem. Soc., Perkin Trans 1 2001, 16, 1929–1938; [Google Scholar]; c) Essers M, Ernet T, Haufe G, J. Fluor. Chem 2003, 121, 163–170; [Google Scholar]; d) Su R, Xie K, Liang Y, Houk KN, Liu F, J. Org. Chem 2023, 88, 893–900. [DOI] [PubMed] [Google Scholar]
- [15].Merzoud L, Saal A, Moussaoui R, Ouamerali O, Morell C, Chermette H, Phys. Chem. Chem. Phys 2018, 20, 16102–16116. [DOI] [PubMed] [Google Scholar]
- [16].a) Ciampi S, Darwish N, Aitken HM, Díez-Perez I, Coote ML, Chem. Soc. Rev 2018, 47, 5146–5164; [DOI] [PubMed] [Google Scholar]; b) Shaik S, Mandal D, Ramanan R, Nat. Chem 2016, 8, 1091–1098; [DOI] [PubMed] [Google Scholar]; c) Shaik S, Ramanan R, Danovich D, Mandal D, Chem. Soc. Rev 2018, 47, 5125–5145. [DOI] [PubMed] [Google Scholar]
- [17].a) Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP,, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F,, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ, Gaussian, Inc., Wallingford CT, 2016;; b) Tomasi J, Mennucci B, Cancès E, J. Mol. Struct.: THEOCHEM 1999, 464, 211–226; [Google Scholar]; c) Marenich AV, Cramer CJ, Truhlar DG, J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- [18].a) Contreras-García J, Johnson ER, Keinan S, Chaudret R, Piquemal J-P, Beratan DN, Yang W, Chem J. Theory Comput. 2011, 7, 625–632; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu T, Chen F, J. Comput. Chem 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
- [19].Bickelhaupt FM, Houk KN, Angew. Chem., Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Fernández I, Bickelhaupt FM, Cossío FP, Chem. – Eur. J 2009, 15, 13022–13032. [DOI] [PubMed] [Google Scholar]; b) Fernández I, Cossío FP, Curr. Org. Chem 2010, 14, 1578–1585; [Google Scholar]; c) Fernández I, Cossío FP, Bickelhaupt FM, J. Org. Chem 2011, 76, 2310–2314; [DOI] [PubMed] [Google Scholar]; d) Fernández I, Bickelhaupt FM J. Comput. Chem 2012, 33, 509–516; [DOI] [PubMed] [Google Scholar]; e) Fernández I, Bickelhaupt FM, Cossío FP, Chem. – Eur. J 2012, 18, 12395–12403; [DOI] [PubMed] [Google Scholar]; f) Nieto Faza O, Silva López C, Fernández I, J. Org. Chem 2013, 78, 5669–5676. [DOI] [PubMed] [Google Scholar]
- [21].Hopffgarten M, von Frenking G, WIREs Comput. Mol. Sci 2011, 2, 43–62. [Google Scholar]
- [22].Jerabek P, Schwerdtfeger P, Frenking G, J. Comput. Chem 2018, 40, 247–264. [DOI] [PubMed] [Google Scholar]
- [23].Bing JA, Schley ND, Johnston JN, Chem. Sci 2022, 13, 2614–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Smith PJ, Wilcox CS, J. Org. Chem 1990, 55, 5675–5678; [Google Scholar]; b) Smith PJ, Wilcox CS, Tetrahedron 1991, 47, 2617–2628; [Google Scholar]; c) Smith PJ, Soose DJ, Wilcox CS, J. Am. Chem. Soc 1991, 113, 7412–7414; [Google Scholar]; d) Smith PJ, Reddington MV, Wilcox CS, Tetrahedron Lett. 1992, 33, 6085–6088; [Google Scholar]; e) Smith PJ, Kim E.-i., Wilcox CS, Angew. Chem. Int. Ed. Engl 1993, 32, 1648–1650; [Google Scholar]; f) Kim E.-i., Paliwal S, Wilcox CS, J. Am. Chem. Soc 1998, 120, 11192–11193; [Google Scholar]; g) Raposo C, Wilcox CS, Tetrahedron Lett. 1999, 40, 1285–1288. [Google Scholar]
