

Abstract
The adenovirus E1A 243R oncoprotein is capable of transactivating the expression of the human proliferating cell nuclear antigen (PCNA) promoter. Mutational analysis of the E1A 243R protein suggested that both its p300/CBP- and p107-binding regions are required for optimal induction of the PCNA promoter (C. Kannabiran, G. F. Morris, C. Labrie, and M. B. Mathews, J. Virol. 67:425–437, 1993). We show that overexpression of p107 antagonizes the induction of PCNA by E1A 243R in transient expression assays. This inhibition is largely independent of p107’s ability to interact with E1A 243R, because p107 mutants unable to bind to E1A 243R retain the ability to repress the E1A-activated PCNA promoter. Electrophoretic mobility shift assays with the PCNA promoter detected the presence of p107 in one of the major DNA-protein complexes, EH1, formed with HeLa cell nuclear extracts. Promoter mutations that disrupt the formation of complex EH1 abrogated p107’s ability to reverse E1A 243R-induced PCNA expression. The same mutations characterize a sequence important for the binding of transcription factor RFX1 (C. Labrie, G. F. Morris, and M. B. Mathews, Nucleic Acids Res. 23:3732–3741, 1995), implying that p107 antagonizes E1A 243R-induced PCNA expression through this RFX1-binding site. Our data are suggestive of a novel cooperative mechanism for transactivation of PCNA expression, in which E1A 243R relieves transcriptional repression exerted by p107 on the promoter.
The adenovirus E1A oncogene products are multifunctional proteins which possess both transforming and transactivating capabilities (6, 51, 55). Alternative splicing of the E1A transcript produces at least five E1A mRNAs, chiefly the 13S and 12S species (7, 11), which are translated into proteins containing 289 and 243 amino acid residues, respectively (referred to as E1A 289R and E1A 243R) (6, 44). While the transforming functions of E1A map to regions common to the 289R and 243R proteins (60), the CR3 domain unique to the 289R species endows it with potent transactivation properties (6, 44, 51, 55).
Concomitant with its ability to induce DNA synthesis and mitosis in quiescent cells (44, 54, 56), the adenovirus E1A gene can activate the expression of the proliferating cell nuclear antigen (PCNA), a highly conserved protein intimately involved in cellular DNA synthesis (47, 66). As an integral component of the eukaryotic DNA replication machinery, PCNA plays a crucial role in normal cellular growth and differentiation. PCNA is believed to function in a manner analogous to a sliding clamp to increase the processivity of DNA synthesis as an auxiliary factor of DNA polymerase-δ and is necessary for the replication of both leading- and lagging-strand DNA in an in vitro simian virus 40 replication system (9, 52, 53, 58, 59, 62). In vivo, PCNA is required for growth and cell cycle progression in both yeast (4) and mammalian cells (31, 40) and is found in quaternary complexes containing cyclins, cyclin-dependent kinases (CDKs), and the inhibitor p21 (63–65, 67). Although PCNA protein and mRNA levels change relatively little in cycling human HeLa (45) and Chinese hamster ovary (40) cells, they increase dramatically upon stimulation of quiescent cells by mitogenic agents, including serum and growth factors or during the course of viral infection (1, 8, 31, 42, 66).
In light of its essential role in normal cell cycle regulation, the induction of PCNA during the transformation of quiescent rodent cells by adenovirus (66) represents an informative system for studying the mechanisms underlying cellular transformation and transactivation by the adenovirus E1A oncogene products. We previously demonstrated that activation of PCNA expression by the E1A oncoproteins occurs at the transcriptional level through an increase in PCNA promoter activity (46). Significantly, the PCNA promoter is induced equally well by the smaller E1A 243R protein and by the larger E1A 289R species, indicating that the CR3 region is dispensable for PCNA activation by E1A (47). Analysis of the PCNA promoter revealed that its induction by E1A 243R occurs through a novel cis-acting PCNA E1A-responsive element (PERE) which resides between nucleotides −59 and −45 relative to the transcriptional start site (35) and can confer induction by the E1A 243R oncoprotein upon a normally E1A-unresponsive heterologous promoter (48). The PERE contains a sequence homologous to an activating transcription factor (ATF) motif (5′-TGACGTCG-3′) at its 3′ end, and the ATF-1 and CREB transcription factors have been shown to be major components of PERE-protein complexes (35–37). In addition, we have shown that RFX1, a ubiquitous transcription factor with both activation and repression properties (33), binds to a site which overlaps with the ATF-CREB consensus motif and extends to sequences immediately downstream of the PERE (36).
Most of E1A’s pleiotropic actions are mediated by its capacity to bind and subvert the function of well-characterized and important cellular regulatory proteins, including the retinoblastoma susceptibility gene product, pRB; pRB-related proteins p107 and p130; and the transcriptional coactivators p300 and CBP (the CREB-binding protein) (2, 3, 5, 19, 20, 41, 43). Our recent investigation of the role played by the p300/CBP coactivators in E1A-induced PCNA expression supported a mechanism whereby E1A 243R can target and transactivate the PCNA promoter via a CBP-CREB-PERE pathway (37). However, results obtained with a panel of E1A mutants argued that E1A 243R displays a functional redundancy in its capacity to activate the PCNA promoter and most likely induces PCNA expression by multiple mechanisms via interactions with more than one E1A-binding protein (32, 50). In particular, optimal activation of PCNA by E1A 243R requires domains of E1A 243R that interact with both the cellular transcriptional coactivators p300/CBP and the retinoblastoma-related tumor suppressor protein p107 (32).
Here we set out to assess the role of the retinoblastoma-related tumor suppressor protein p107 in transactivation of the PCNA promoter. First, we examined the consequences of overexpressing p107 on PCNA promoter activity in transient transfection assays. Our data indicate that p107 antagonizes the ability of E1A 243R to transactivate a PCNA-chloramphenicol acetyltransferase (CAT) reporter gene and that this inhibitory effect is independent of p107’s ability to associate with E1A 243R, but dependent on an RFX1-binding site contained in the promoter. In addition, we detected an association between p107 and the PCNA promoter, finding that RFX1 and p107 are present in the same DNA-protein complex in a mobility shift assay. These results provide direct evidence for the involvement of p107 in the regulation of the PCNA promoter by E1A 243R and suggest novel transcriptional regulatory roles for p107 in gene expression and growth control in normal cells.
