

Abstract
About one-third of the MA protein in Rous sarcoma virus (RSV) is phosphorylated. Previous analyses of this fraction have suggested that serine residues 68 and 106 are the major sites of phosphorylation. As a follow-up to that study, we have characterized mutants which have these putative phosphorylation sites changed to alanine, either separately or together. None of the substitutions (S68A, S106A, or S68/106A) had an effect on the budding efficiency or infectivity of the virus. Upon examination of the 32P-labeled viral proteins, we found that the S68A substitution did not affect phosphorylation in vivo at all. In contrast, the S106A substitution prevented all detectable phosphorylation of MA, suggesting that there is only one major site of phosphorylation in MA. We also found that the RSV MA protein is phosphorylated on tyrosine, but the amount was low and detectable only with large numbers of virions and an antibody specific for phosphotyrosine.
Rous sarcoma virus (RSV) is known to contain four phosphoproteins: the β subunit of reverse transcriptase (RT), integrase (IN), matrix (MA), and nucleocapsid (NC) proteins (10, 16, 18, 26). The latter two are initially synthesized as a part of the Gag polyprotein, where they provide functions needed for the process of budding from the plasma membrane (for a review, see reference 31). Shortly after a particle is formed, these proteins (as well as the other products of Gag) are released from the precursor by action of the viral protease (PR), allowing the particles to mature and become infectious.
About one-third of the MA protein found within RSV is phosphorylated (10). This causes MA to migrate as a doublet during sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the slower-migrating band being the phosphorylated species (10). While phosphoamino acid analysis by Lai demonstrated that a majority of the phosphate is attached to serine (18), it was not until mapping studies by Leis et al. that the putative sites of phosphorylation were identified as serine residues 68 and 106 (20). However, a biological role for this modification has not been reported.
We have attempted to confirm the putative sites of phosphorylation in MA and investigate their role in RSV replication by constructing mutants which have either one or both changed to alanine. Unexpectedly, only the serine 106 substitution abolished phosphorylation in the mature MA protein. Moreover, this phosphorylation was found to be nonessential for both budding and infectivity. Additionally, a very small amount of tyrosine phosphorylation was identified on the wild-type MA protein, leaving open the possibility that other phosphorylation events might play a role in virus replication.
MATERIALS AND METHODS
DNAs and cells.
The gag gene used for this study is from pATV-8, an infectious molecular clone of the RSV Prague C genome (27). M13mp19P12, a M13mp19 recombinant containing this gag gene, was used for oligonucleotide-directed mutagenesis and has been described previously (12). RSV gag alleles were expressed in COS-1 (simian) cells with pSV.Myr0 (32). To study infectivity, the gag alleles were cloned either into pBH-RCAN-HiSV (8) for studies in QT6 (quail) cells or into pJD100 (9) for studies in TEF (turkey embryo fibroblast) cells. Standard protocols were used for all DNA manipulations (25).
COS-1 cells were grown in Dulbecco’s modified Eagle’s medium (GIBCO Laboratories) supplemented with 3% fetal bovine serum and 7% calf bovine serum (Hyclone, Inc.). QT6 cells (kindly provided by Paul Bates, University of Pennsylvania, Philadelphia, Pa.) were cultured in F10 medium (GIBCO Laboratories) supplemented with 10% tryptose phosphate broth, 5% fetal calf serum, and 1% chicken serum. TEFs were isolated from fertile eggs (Hudson Farms, Muskogee, Okla. or Clearview Hatchery, Gratz, Pa.) and propagated in supplemented F10 medium as previously described (17).
Construction of mutants.
S68A, S106A, and R121L were created by oligonucleotide-directed mutagenesis with a single-stranded, uracil-containing template DNA isolated from M13mp19P12 and oligonucleotides for introducing point mutations at codons 68 (TCG to GCG), 106 (TCG to GCT), or 121 (CGA to CTA). Mutations were confirmed by DNA sequencing by the dideoxy method and moved from the replicative form DNA into the pSV.Myr0 plasmid by transfer of an SstII fragment. pSV.S68/106A was constructed by moving the XhoI-EcoRV fragment (and the S106 mutation) from pSV.S106A into pSV.S68A.
All mutations (S68A, S106A, S68/106A, and R121L) were moved from the pSV.Myr0 plasmids into pBH-RCAN-HiSV and pJD100 by first transferring their SacI-EcoRI fragments into pSV.G1P (4). This intermediate step allowed subsequent transfer of the mutations into the RSV genome with the same upstream SacI site and the unique downstream HpaI site obtained from pSV.G1P.