- [25].a) Shaik S, de Visser SP, Kumar D, J. Am. Chem. Soc 2004, 126, 11746–11749; [DOI] [PubMed] [Google Scholar]; b) Hirao H, Chen H, Carvajal MA, Wang Y, Shaik S, J. Am. Chem. Soc 2008, 130, 3319–3327; [DOI] [PubMed] [Google Scholar]; c) Lai W, Chen H, Cho K-B, Shaik S, J. Phys. Chem. Lett 2010, 1, 2082–2087; [Google Scholar]; d) Meir R, Chen H, Lai W, Shaik S, ChemPhysChem. 2010, 11, 301–310; [DOI] [PubMed] [Google Scholar]; e) Gryn’ova G, Coote ML, J. Am. Chem. Soc 2013, 135, 15392–15403; [DOI] [PubMed] [Google Scholar]; f) Gryn’ova G, Marshall DL, Blanksby SJ, Coote ML, Nat. Chem 2013, 5, 474–481; [DOI] [PubMed] [Google Scholar]; g) Lau VM, Gorin CF, Kanan MW, Chem. Sci 2014, 5, 4975–4979; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Lau VM, Pfalzgraff WC, Markland TE, Kanan MW, J. Am. Chem. Soc 2017, 139, 4035–4041; [DOI] [PubMed] [Google Scholar]; i) Wang Z, Danovich D, Ramanan R, Shaik S, J. Am. Chem. Soc 2018, 140, 13350–13359. [DOI] [PubMed] [Google Scholar]
- [26].For recent experimental examples, see:; a) Aragones AC, Haworth NL, Darwish N, Ciampi S, Bloomfield NJ, Wallace CG, Diez-Perez I, Coote ML, Nature 2016, 531, 88–91; [DOI] [PubMed] [Google Scholar]; b) Che F, Gray JT, Ha S, McEwen J-S, ACS Catal. 2017, 7, 551–562; [Google Scholar]; c) Akamatsu M, Sakai N, Matile S, J. Am. Chem. Soc 2017, 139, 6558–6561; [DOI] [PubMed] [Google Scholar]; d) Huang X, Tang C, Li J, Chen L-C, Zheng J, Zhang P, Le J, Li R, Li X, Liu J, Yang Y, Shi J, Chen Z, Bai M, Zhang H-L, Xia H, Cheng J, Tian Z-Q, Hong W, Sci. Adv 2019, 5, No. eaaw3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].He CQ, Lam CC, Yu P, Song Z, Chen M, Lam Y, Chen S, Houk KN, J. Org. Chem 2020, 85, 2618–2625. [DOI] [PubMed] [Google Scholar]
- [28].a) Kumatabara Y, Kaneko S, Nakata S, Shirakawa S, Maruoka K, Chem. Asian J 2016, 11, 2126–2129; [DOI] [PubMed] [Google Scholar]; b) Kaneko S, Kumatabara Y, Shimizu S, Maruok S Shirakawa K, Chem. Commun 2017, 53, 119–122. [DOI] [PubMed] [Google Scholar]
- [29].a) Aitken HM, Coote ML, Phys. Chem. Chem. Phys 2018, 20, 10671–10676; [DOI] [PubMed] [Google Scholar]; b) Blyth MT, Coote MA, J. Org. Chem 2019, 84, 1517–1522. [DOI] [PubMed] [Google Scholar]
- [30].Blyth MT, Coote ML, in Effects of Electric Fields on Structure and Reactivity: New Horizons in Chemistry, (Eds.: Shaik S and Stuyver T), The Royal Society of Chemistry, 2021, pp. 119–146. [Google Scholar]
- [31].a) Pietruś W, Kurczab R, Kafel R, Machalska E, Kalinowska-Tłuścik J, Hogendorf A, Żylewski M, Baranska M, Bojarski AJ, Spectrochim. Acta Part A 2021, 252, 1386–1425; [DOI] [PubMed] [Google Scholar]; j) Dalvit C, Invernizzi C, Vulpetti A, Chem. Eur. J 2014, 20, 11058–11068. [DOI] [PubMed] [Google Scholar]
- [32].He CQ, Lam CC, Yu P, Song Z, Chen M, Lam Y, Chen S, Houk KN, J. Org. Chem 2020, 85, 2618–2625. [DOI] [PubMed] [Google Scholar]
- [33].a) Bader RFW, Atoms in Molecules: A Quantum Theory, Oxford University Press: Oxford, 1990; [Google Scholar]; b) Popelier P, Atoms in Molecules, An Introduction, Prentice Hall: Harlow, 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.