MATERIALS AND METHODS
Plasmids.
Wild-type PCNA−87 CAT and constructs −46/−39, ATF-BAM, −56GA, and −59/−56 CAT were described previously (35, 46). Reporter plasmids −53GT and −44/−40 PCNA-CAT contain single or multiple mutations in the PCNA promoter and were created by using an oligonucleotide-directed mutagenesis kit (Amersham, Buckinghamshire, United Kingdom). 44/−40 PCNA-CAT contains a 5-bp clustered mutation (5′-CAACG-3′→5′-ATCGA-3′) between sites −44 and −40 of the PCNA promoter, while −53GT PCNA-CAT contains a single G-to-T point mutation at position −53 of the promoter. Plasmids expressing the E1B 19-kDa protein (pCMV19K), β-galactosidase (pCMVβ-gal), E1A243R (pCMV12S), and a truncated form of E1A 243R product (pCMV12S.FS, predicted to encode only the first 22 amino acids of E1A 243R), under the control of the cytomegalovirus promoter have been described previously (47). Plasmid pCH110 (25), which also expresses β-galactosidase, was obtained from M. Gilman (Ariad Pharmaceuticals), and constructs expressing wild-type p107 (pCMV107) and p107DE (pCMV107DE) (68), a mutant form of p107 which does not bind to E1A 243R, were generously provided by M. Ewen (Dana-Farber Cancer Institute). All plasmids were purified by two cesium chloride gradient ultracentrifugations followed by polyethylene glycol precipitation to remove low-molecular-weight RNA.
DNA probes.
PCNA promoter fragments (∼150 bp) used as electrophoretic mobility shift assay (EMSA) probes encompass nucleotides from −87 to +62 relative to the transcription start site of the human PCNA promoter. They include wild-type EH87, EH−46/−39, EH−44/−40, and EHATF-BAM probes. Sequences of wild-type EH87 and mutants EH−46/−39 and EHATF-BAM have been reported previously (35, 36). DNA fragments EH−44/−40 and EH−53GT contain the mutations described in the corresponding −44/−40 and −53GT PCNA-CAT plasmids described above. All DNA fragments (wild type or mutant derivatives) were prepared by digestion of wild-type PCNA−87 CAT plasmid or mutant reporter plasmids with EcoRI and HindIII and were radiolabeled with [α-32P]dATP (3,000 Ci/mmol) and cold deoxynucleoside triphosphates with DNA polymerase I (Klenow fragment). Probes were isolated by electrophoresis in native 6% polyacrylamide gels and eluted from gel slices in oligonucleotide buffer (10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA) containing 0.5% sodium dodecyl sulfate. After phenol extraction and ethanol precipitation, purified probes were dissolved in oligonucleotide buffer.
EMSAs.
Nuclear extracts were prepared according to a modified version of the Dignam procedure described previously (17, 36). EMSA was carried out as before (36). Typically, assays were performed with a 20-μl reaction mixture containing 1× EMSA buffer (12 mM HEPES [pH 7.6], 50 mM NaCl, 1 mM dithiothreitol, 5% [vol/vol] glycerol), 1.5 μg of poly(dI-dC)-poly(dI-dC) as nonspecific DNA competitor, 5 μg of HeLa cell nuclear extract, and 20,000 cpm of DNA fragment probe. Incubations were conducted on ice for 15 min in the absence of probe, followed by incubation at 16°C for 15 min after addition of the probe. When indicated, antibody was added to the binding reaction mixture at least 1 h prior to the addition of probes, and the incubation was performed at 4°C. Protein-DNA complexes were resolved in 4.5% polyacrylamide (39:1 N,N′-methylenebisacrylamide)–0.25× Tris-borate-EDTA gels.
Antibodies.
Anti-RFX1 antibody was a generous gift from P. Hearing (SUNY—Stony Brook), and p107 (SD9), anti-p53 (D0-1), and anti-ATF-1 (C41-5.1) monoclonal antibodies were obtained from Santa Cruz Biotechnology, Inc.
Transfections.
Transient expression assays were performed in duplicate in American Type Culture Collection HeLa cells (passages 6 to 13) as described previously (47). Briefly, cells at 40 to 50% confluence were transfected by the calcium phosphate coprecipitation technique (61). Unless otherwise indicated, each 6-cm-diameter plate received a total of 20 μg of DNA, including 10 μg of reporter construct, 0.5 μg of either pCMV12S or pCMV12S.FS, 0.5 μg of pCMV19K, and 1 μg of pCMVβ-gal or pCH110 as a control for transfection efficiency and salmon sperm carrier DNA. The plasmid pCMV19K encodes the E1B 19-kDa protein, which stabilizes transfected DNAs (28, 47) and was included to enhance the level of transfected reporter plasmid and improve sensitivity. The cells were washed twice with phosphate-buffered saline, and fresh medium was added 16 h after the transfection. Cells were harvested 48 h after the transfection.
CAT and β-galactosidase assays.
Cell extracts were prepared by freezing and thawing cells in 0.25 M Tris-HCl (pH 8.0). CAT assays were performed so that they yielded values within the linear range of the assay. Usually, up to 50 μl of cell extract was added to the 100-μl CAT assay reaction mixture. Thin-layer chromatograms were quantified with a Betascan System (AMBIS, San Diego, Calif.), and CAT activity was expressed as the percentage of chloramphenicol acetylated by 50 μl of cell extract incubated at 37°C for 1 h. β-Galactosidase assays were performed as described previously (27). Reaction mixtures were incubated at 37°C, and reactions were stopped by addition of 1 M Na2CO3. β-Galactosidase activity was expressed as the optical density at 420 nm obtained with 50 μl of extract incubated at 37°C for 1 h. All CAT assay results were normalized to the levels of β-galactosidase activity.