To avoid inadvertent mutations that may arise during the mutagenesis and cloning steps, multiple independent clones of each mutant were sequenced and characterized in transfection experiments to be sure they exhibited the same phenotype.
Transfection and labeling of mammalian cells.
COS-1 cells were transfected by the DEAE-dextran-chloroquine method as described previously (2, 32, 33). Plasmid DNAs were digested with XbaI and ligated at a concentration of 25 μg/ml prior to transfection. This step removes the bacterial plasmid sequence and joins the 3′ end of gag to the simian virus 40 polyadenylation signal for high-level expression.
Two days posttransfection, COS-1 cells were metabolically labeled with l-[35S]methionine for 2.5 h (50 μCi, >1,000 Ci/mmol), as previously described (2, 32, 33). The cells and growth medium were collected and mixed with lysis buffer containing protease inhibitors. Gag proteins were collected from the samples by immunoprecipitation with a rabbit antiserum against whole RSV (30) by methods previously described (2, 32, 33).
SDS-PAGE.
Proteins were separated by electrophoresis in SDS–12% polyacrylamide gels, as described before (2, 32, 33). Gels were fixed in a solution of 5% methanol and 7% acetic acid, and radiolabeled proteins were detected by autoradiography with Kodak X-Omat AR5 film at −80°C.
Relative amounts of Gag proteins in medium and cell lysate samples were measured by laser scanning densitometry of fluorograms. The budding efficiency of each mutant was calculated by dividing the amount of Gag protein present in the medium sample by the total amount of Gag protein found in both medium and lysate samples.
Transfection and infection of avian cells.
Recombinants of pBH-RCAN-HiSV and pJD100 carrying the mutant gag alleles were initially tested for infectivity by a qualitative method in which persistent expression of viral proteins is assayed (21). Because transfected DNA exists transiently, only those mutants that can integrate their proviral genomes (and therefore are infectious) will continue to express viral proteins after several passages. DNA clones that are noninfectious will not spread throughout the culture, and expression of their viral proteins will be only transient. For these experiments, duplicate plates of avian (QT6) cells were transfected by the calcium phosphate method (5, 6). One plate was used to monitor the transfection efficiency by measuring the level of Pr76gag expression immediately following the 18-h transfection period. The cells in the second plate were passaged (1:5) into two plates 2 days posttransfection and then every 3rd day thereafter. Forty-eight hours after each passage, infectivity was assessed by measuring expression of Pr76gag. This was accomplished by labeling with l-[35S]methionine (100 μCi, >1,000 Ci/mmol) for 2 h, collecting the labeled Gag proteins from cell lysates by immunoprecipitation with polyclonal anti-RSV serum, and separating them by SDS-PAGE, as described above.
Focus assays.
Focus assays were performed with the pJD100 recombinants, as previously described (29). Briefly, freshly harvested virions were normalized by RT activity (4, 9) and used to infect 60-mm plates of uninfected TEF cells for 2 h. Immediately following infection, the plates were washed four times before the addition of 5 ml of a soft agar overlay (29). Cultures were fed every 3 days with fresh media until foci were clearly visible and easily countable (14 to 21 days).
Analysis of phosphorylation.
For examining serine phosphorylation, 100-mm plates of TEFs infected with recombinants of pJD100 bearing the wild-type (myr0) or mutant Prague C gag alleles were metabolically labeled with [32P]ortho-phosphate (1 mCi) in 5 ml of phosphate-free Dulbecco’s modified Eagle’s medium for 24 h. The media were collected, and loose cells were removed by centrifugation, and then the viral particles were allowed to mature at 37°C for 24 h. The mature virions were passed through a syringe-tip filter (45 μm), pelleted through a 25% sucrose cushion, and dissolved in loading buffer. The viral proteins were separated by SDS-PAGE and transferred to nitrocellulose by electroblotting, and 32P-labeled proteins were visualized by autoradiography. To visualize all the viral proteins, the blots were probed with antiserum against RSV, and the resulting antigen-antibody complexes were detected by enhanced chemiluminescence (Amersham) as previously described (1).
For detection of tyrosine phosphorylation, 300 μg of sucrose-purified Prague C RSV was disrupted in gel loading buffer, subjected to SDS-PAGE, and transferred to nitrocellulose. Phosphotyrosine-containing proteins were detected with PY20, a monoclonal antibody that is specific for phosphotyrosine (Transduction Labs).
RESULTS
Previous data have suggested that RSV MA is phosphorylated on the hydroxyl groups of serines 68 and 106. To investigate the importance of this modification in RSV replication, we constructed mutants in which either one or both of these residues are changed to alanine (Fig. 1), an amino acid that differs from serine in lacking the hydroxyl group. To examine the effects of these substitutions on particle production, the corresponding gag alleles were introduced into pSV.Myr0, a previously described plasmid that permits expression and budding of the wild-type Gag protein in mammalian cells (32). Budding efficiencies were compared to the parental construct and a mutant, Myr0.dMA4, which is defective for budding (21). All three serine mutants exhibited budding efficiencies which were similar to that of the wild type (Fig. 2). While the efficiencies varied slightly from experiment to experiment, they were typically 5 to 20% less than that of the control. Such minor differences indicate that serine phosphorylation is not important for particle release.
FIG. 1.

RSV MA sequence. Serine residues previously reported as the sites of phosphorylation are denoted by circles. Positions of tyrosine residues are highlighted with boxes. Numbers refer to the positions of individual residues with respect to the amino terminus of MA.
FIG. 2.
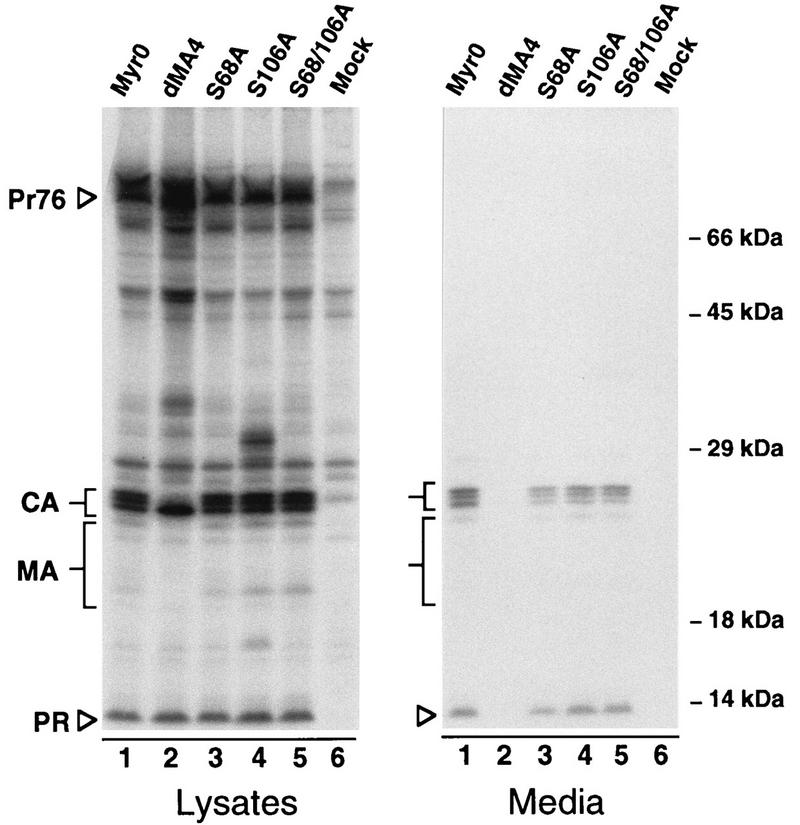
Expression and release of serine mutants. COS-1 cells were metabolically labeled for 2.5 h with [35S]methionine 48 h after transfection with the indicated mutants. Gag proteins were immunoprecipitated from cell and medium lysates with polyclonal anti-RSV serum, resolved by SDS-PAGE, and visualized by fluorography. Positions of molecular size markers are indicated on the right. The positions of the full-length Gag protein (Pr76gag) and those of its mature cleavage products are indicated on the left. The heterogeneous MA proteins are marked by larger brackets. Myr0 and dMA4 are wild-type and negative controls, respectively.
To determine whether phosphorylation is essential for infectivity, the mutations were cloned into pBH.RCAN, a plasmid which carries an infectious proviral genome and an hygromycin resistance gene in lieu of the Src-coding sequence normally present in RSV. The infectivity of these recombinants was tested in quail (QT6) cells. All the mutants appeared to be infectious, as indicated by both the spread of hygromycin resistance throughout transfected cultures and the ability of the mutants to maintain viral protein expression weeks after transfection (data not shown).
The infectivity of the MA mutants was also assessed with pJD100, a plasmid which retains the v-src oncogene within the proviral genome (9). Each recombinant was transfected into duplicate plates of QT6 cells. One set of plates was used to monitor transient Gag expression, confirming that all of the transfections had been successful (Fig. 3, transient panel). The cells in the remaining plates were split (1:5) into two fresh plates 48 h posttransfection and every 3rd day thereafter. Attempts were made to immunoprecipitate Gag proteins from [35S]methionine-labeled cells 48 h after each passage. By the third passage, expression from the negative control (pRC.dMA10 [21]) had been lost, but expression of all of the serine mutants continued (Fig. 3, passage 3).