RESULTS
p107 antagonizes E1A 243R-transactivated PCNA promoter activity.
Analysis of a panel of E1A 243R deletion and point mutants for their abilities to activate PCNA promoter-directed reporter gene expression in HeLa cells suggested that E1A activates transcription of the PCNA gene by multiple mechanisms and that of the known E1A 243R-associated cellular proteins, p300/CBP and p107 are good candidates for mediating transactivation of the PCNA promoter by E1A 243R (32). The involvement of the p300/CBP pathway has recently been confirmed (37). To characterize the putative role of p107 in PCNA gene expression, we began by examining the effects on basal and E1A 243R-induced PCNA levels of overexpression of p107 in transient expression assays. While the overexpression of p107 (2 μg of plasmid) in HeLa cells did not have any detectable effect on basal transcription from the wild-type PCNA−87 CAT reporter, E1A 243R-induced PCNA-CAT expression was significantly reduced compared to reporter levels in the absence of overexpressed p107 at all concentrations of cotransfected E1A 243R tested (from 0.1 to 2.5 μg of E1A 12S plasmid) (Fig. 1). This inhibitory effect of p107 on E1A-induced PCNA expression was confirmed in reciprocal experiments with various concentrations of cotransfected p107 plasmid. HeLa cells were cotransfected with increasing amounts of p107 (from 0 to 6 μg), and a constant amount of wild-type E1A 12S (0.5 μg) or a control plasmid, E1A 12S.FS (0.5 μg). Again, overexpression of p107 did not exert any detectable effect on basal transcription levels from the PCNA−87 CAT reporter construct, even at the largest amounts of cotransfected p107 plasmid (Fig. 2). In the absence of transiently expressed p107, the E1A 12S plasmid stimulated PCNA-CAT expression by 30- to 35-fold. However, overexpression of p107 dramatically reduced E1A 243R’s ability to stimulate PCNA-CAT levels to less than 10-fold in a dose-dependent fashion (Fig. 2).
FIG. 1.

p107 inhibits E1A 243R-induced PCNA-CAT activity. PCNA−87 CAT (10 μg) reporter was transfected into HeLa cells with either pCMV12S.FS (0.5 μg) or increasing amounts of pCMV12S (E1A 243R) plasmid (0.1, 0.5, or 2.5 μg) and a constant amount (2 μg) of wild-type pCMV107 expression plasmid. The results are the average of two independent transfections performed in duplicate with standard deviations indicated.
FIG. 2.

p107 antagonizes E1A 243R-induced PCNA expression independent of its E1A-binding ability. Wild-type (WT) PCNA−87 CAT plasmid reporter (10 μg) was cotransfected into HeLa cells with either pCMV12S.FS as a control or pCMV12S (E1A 243R) and increasing amounts (0, 2, 4, or 6 μg) of either wild-type pCMV107 or mutant pCMV107DE expression plasmids. CAT activity was corrected for β-galactosidase activity generated from a cotransfected reporter plasmid and expressed as the means ± the standard deviation relative to the level of CAT activity obtained by cotransfection of wild-type PCNA−87 CAT with pCMV12S.FS. These data represent the means of three independent transfections performed in duplicate.
To determine whether the inhibitory effect of p107 was simply attributable to squelching (the sequestration of E1A 243R by p107 in an inactive complex), we conducted similar experiments with a p107 mutant (p107DE) deficient in its capacity to bind to the E1A protein. Like wild-type p107 protein, expression of p107DE (from 0 to 6 μg) had no significant effect on basal PCNA-CAT levels but inhibited the E1A 243R transactivation of PCNA-CAT levels in a dose-dependent manner, although it was somewhat less effective than wild-type p107 overexpression (Fig. 2). This dose-dependent inhibition of E1A 243R-induced PCNA-CAT levels was also observed with two other p107 mutants, p107EC and p107F846 (68), that are deficient in binding to E1A 243R (data not shown). Thus, p107 appears to exert an inhibitory effect on E1A-transactivated PCNA expression that is independent of its binding to E1A 243R.
Association of p107 with the PCNA promoter in vitro.
The reversal of E1A 243R-induced PCNA promoter activity by p107 in vivo raised the possibility that p107 might associate with the promoter. We previously used an EMSA to characterize the interaction of cellular factors with the promoter (36). When the DNA probe (EH87), containing wild-type PCNA promoter sequences from −87 to +62 relative to the transcription initiation site, was incubated with HeLa cell nuclear extract, five major DNA-protein complexes (EH1 to EH5) were formed (36) (Fig. 3A, lanes 1 and 7). To determine whether any one of these DNA-protein complexes contains p107, we performed an antibody interference experiment with an antibody directed against the p107 cellular factor. Preincubation of HeLa cell nuclear extracts with p107 monoclonal antibody SD9 specifically supershifted the EH1 complex (lane 5). The supershifted band was more intense than the EH1 band in control lanes, suggesting that the p107 antibody stabilizes the EH1 complex. None of the EH87 protein complexes was altered by an unrelated monoclonal antibody directed against p53 (lane 4), consistent with our previous observation that p53 binds to upstream PCNA promoter sequences not contained in the EH87 probe (49). In contrast, a monoclonal antibody against the ATF-1 transcription factor previously shown to disrupt PCNA promoter-protein complexes (36) disrupted EH87 complexes EH3 and EH4 (lane 6), demonstrating the specificity of the p107 antibody for complex EH1. Although p107 associates with cyclin A and CDK2 in vivo (10, 16, 22, 23), antibodies against these proteins did not perturb any of the EMSA complexes (data not shown). These data suggest that p107 is a component of complex EH1.
FIG. 3.
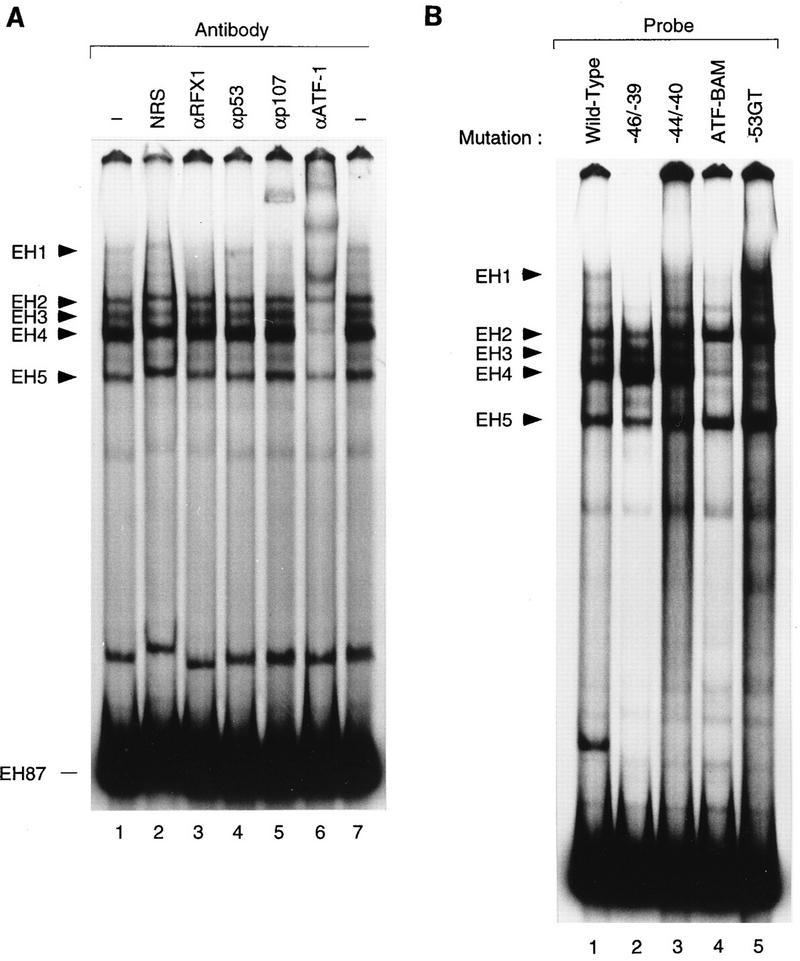
EMSA complex EH1 contains both p107 and RFX1. (A) An end-labeled PCNA promoter fragment from −87 to +62 relative to the transcription initiation site (EH87) was incubated with 5 μg of HeLa cell nuclear extract, and the protein-DNA complexes were resolved on a 4.5% native polyacrylamide gel. Extracts were incubated in the absence (lanes 1 and 7) or the presence of antibodies (α) against either RFX1 (lane 3), p53 (lane 4), p107 (lane 5), or ATF-1 (lane 6). As a control, nuclear extract was incubated with normal rabbit serum (NRS) (lane 2). Complexes EH1 to EH5 are denoted in order of increasing mobility. (B) Formation of complex EH1 depends on an intact RFX1 site. Probes derived from the wild-type PCNA−87 CAT construct (EH87) or from promoter constructs harboring mutations from −50 to −47 (EH ATF-BAM), −46 to −39 (EH −46/−39), −44 to −44 (EH −44/−40), or a G-to-T point mutation at position −53 (EH −53GT) were incubated with HeLa nuclear extract (5 μg), and the protein-DNA complexes were resolved in a native gel.
Complex EH1 formation is dependent on RFX1-binding site sequences and contains RFX1.
Previous studies led us to suspect that nucleotide sequences important for the formation of EH1 are also essential for the binding of the transcription factor RFX1 to the PCNA promoter (36). This idea was strengthened by the results of EMSA with EH probes containing mutations within the RFX1-binding site, namely EH−46/−39, EH ATF-BAM, and EH−44/−40. As shown in Fig. 3B, the formation of complex EH1 was reduced in all cases (Fig. 3B, lanes 2 to 4). These results indicated that sequences between −50 and −40 are important for EH1 complex formation. Moreover, a mutation further upstream (EH−53GT) that enhances the binding of RFX1 (39a) increased the formation of the EH1 complex (lane 5). To determine directly whether RFX1 is a component of complex EH1, HeLa cell nuclear extract was preincubated with an antibody raised against RFX1. The RFX1 antibody specifically affected complex EH1 (Fig. 3A, lane 3), either causing a weak supershift or abrogating the band, whereas normal rabbit serum had no effect (Fig. 3A, lane 2). These data show that EH1 is a multiprotein complex, containing both the RFX1 transcription factor and p107.
Inhibition of E1A 243R-transactivated PCNA expression requires an intact RFX1 site.
Since p107 and RFX1 are components of complex EH1 and EH1 formation is dependent upon sequences that are important for RFX1 binding, it seemed possible that p107 inhibition might be effected via the RFX1 site. We therefore determined whether p107 overexpression can reverse the E1A 243R-induced expression from the PCNA promoter containing mutations in the RFX1-binding site. HeLa cells were cotransfected with either wild-type PCNA−87 CAT or −44/−40 CAT and increasing amounts of p107 to assess the effect of this promoter mutation on the inhibitory action of overexpressed p107. The −44/−40 mutation increased basal transcription levels slightly compared to those of the wild-type promoter, but overexpression of p107 had no significant effect on basal expression in either case. As in Fig. 1 and 2, p107 overexpression greatly reduced E1A 243R-induced PCNA-CAT levels from those of wild-type PCNA−87 CAT (Fig. 4), but was much less inhibitory with the −44/−40 CAT reporter (Fig. 4). To eliminate confounding effects of squelching, we repeated this experiment with the mutant p107DE protein. The results of Fig. 5 show that p107DE behaved similarly to wild-type p107 in that it antagonized E1A 243R-transactivated PCNA-CAT levels with the −44/−40 promoter mutation only weakly compared to the wild-type promoter. Thus, the −44/−40 promoter mutation significantly relieves the inhibition of E1A-induced transactivation by p107.
FIG. 4.