FIG. 3.
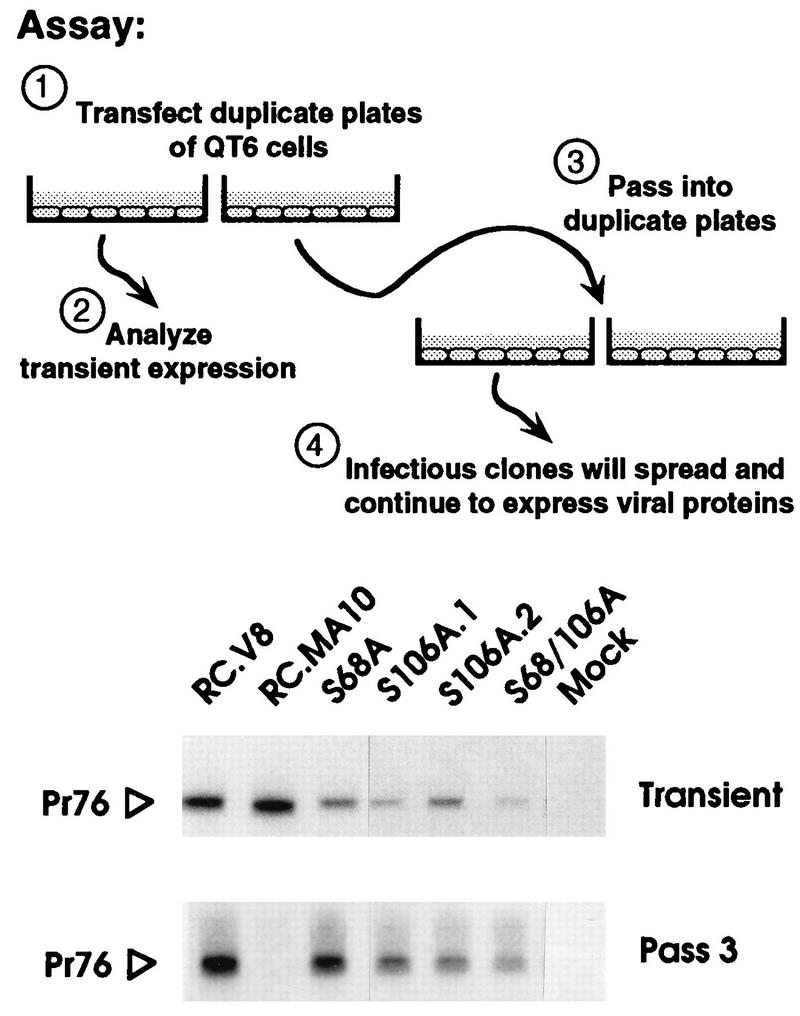
Qualitative analysis of infectivity. Each indicated clone was used to transfect duplicate plates of QT6 cells. One plate from each set was metabolically labeled for 2 h with [35S]methionine immediately following transfection, and the Gag proteins were collected by immunoprecipitation (Transient). The remaining plate of cells was passaged 2 days posttransfection and every 3rd day thereafter. On the 3rd day of the third passage, the cells were metabolically labeled with [35S]methionine, and again the Gag proteins were collected by immunoprecipitation (Pass 3). Proteins were separated by SDS-PAGE and visualized by fluorography. Only the region of the gel containing the Gag precursors is shown. RC.V8 and RC.MA10 are wild-type and negative controls, respectively. S106A.1 and S106A.2 are independent clones containing the same mutation.
The infectivity of each of the MA mutants was quantified in focus assays. Virus particles were obtained from transfected QT6 cells, the relative concentrations were measured by RT activity, and equal amounts were placed on uninfected TEF cells. Dense foci were scored 14 to 21 days later. As summarized in Table 1, neither the S68A nor the S106A mutation alone had detectable effects on infectivity, while the infectivity of virus harboring both mutations was reduced to only about half that of the wild type.
TABLE 1.
Quantitative analysis of infectivity
| Virus | No. of foci/mla
|
% Infectivity (mean ± SD)b | ||
|---|---|---|---|---|
| Exp 1 | Exp 2 | Exp 3 | ||
| JD.MyrO | 3.6 × 103 | 2.7 × 102 | 1.4 × 102 | 100 |
| JD.S68A | 3.8 × 103 | 6.1 × 102 | 1.1 × 102 | 136 ± 36 |
| JD.S106A | 3.2 × 103 | 3.0 × 102 | 1.0 × 102 | 90 ± 9 |
| JD.S68/106A | 1.6 × 103 | 1.9 × 102 | 0.4 × 102 | 48 ± 10 |
| Mock | 0 | 0 | 0 | 0 |
Experiments 1, 2, and 3 indicate results from three independent experiments.