p107 inhibition of E1A 243R-induced PCNA expression is dependent on the RFX1-binding site. HeLa cells were transfected with either wild-type PCNA−87 CAT or −44/−40 CAT reporter plasmid (10 μg), pCMV12S.FS or pCMV12S, and increasing amounts of pCMV107 (0, 2, or 4 μg) or pCMV107DE (0, 2, 4, or 6 μg) expression plasmid. CAT activity was calculated as described in the legend to Fig. 1 and represents the average of three independent experiments performed in duplicate with standard deviations indicated.
FIG. 5.

Inhibition of E1A-induced PCNA activity by p107DE is also dependent upon RFX1-binding site sequences. HeLa cells were transfected with either wild-type (WT) PCNA−87 CAT or −44/−40 CAT reporter plasmid (10 μg), pCMV12S.FS or pCMV12S, and increasing amounts of pCMV107DE (0, 2, 4, or 6 μg) expression plasmid. CAT activity was calculated as described in the legend to Fig. 1 and represents the average of three independent experiments performed in duplicate with standard deviations indicated.
We extended these experiments to further promoter mutants to determine whether reversal of E1A 243R-induced transactivation by p107 requires an intact RFX1 site. The −44/−40 and −46/−39 mutations, which both adversely affect RFX1 binding and EH1 complex formation in vitro (36) (Fig. 3B), eliminated the ability of p107 to antagonize E1A 243R transactivation of the PCNA promoter (Fig. 6). In contrast, p107 could still reverse E1A 243R-induced expression directed by PCNA promoters containing mutations −53GT and −56GA. These point mutations lie within the PERE and reduce transactivation by E1A 243R but reside outside the RFX1-binding site and do not adversely affect RFX1 binding and EH1 complex formation in vitro (Fig. 3B, lane 5) (39a). These data support the conclusion that the repressive effect of p107 on E1A-activated PCNA expression is exerted through PCNA promoter sequences which constitute part of an RFX1 consensus binding site.
FIG. 6.

Mutations that specifically affect the RFX1-binding site abrogate reversal of E1A 243R-induced PCNA expression by p107. Wild-type (WT) and mutant (−44/−40, −46/−39, −53GT, −56GA CAT) PCNA-CAT reporter constructs (10 μg) were cotransfected with either 0.5 μg of pCMV12S.FS (control) or pCMV12S (E1A 243R) and with or without pCMV107 expression plasmid (2 μg). The fold increase ± standard deviation in CAT expression was normalized for β-galactosidase activity and represents the average of three independent experiments done in duplicate.
DISCUSSION
p107 is a member of the pocket family of proteins that includes the retinoblastoma tumor suppressor pRB and the p130 protein (15, 18, 21, 26, 29, 30, 39). Overexpression of p107 arrests cells in the G1 phase of the cell cycle (12, 68), suggesting that it plays an important role in cell cycle progression. Because p107 is a nuclear phosphoprotein that can bind to and modulate the activity of transcription factors, including E2F (16, 22, 38), one of its biological functions likely involves the transcriptional regulation of genes involved in the control of cell proliferation. Therefore, the downregulation of PCNA expression by p107 may constitute one of the pathways by which this E1A-associated protein can influence the cell cycle.
In addition to the well-characterized regulatory mechanism mediated by E2F (16, 22, 38), p107 can also repress transcription independently of its ability to interact with the E2F family of transcription factors (13). In fact, p107 has also been shown to bind to and inhibit the transactivation domain of the c-myc transcription factor (24). However, we did not observe any effect of E2F overexpression in transient expression assays with the minimal PCNA E1A-responsive promoter (data not shown). The lack of clearly defined E2F or c-myc binding sites in the minimal PCNA E1A-responsive promoter, and the failure to detect E2F in PCNA promoter mobility shift assays by either oligonucleotide competition or antibody interference (data not shown), also argued that antagonism of E1A-induced PCNA expression by p107 occurs via a novel inhibitory mechanism. This reversal of E1A-induced PCNA expression by p107 is congruous with p107’s known transcriptional repression properties. While p107 overexpression has no significant effect on basal transcription from the PCNA promoter, it antagonizes dramatically the transactivation of the PCNA promoter by E1A 243R. Since p107DE and other derivatives which cannot bind E1A 243R retain the ability to antagonize E1A 243R-induced promoter expression, only a minor portion of this inhibition occurs via a squelching mechanism. Evidently, p107’s ability to reverse E1A 243R-transactivated PCNA expression resides in domains outside its pocket region.
Antibody interference experiments demonstrated that the retinoblastoma-related protein p107 and RFX1 participate in the same DNA-protein complex in vitro, but the existence and nature of any physical or functional interaction between the two proteins remain to be determined. Moreover, in contrast to the clear-cut effects of p107 overexpression, in vivo assays with RFX1 have been inconclusive to date. RFX1 overexpression nonspecifically increased both basal and E1A-transactivated PCNA promoter activity as well as the expression of β-galactosidase from control plasmids, so no firm conclusions could be drawn (data not shown). Nevertheless, mutations that abolish the RFX1-binding site and adversely affect the formation of complex EH1 abrogated the ability of p107 to reverse E1A-induced PCNA promoter activity, suggesting that the inhibition by p107 requires these cis-acting sequences. Taken together, these results argue that p107 associates with the human PCNA promoter via the RFX1-binding site (through some as yet uncharacterized protein-protein interaction with RFX1 or another factor) to reverse E1A 243R transactivation of PCNA. In support of this notion, p107 can act as a general transcriptional repressor protein when tethered to DNA either as a GAL4 fusion protein or by virtue of its capacity to bind to E2F (57). Thus, it is possible that p107 can interfere with the function of one or several of the components of the basal transcription machinery (57). Our findings with the PCNA promoter imply that the p107-sensitive step is rate limiting for E1A-activated transcription, but not for basal transcription, at least in HeLa cells. Although it is possible that the human papillomavirus E7 protein (which is present in these cells and can also bind p107) participates in the regulation of the PCNA promoter, no significant effects have been detected in transfection experiments carried out to date (not shown).
Recently, we demonstrated that E1A 243R can target and transactivate the PCNA promoter through interactions of the PERE with the cellular coactivator protein CBP and CREB (37). In view of complementation studies with different E1A mutants suggesting the existence of functional domains within E1A that simultaneously associate with both p107 and p300/CBP to optimally transactivate PCNA expression (32), we propose that transactivation of the PCNA promoter by E1A occurs via a multistep mechanism (Fig. 7). According to this model, E1A 243R initially targets the PERE via a CBP-CREB-DNA interaction (Fig. 7A). The E1A 243R-CBP interaction by itself may suffice to stimulate the PCNA promoter (Fig. 7B), because CBP is known to interact with components of the basal transcription machinery, in particular, TFIIB (14, 34). Once targeted to the promoter, however, E1A 243R may exert additional effects which contribute to PCNA activation: as illustrated in Fig. 7C, E1A 243R may simultaneously interact with p107, either directly or indirectly, to mediate the relief of transcriptional repression exerted by p107 on the PCNA promoter.
FIG. 7.

Model for activation of the human PCNA promoter by E1A 243R. Schematic diagram of the minimal PCNA-E1A-responsive promoter is illustrated with the cis-acting PERE boxed in gray, the RFX1-binding site, and the cellular factors indicated. A possible mechanism by which E1A 243R might transactivate the PCNA promoter is depicted sequentially. (A) E1A 243R first targets the PCNA promoter at the PERE site via a CBP-CREB-PERE pathway (37). (B) The binding between E1A and CBP activates the promoter by itself through an interaction between CBP and components of the general transcription machinery (general transcription factors [GTFs]). (C) E1A 243R increases PCNA promoter expression by mediating the relief of a transcriptional repression exerted on the promoter by p107. Note that the events depicted in panels B and C might take place simultaneously.
This model provides a link between the p107 protein and the human PCNA promoter, a relationship which had previously been inferred only from mutational studies of E1A 243R and correlation with the known E1A-binding proteins (32). It readily explains the observations reported here which implicate p107 in the activation of PCNA by E1A. Presumably, E1A 243R that is transfected into cells relieves transcriptional repression of the PCNA promoter mediated by endogenous p107 present in these cells. When p107 is overexpressed, however, the amounts of E1A present in the cell are insufficient to wholly overcome the inhibition and relieve the repression (Fig. 1 and 2). Therefore, overexpression of p107 appears to antagonize the capacity of E1A to transactivate the PCNA promoter in these transient expression assays. Furthermore, the coordinate action of E1A 243R mediated via p300/CBP and p107 at the PCNA promoter (Fig. 7C) can account for observations implying that the p300/CBP and p107 binding regions have to be on the same E1A protein molecule for full activation of the PCNA promoter by E1A 243R (32). This synergy between p107 and p300/CBP in the regulation of PCNA gene expression sheds new light on the pathways and networks of interactions which control the cell cycle and normal cellular growth.
ACKNOWLEDGMENTS
B.H.L. and M.L. contributed equally to this work.
We thank P. Wendel for excellent technical assistance. We are grateful to P. Hearing for RFX1 reagents and M. Ewen for p107 expression plasmids and to C. Labrie for helpful discussions and contributions at the initial stages of this study.
This work was supported by National Institutes of Health grant CA 13106.
REFERENCES
- 1.Almendral J M, Huebusch D, Blundell P A, Macdonald-Bravo H, Bravo R. Cloning and sequence of the human nuclear protein cyclin: homology with DNA-binding proteins. Proc Natl Acad Sci USA. 1987;84:1575–1579. doi: 10.1073/pnas.84.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arany Z, Sellers W R, Livingston D M, Eckner R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell. 1994;77:799–800. doi: 10.1016/0092-8674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 3.Arany Z, Newsome D, Oldread E, Livingston D M, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 4.Bauer G A, Burgers P M J. Molecular cloning, structure, and expression of the yeast proliferating cell nuclear antigen gene. Nucleic Acids Res. 1990;18:261–265. doi: 10.1093/nar/18.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayley S T, Mymryk J S. Adenovirus E1A proteins and transformation. Int J Oncol. 1994;5:425–444. doi: 10.3892/ijo.5.3.425. [DOI] [PubMed] [Google Scholar]
- 6.Berk A J. Adenovirus promoters and E1A transactivation. Annu Rev Genet. 1986;20:45–79. doi: 10.1146/annurev.ge.20.120186.000401. [DOI] [PubMed] [Google Scholar]
- 7.Berk A J, Sharp P A. Structure of adenovirus 2 early mRNAs. Cell. 1978;14:695–711. doi: 10.1016/0092-8674(78)90252-0. [DOI] [PubMed] [Google Scholar]
- 8.Bravo R, Macdonald-Bravo H. Induction of the nuclear protein ‘cyclin’ in quiescent mouse 3T3 cells stimulated by serum and growth factors. Correlation with DNA synthesis. EMBO J. 1984;3:3177–3181. doi: 10.1002/j.1460-2075.1984.tb02276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bravo R, Frank R, Blundell P A, Macdonald-Bravo H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-d. Nature. 1987;326:515–517. doi: 10.1038/326515a0. [DOI] [PubMed] [Google Scholar]
- 10.Cao L, Faha B, Dembski M, Tsai L-H, Harlow E, Dyson N. Independent binding of the retinoblastoma protein and p107 to the transcription factor E2F. Nature (London) 1992;355:176–179. doi: 10.1038/355176a0. [DOI] [PubMed] [Google Scholar]
- 11.Chow L T, Broker T R, Lewis J B. Complex splicing patterns of RNAs from the early regions of adenovirus-2. J Mol Biol. 1979;134:265–303. doi: 10.1016/0022-2836(79)90036-6. [DOI] [PubMed] [Google Scholar]
- 12.Claudio P P, Howard C M, Baldi A, De Luca A, Fu Y, Condorelli G, Sun Y, Colburn N, Calabretta B, Giordano A. p130/pRb2 has growth suppressive properties similar to yet distinctive from those of retinoblastoma family members pRb and p107. Cancer Res. 1994;54:5556–5560. [PubMed] [Google Scholar]