Average percentage of infectivity normalized to wild type.
Before we could conclude that RSV infectivity is independent of serine phosphorylation of MA, it was necessary to assess the phosphorylation state of the mutants. This was accomplished by labeling infected TEFs with H332PO4 as described in Materials and Methods. Labeled virus was pelleted through a 25% sucrose cushion and then dissolved in sample buffer. Viral proteins were separated by SDS-PAGE and subsequently transferred to nitrocellulose by electroblotting. Visualization of 32P-labeled proteins by autoradiography showed that the wild-type virus contains two major phosphoproteins of 19 and 12 kDa, consistent with the sizes of MA and NC, respectively (Fig. 4B, lane 1, bands marked PP1 and PP4). Two additional phosphoproteins were detected which together accounted for less than 20% of the incorporated label (Fig. 4B, lane 1, bands marked PP2 and PP3). The migration of one of these (PP2) is consistent with a degradation product of MA which lacks the final four amino acids (23). We suspect that PP3 is also a breakdown product of MA, since substitutions that block phosphorylation of MA also prevent detection of this phosphoprotein (see below). The origin of PP5 is not clear. It migrates faster than the other known viral phosphoproteins during SDS-PAGE, and its intensity varied in repeats of this experiment. While this band appears to comigrate with PR under the SDS-PAGE conditions used here and those used by Lai, clearly it is not PR (18).
FIG. 4.

In vivo phosphorylation. Infected TEFs were labeled with 32P for 24 h. Virus was pelleted through a 25% sucrose cushion and dissolved in sample buffer. Viral proteins were separated by SDS–12% PAGE and then transferred to nitrocellulose. (A) Immunoblot with anti-RSV serum. (B) Autoradiograph of the same blot. The positions of Gag cleavage products and molecular size markers are indicated on the left and right, respectively. Phosphoproteins detected in this experiment are labeled PP1 to PP5 and IN∗ (predicted to be phosphorylated IN based on apparent size).
With regard to the mutants, the S68A substitution appeared to have little if any effect on the phosphoprotein profile, while the S106A substitution abolished almost all of the detectable phosphorylation (Fig. 4B, lanes 2 and 3). The absence of 32P-labeled proteins for S106A was not due to a sample loading error, because immunoblot analysis of the same nitrocellulose filter revealed that all the lanes contained approximately the same amount of protein (Fig. 4A, lanes 1 to 4). Furthermore, the S106A sample contained a single MA species which comigrated with the lower band of the wild-type MA doublet, the position of the unphosphorylated form of MA (10). These results demonstrate that the major site of phosphorylation within RSV MA is serine 106 and allow us to conclude that serine phosphorylation is not important for infectivity.
We were surprised to find that the S106A mutation seems to prevent almost all of the phosphorylation of the 12-kDa protein (PP4), previously reported to be NC. This finding could indicate that MA phosphorylation is a prerequisite for phosphorylation of NC, possibly through recruitment of a specific kinase or cofactor which is required for NC phosphorylation. However, it seemed possible that the 12-kDa phosphoprotein was not NC but was actually a comigrating breakdown product of phosphorylated MA previously referred to as p19f (23, 28). To explore this possibility, mutants which produce either a faster- or a slower-migrating MA protein (RC.ΔMA6 [21] and RC.R121L, respectively) were labeled with H332PO4 and analyzed as described above. For both of these mutants, the altered migration of a portion of the 12-kDa phosphoprotein matched that of the mutant MA species (Fig. 5), indicating that the 12-kDa phosphoprotein is actually a mixture of phosphorylated p19f and NC proteins. Since serine phosphorylation is not important for infectivity, we did not explore this any further.
FIG. 5.

Analysis of pp12 species. 32P-labeled virions produced from infected TEFs were pelleted through a 25% sucrose cushion. Following suspension in sample buffer, the viral proteins were separated by SDS–12% PAGE and transferred to nitrocellulose. (A) Immunoblot with anti-RSV serum. (B) Autoradiograph of the same blot. R121L and ΔMA6 contain mutations which cause MA-related proteins to migrate either slower or faster, respectively, during electrophoresis. The locations of the wild-type and slower-migrating forms of p19f are indicated on the left side of panel A.