- 13.Dagnino L, Zhu L, Skorecki K L, Moses H L. E2F-independent transcriptional repression by p107, a member of the retinoblastoma family of proteins. Cell Growth Differ. 1995;6:191–198. [PubMed] [Google Scholar]
- 14.Dallas P B, Yaciuk P, Moran E. Characterization of monoclonal antibodies raised against p300: both p300 and CBP are present in intracellular TBP complexes. J Virol. 1997;71:1726–1731. doi: 10.1128/jvi.71.2.1726-1731.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies R, Hicks R, Crook R, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devoto S H, Mudryj M, Pines J, Hunter T, Nevins J R. A cyclin A-protein kinase complex possesses sequence-specific DNA binding activity: p33cdk2 is a component of the E2F-cyclin A complex. Cell. 1992;68:167–176. doi: 10.1016/0092-8674(92)90215-x. [DOI] [PubMed] [Google Scholar]
- 17.Dignam D J, Lebovitz R M, Roeder R G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dyson N, Howley P M, Münger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 19.Dyson N, Harlow E. Adenovirus E1A targets key regulators of cell proliferation. Cancer Surv. 1992;12:161–195. [PubMed] [Google Scholar]
- 20.Eckner R, Ewen M E, Newsome D, Gerdes M, deCaprio J A, Lawrence J B, Livingston D M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 21.Ewen M E, Xing Y, Lawrence J B, Livingston D M. Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell. 1991;66:1155–1164. doi: 10.1016/0092-8674(91)90038-z. [DOI] [PubMed] [Google Scholar]
- 22.Ewen M E, Faha B, Harlow E, Livingston D M. Interaction of p107 with cyclin A independent of complex formation with viral oncoproteins. Science. 1992;255:85–87. doi: 10.1126/science.1532457. [DOI] [PubMed] [Google Scholar]
- 23.Faha B, Ewen M E, Tsai L-H, Livingston D M, Harlow E. Interaction between human cyclin A and adenovirus E1A-associated p107 protein. Science. 1992;255:87–90. doi: 10.1126/science.1532458. [DOI] [PubMed] [Google Scholar]
- 24.Gu W, Bhatia K, Magrath I T, Dang C V, Dalla-Favera R. Binding and suppression of the myc transcriptional activation domain by p107. Science. 1994;264:251–254. doi: 10.1126/science.8146655. [DOI] [PubMed] [Google Scholar]
- 25.Hall C V, Jacob P E, Ringold G M, Lee F. Expression and regulation of Escherichia coli lacZ fusions in mammalian cells. J Mol Appl Genet. 1983;2:101–109. [PubMed] [Google Scholar]
- 26.Hannon G J, Demetrick D, Beach D. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev. 1993;7:2378–2391. doi: 10.1101/gad.7.12a.2378. [DOI] [PubMed] [Google Scholar]
- 27.Herbomel P, Bourachot B, Yaniv M. Two distinct enhancers with different cell specificities coexist in the regulatory region of polyoma. Cell. 1984;39:653–662. doi: 10.1016/0092-8674(84)90472-0. [DOI] [PubMed] [Google Scholar]
- 28.Herrmann C H, Mathews M B. The adenovirus E1B 19-kilodalton protein stimulates gene expression by increasing DNA levels. Mol Cell Biol. 1989;9:5412–5423. doi: 10.1128/mcb.9.12.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Q, Dyson N, Harlow E. The regions of the retinoblastoma protein needed for binding to adenovirus E1A or SV40 large T antigen are common sites for mutations. EMBO J. 1990;9:1147–1155. doi: 10.1002/j.1460-2075.1990.tb08221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S, Wang N P, Tseng B Y, Lee W-H, Lee E Y. Two distinct and frequently mutated regions of the retinoblastoma protein are required for binding to SV40 T antigen. EMBO J. 1990;9:1815–1822. doi: 10.1002/j.1460-2075.1990.tb08306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaskulski D, deRiel J K, Mercer W E, Calabretta B, Baserga R. Inhibition of cellular proliferation by antisense oligodeoxynucleotides to PCNA cyclin. Science. 1988;240:1544–1546. doi: 10.1126/science.2897717. [DOI] [PubMed] [Google Scholar]
- 32.Kannabiran C, Morris G F, Labrie C, Mathews M B. The adenovirus E1A 12S product displays functional redundancy in activating the human proliferating cell nuclear antigen promoter. J Virol. 1993;67:507–515. doi: 10.1128/jvi.67.1.507-515.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katan Y, Agami R, Shaul Y. The transcriptional activation and repression domains of RFX1, a context-dependent regulator, can mutually neutralize their activities. Nucleic Acids Res. 1997;25:3621–3628. doi: 10.1093/nar/25.18.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwok R P S, Lundblad J R, Chrivia J C, Richards J P, Bächinger H P, Brennan R G, Roberts S G E, Green M R, Goodman R H. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 35.Labrie C, Morris G F, Mathews M B. A complex promoter element mediates transactivation of the human proliferating cell nuclear antigen promoter by the 243-residue adenovirus E1A oncoprotein. Mol Cell Biol. 1993;13:1697–1707. doi: 10.1128/mcb.13.3.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labrie C, Lee B H, Mathews M B. Transcription factors RFX1/EF-C and ATF-1 associate with the E1A-responsive element of the human proliferating cell nuclear antigen promoter. Nucleic Acids Res. 1995;23:3732–3741. doi: 10.1093/nar/23.18.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee B H, Mathews M B. Transcriptional coactivator cAMP response element binding protein mediates induction of the human proliferating cell nuclear antigen promoter by the adenovirus E1A oncoprotein. Proc Natl Acad Sci USA. 1997;94:4481–4486. doi: 10.1073/pnas.94.9.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lees J A, Saito M, Vidal M, Valentine M, Look T, Harlow E, Dyson N, Helin K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 1992;13:7813–7825. doi: 10.1128/mcb.13.12.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Graham C, Lacy S, Duncan A M V, Whyte P. The adenovirus E1A-associated 130-kd protein is encoded by a member of the retinoblastoma gene family and physically interacts with cyclins A and E in a temporally distinct manner. Genes Dev. 1993;7:2366–2377. doi: 10.1101/gad.7.12a.2366. [DOI] [PubMed] [Google Scholar]
- 39a.Liu, M., B. H. Lee, and M. B. Mathews. Submitted for publication.
- 40.Liu Y-C, Marraccino R L, Keng P C, Bambara R A, Lord E M, Chou W-G, Zain S B. Requirement for proliferating cell nuclear antigen expression during stages of the Chinese hamster ovary cell cycle. Biochemistry. 1989;28:2967–2974. doi: 10.1021/bi00433a034. [DOI] [PubMed] [Google Scholar]
- 41.Lundblad J R, Kwok R P, Laurance M E, Harter M L, Goodman R H. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional c-activator CBP. Nature. 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto K, Moriuchi T, Koji T, Nakane P K. Molecular cloning of cDNA coding for rat proliferating cell nuclear antigen (PCNA)/cyclin. EMBO J. 1987;6:637–642. doi: 10.1002/j.1460-2075.1987.tb04802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moran E. DNA tumor virus transforming proteins and the cell cycle. Curr Opin Genet Dev. 1993;3:63–70. doi: 10.1016/s0959-437x(05)80342-9. [DOI] [PubMed] [Google Scholar]
- 44.Moran E, Mathews M B. Multiple functional domains in the adenovirus E1A gene. Cell. 1987;48:177–188. doi: 10.1016/0092-8674(87)90418-1. [DOI] [PubMed] [Google Scholar]
- 45.Morris G F, Mathews M B. Regulation of the proliferating cell nuclear antigen during the cell cycle. J Biol Chem. 1989;264:13856–13864. [PubMed] [Google Scholar]
- 46.Morris G F, Mathews M B. Analysis of the proliferating cell nuclear antigen promoter and its response to adenovirus early region 1. J Biol Chem. 1990;265:16116–16125. [PubMed] [Google Scholar]
- 47.Morris G F, Mathews M B. The adenovirus E1A transforming protein activates the proliferating cell nuclear antigen promoter via an activating transcription factor site. J Virol. 1991;65:6397–6406. doi: 10.1128/jvi.65.12.6397-6406.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris G F, Labrie C, Mathews M B. Modulation of transcriptional activation of the proliferating cell nuclear antigen promoter by the adenovirus E1A 243-residue oncoprotein depends on proximal activators. Mol Cell Biol. 1994;14:543–553. doi: 10.1128/mcb.14.1.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morris G F, Bischoff J R, Mathews M B. Transcriptional activation of the human proliferating cell nuclear antigen promoter by p53. Proc Natl Acad Sci USA. 1996;93:895–899. doi: 10.1073/pnas.93.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mymryk J S, Bayley S T. Multiple pathways for gene activation in rodent cells by the smaller adenovirus 5 E1A protein and their relevance to growth and transformation. J Gen Virol. 1993;74:2131–2141. doi: 10.1099/0022-1317-74-10-2131. [DOI] [PubMed] [Google Scholar]
- 51.Nevins J R. Mechanisms of viral-mediated trans-activation of transcription. Adv Virus Res. 1989;37:35–83. doi: 10.1016/s0065-3527(08)60832-5. [DOI] [PubMed] [Google Scholar]
- 52.Prelich G, Kostura M, Marshak D R, Mathews M B, Stillman B. The cell-cycle regulated proliferating cell nuclear antigen is required for SV40 DNA replication in vitro. Nature. 1987;326:471–475. doi: 10.1038/326471a0. [DOI] [PubMed] [Google Scholar]
- 53.Prelich G, Tan C-K, Kostura M, Mathews M B, So A G, Downey K M, Stillman B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-d auxiliary protein. Nature. 1987;326:517–520. doi: 10.1038/326517a0. [DOI] [PubMed] [Google Scholar]
- 54.Ruley E. Transforming collaborations between ras and nuclear oncogenes. Cancer Cells. 1990;2:258–268. [PubMed] [Google Scholar]
- 55.Shenk T, Flint J. Transcriptional and transforming activities of the adenovirus E1A proteins. Adv Cancer Res. 1991;57:47–85. doi: 10.1016/s0065-230x(08)60995-1. [DOI] [PubMed] [Google Scholar]
- 56.Spindler K R, Eng C Y, Berk A J. An adenovirus early region 1A protein is required for maximal viral DNA replication in growth-arrested human cells. J Virol. 1985;53:742–750. doi: 10.1128/jvi.53.3.742-750.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Starostik P, Chow K N B, Dean D C. Transcriptional repression and growth suppression by the p107 pocket protein. Mol Cell Biol. 1996;16:3606–3614. doi: 10.1128/mcb.16.7.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan C-K, Castillo C, So A G, Downey K M. An auxiliary protein for DNA polymerase-d from fetal calf thymus. J Biol Chem. 1986;261:12310–12316. [PubMed] [Google Scholar]
- 59.Waga S, Stillman B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature. 1994;369:207–212. doi: 10.1038/369207a0. [DOI] [PubMed] [Google Scholar]
- 60.Whyte P, Williamson N M, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 61.Wigler M, Pellicer A, Silverstein S, Axel R. Biochemical transfer of single-copy eukaryotic genes using total cellular DNA as donor. Cell. 1978;14:725–731. doi: 10.1016/0092-8674(78)90254-4. [DOI] [PubMed] [Google Scholar]
- 62.Wold M S, Li J, Weinberg D H, Varshup D M, Verkeyen E, Kelly T. Cellular proteins required for SV40 DNA replication in vitro. Cancer Cells. 1988;6:133–141. [Google Scholar]
- 63.Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:505–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- 64.Xiong Y, Hannon G J, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 65.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 1993;7:1572–1583. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 66.Zerler B, Roberts R J, Mathews M B, Moran E. Different functional domains of the adenovirus E1A gene are involved in the regulation of host cell cycle products. Mol Cell Biol. 1987;7:821–829. doi: 10.1128/mcb.7.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Xiong Y, Beach D. Proliferating cell nuclear antigen and p21 are components of multiple cell cycle kinase complexes. Mol Biol Cell. 1993;4:897–906. doi: 10.1091/mbc.4.9.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]