Given the lack of an apparent role for serine phosphorylation, we wondered whether other minor sites of phosphorylation (such as the tyrosine phosphorylation found in human immunodeficiency virus [HIV] MA [14, 15]) might also exist in RSV MA. To explore this possibility, we probed blots of purified RSV with an antibody that recognizes phosphotyrosine in a manner that is insensitive to the adjacent amino acids. Three viral proteins were detected (Fig. 6, lanes 3). These proteins correspond to the positions of the MA-p2 cleavage intermediate, serine-phosphorylated MA, and MA lacking phosphoserine. Of the three tyrosines contained within the MA sequence (Fig. 1, residues 15, 46, and 155), Y46 is most likely the site recognized by a protein tyrosine kinase, based on its surrounding sequences which have characteristics of known tyrosine phosphorylation sites (24). The lack of detectable phosphotyrosine on the smaller species of MA (including p19f, which consists of a mixture of two species corresponding to residues 1 to 123 and 1 to 129 [23]) suggests either that these are derived from MA species lacking phosphotyrosine or that Y155 is actually the site of modification. Further experimentation will be required to determine which residue(s) is modified.
FIG. 6.

Detection of tyrosine-phosphorylated MA. (A) Proteins from sucrose gradient-purified RSV were separated by SDS–12% PAGE, transferred to nitrocellulose, and probed with an antiphosphotyrosine antibody and visualized by enhanced chemiluminescence. (B) Later, the same blot was stripped of antibodies and reprobed with anti-RSV serum. Lanes: 1, Src-transformed Rat-1 cell extract (positive control); 2, Rat-1 cell extract (negative control); 3, RSV.
DISCUSSION
The experiments described in this study identify serine 106 as the major site of phosphorylation within RSV MA and are in agreement with recent electronic spray mass spectroscopy measurements of MA, indicating that the phosphorylated form of MA contains only one phosphate molecule (23). While it appears that serine 68 is not a major site of phosphorylation, methods employed in this study are not sensitive enough to determine whether a small amount of phosphate (3% or less, as determined by laser scanning densitometry) is attached to this residue. Likewise, we cannot address the possibility that serine 68 may be phosphorylated in a transient manner, since labeling was performed only under steady-state conditions. However, since phosphorylation of either site is nonessential for virus replication, this issue seems to be of little significance. Furthermore, infectivity of the serine mutants was observed in two different cell types (TEF and QT6) and thus does not appear to have a cell-type dependent function. Nevertheless, we cannot eliminate the possibility that this region plays some important role in nature (i.e., in chickens).
pp12 proteins.
The fact that the S106A mutation reduces the phosphorylation of NC is consistent with phosphorylation of MA being a prerequisite for phosphorylation of NC. The mechanism by which this would occur is unclear, but it is possible that modification of MA induces a conformational change in Gag which then allows NC to be recognized by a kinase. Alternatively, phosphorylation of serine 106 may be necessary for the recruitment of factors which directly or indirectly promote NC phosphorylation. Regardless of the mechanism, the need for such a regulatory event is not clear. Although previous studies indicate that the dephosphorylation of NC results in a conformational change and a 100-fold decrease in affinity for RNA (11, 19), the phosphorylation of NC, like that of MA, can be prevented without affecting infectivity (12), suggesting that if the phosphorylation of either protein is important, it is not rate limiting. It is possible that the phosphorylation of both proteins is fortuitous—the result of Gag proteins assembling into virus particles in the vicinity of a host of protein kinases on the plasma membrane.
Why are only a third of the RSV MA molecules phosphorylated?
When MA protein from RSV particles is subjected to SDS-PAGE, it migrates as a doublet, with the upper band containing the phosphorylated species. Assuming that this band contains only phosphorylated MA and that the lower band contains only unphosphorylated MA, it has been estimated that approximately one-third of the MA found in the virion is phosphorylated (10). With the same assumptions, MA molecules associated with the cell appear to be phosphorylated at levels similar to that found within the virion, suggesting that phosphorylation occurs either before or during particle formation. Therefore, at least three possible mechanisms can account for this partial modification. (i) The responsible kinase is present in limited quantities or is sequestered in a cellular location that some Gag molecules bypass. (ii) Gag molecules form multimers before phosphorylation occurs, thereby limiting access to the internal molecules. (iii) All Gag molecules may be equally capable of being phosphorylated, but competition between kinases and phosphatases determines what portion of the Gag molecules are phosphorylated (7).
Minor sites of phosphorylation.
While the experiments presented here show that serine phosphorylation is not needed for infectivity of RSV, they do not rule out the possibility that other minor sites of phosphorylation are essential. Recent studies of the HIV MA protein have suggested that such minor modifications are important. Specifically, the phosphorylation of a small fraction of MA molecules (1% percent) on the C-terminal tyrosine enables them to shift from being membrane associated to being bound to IN within the core of the virion (14, 15). Once this core is released into a cell, the phosphorylated MA molecules appear to be able to direct the complex to the nucleus via its nuclear localization sequence prior to integration, a feature which may be required by HIV to infect nondividing cells in some cases (3, 13). As demonstrated here, a minor population of RSV MA also contains phosphotyrosine. While the role of this modification, if any, is unknown at this time, clearly it cannot be the same as in HIV, since RSV does not infect nondividing cells. Nevertheless, recently published work suggests that the MA protein of RSV plays a role of some sort during the establishment phase of the replication cycle (22). Efforts to elucidate this function are currently in progress.
ACKNOWLEDGMENTS
We thank Anu Raman for creating the point mutations in M13mp19P12.
This work was supported by NIH grants to T.D.N. (training grant CA60395), M.F.V. (CA52791), J.L. (CA38046), and J.W.W. (CA47482) and by a grant from the American Cancer Society to J.W.W. (FRA-427).
REFERENCES
- 1.Bennett R P, Nelle T D, Wills J W. Functional chimeras of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol. 1993;67:6487–6498. doi: 10.1128/jvi.67.11.6487-6498.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett R P, Rhee S, Craven R C, Hunter E, Wills J W. Amino acids encoded downstream of gag are not required by Rous sarcoma virus protease during Gag-mediated assembly. J Virol. 1991;65:272–280. doi: 10.1128/jvi.65.1.272-280.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bukrinsky M I, Haggerty S, Dempsey M P, Sharova N, Adzhubel A, Spitz L, Lewis P, Goldfarb D, Emerman M, Stevenson M. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature. 1993;365:666–669. doi: 10.1038/365666a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craven R C, Bennett R P, Wills J W. Role of the avian retroviral protease in the activation of reverse transcriptase during virion assembly. J Virol. 1991;65:6205–6217. doi: 10.1128/jvi.65.11.6205-6217.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craven R C, Leure D A, Erdie C R, Wilson C B, Wills J W. Necessity of the spacer peptide between CA and NC in the Rous sarcoma virus Gag protein. J Virol. 1993;67:6246–6252. doi: 10.1128/jvi.67.10.6246-6252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craven R C, Leure D A, Weldon R J, Wills J W. Genetic analysis of the major homology region of the Rous sarcoma virus Gag protein. J Virol. 1995;69:4213–4227. doi: 10.1128/jvi.69.7.4213-4227.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Depaoli-Roach A A, Park I K, Cerovsky V, Csortos C, Durbin S D, Kuntz M J, Sitikov A, Tang P M, Verin A, Zolnierowicz S. Serine/threonine protein phosphatases in the control of cell function. Adv Enzyme Regul. 1994;34:199–224. doi: 10.1016/0065-2571(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 8.Dong J Y, Dubay J W, Perez L G, Hunter E. Mutations within the proteolytic cleavage site of the Rous sarcoma virus glycoprotein define a requirement for dibasic residues for intracellular cleavage. J Virol. 1992;66:865–874. doi: 10.1128/jvi.66.2.865-874.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erdie C R, Wills J W. Myristylation of Rous sarcoma virus Gag protein does not prevent replication in avian cells. J Virol. 1990;64:5204–5208. doi: 10.1128/jvi.64.10.5204-5208.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erikson E, Brugge J S, Erikson R L. Phosphorylated and nonphosphorylated forms of avian sarcoma virus polypeptide p19. Virology. 1977;80:177–185. doi: 10.1016/0042-6822(77)90390-7. [DOI] [PubMed] [Google Scholar]
- 11.Fu X, Phillips N, Jentoft J, Tuazon P T, Traugh J A, Leis J. Site-specific phosphorylation of avian retrovirus nucleocapsid protein pp12 regulates binding to viral RNA. Evidence for different protein conformations. J Biol Chem. 1985;260:9941–9947. [PubMed] [Google Scholar]
- 12.Fu X D, Tuazon P T, Traugh J A, Leis J. Site-directed mutagenesis of the avian retrovirus nucleocapsid protein, pp12, at serine 40, the primary site of phosphorylation in vivo. J Biol Chem. 1988;263:2134–2139. [PubMed] [Google Scholar]
- 13.Gallay P, Stitt V, Mundy C, Oettinger M, Trono D. Role of the karyopherin pathway in human immunodeficiency virus type 1 nuclear import. J Virol. 1996;70:1027–1032. doi: 10.1128/jvi.70.2.1027-1032.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallay P, Swingler S, Aiken C, Trono D. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell. 1995;80:379–388. doi: 10.1016/0092-8674(95)90488-3. [DOI] [PubMed] [Google Scholar]
- 15.Gallay P, Swingler S, Song J P, Bushman F, Trono D. HIV nuclear import is governed by the phosphotyrosine-mediated binding of matrix to the core domain of integrase. Cell. 1995;83:569–576. doi: 10.1016/0092-8674(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 16.Hizi A, Joklik W K. The beta subunit of the DNA polymerase of avian sarcoma virus strain B77 is a phosphoprotein. Virology. 1977;78:571–575. doi: 10.1016/0042-6822(77)90132-5. [DOI] [PubMed] [Google Scholar]
- 17.Hunter E. Biological techniques for avian sarcoma viruses. Methods Enzymol. 1994;58:379–392. doi: 10.1016/s0076-6879(79)58153-1. [DOI] [PubMed] [Google Scholar]
- 18.Lai M M. Phosphoproteins of Rous sarcoma viruses. Virology. 1976;74:287–301. doi: 10.1016/0042-6822(76)90336-6. [DOI] [PubMed] [Google Scholar]
- 19.Leis J, Johnson S, Collins L S, Traugh J A. Effects of phosphorylation of avian retrovirus nucleocapsid protein pp12 on binding of viral RNA. J Biol Chem. 1984;259:7726–7732. [PubMed] [Google Scholar]
- 20.Leis J, Phillips N, Fu X, Tuazon P T, Traugh J A. Phosphorylation of avian retrovirus matrix protein by Ca2+/phospholipid-dependent protein kinase. Eur J Biochem. 1989;179:415–422. doi: 10.1111/j.1432-1033.1989.tb14569.x. [DOI] [PubMed] [Google Scholar]
- 21.Nelle T D, Wills J W. A large region within the Rous sarcoma virus matrix protein is dispensable for budding and infectivity. J Virol. 1996;70:2269–2276. doi: 10.1128/jvi.70.4.2269-2276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parent L J, Wilson C B, Resh M D, Wills J W. Evidence for a second function of the MA sequence in the Rous sarcoma virus Gag protein. J Virol. 1996;70:1016–1026. doi: 10.1128/jvi.70.2.1016-1026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pepinsky R B, Papayannopoulos I A, Campbell S, Vogt V M. Analysis of Rous sarcoma virus Gag protein by mass spectrometry indicates trimming by host exopeptidase. J Virol. 1996;70:3313–3318. doi: 10.1128/jvi.70.5.3313-3318.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinna L A, Ruzzene M. How do protein kinases recognize their substrates? Biochim Biophys Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 26.Schiff R D, Grandgenett D P. Partial phosphorylation in vivo of the avian retrovirus pp32 DNA endonuclease. J Virol. 1980;36:889–893. doi: 10.1128/jvi.36.3.889-893.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz D E, Tizard R, Gilbert W. Nucleotide sequence of Rous sarcoma virus. Cell. 1983;32:853–869. doi: 10.1016/0092-8674(83)90071-5. [DOI] [PubMed] [Google Scholar]
- 28.Shealy D J, Mosser A G, Rueckert R R. Novel p19-related protein in Rous-associated virus type 61: implications for avian gag gene order. J Virol. 1980;34:431–437. doi: 10.1128/jvi.34.2.431-437.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogt P K. Focus assay of Rous Sarcoma virus. In: Salzman H K and N P., editor. Fundamental techniques of virology. New York, N.Y: Academic Press; 1969. pp. 198–211. [Google Scholar]
- 30.Weldon R J, Erdie C R, Oliver M G, Wills J W. Incorporation of chimeric Gag protein into retroviral particles. J Virol. 1990;64:4169–4179. doi: 10.1128/jvi.64.9.4169-4179.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wills J W, Craven R C. Form, function, and use of retroviral Gag proteins. AIDS. 1991;5:639–654. doi: 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Wills J W, Craven R C, Achacoso J A. Creation and expression of myristylated forms of Rous sarcoma virus Gag protein in mammalian cells. J Virol. 1989;63:4331–4343. doi: 10.1128/jvi.63.10.4331-4343.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wills J W, Craven R C, Weldon R J, Nelle T D, Erdie C R. Suppression of retroviral MA deletions by the amino-terminal membrane-binding domain of p60src. J Virol. 1991;65:3804–3812. doi: 10.1128/jvi.65.7.3804-3812.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]