

Abstract
Herpes simplex virus (HSV-1) employs heparan sulfate (HS) as receptor for cell attachment and entry. During late-stage infection, the virus induces the upregulation of human heparanase (Hpse) to remove cell surface HS allowing viral spread. We hypothesized that inhibition of Hpse will prevent viral release thereby representing a new therapeutic strategy for HSV-1. A range of HS-oligosaccharides was prepared to examine the importance of chain length and 2-O-sulfation of iduronic moieties for Hpse inhibition. It was found that hexa- and octasaccharides potently inhibited the enzymes and that 2-O-sulfation of iduronic acid is tolerated. Computational studies provided a rationale for the observed structure-activity relationship. Treatment of human corneal epithelial cells (HCEs) infected with HSV-1 with the hexa- and octasaccharide blocked viral induced shedding of HS which significantly reduced spread of virions. The compounds also inhibited migration and proliferation of immortalized HCEs thereby providing additional therapeutic properties.
Keywords: Drug discovery, Heparin, Heparanase, Herpes simplex virus type 1, Antivirals
Graphical Abstract
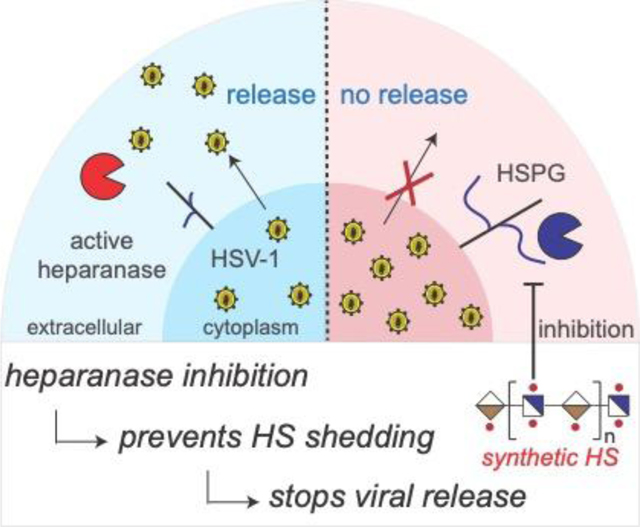
Table of Contents Text: A range of heparan sulfate (HS) oligossacharides was synthesized by a modular synthetic approach and evaluated as human heparanase (Hpse) inhibitors. Treatment of herpes simplex-1 virus (HSV-1) infected corneal epithelial cells with synthetic HS oligosaccharide inhibitors prevented shedding of cell-surface HS, thereby impeding viral release and spread to other cells.
Introduction
Human heparanase (Hpse) is an endo-β-D-glucuronidase that orchestrates the remodelling of extracellular matrix (ECM) and basement membrane by hydrolysing heparan sulfate proteoglycans (HSPGs),[1] which are protein-anchored anionic polysaccharides of alternating glucosamine (GlcN) and uronic acids (glucuronic acid, GlcA or iduronic acid, IdoA) with varying levels of sulfation.[2] The substrate specificity of Hpse is rather broad and primarily defined by the pattern of sulfation around a potential cleavage site and appropriate substrates are thought to be at the border of highly sulfated and lesser sulfated regions.[3] Cleavage of HS chains results in the release of sequestered growth factors, cytokines, and lipoproteins thereby mediating many biological processes.[4] Hpse has been implicated in various diseases such as tumour metastasis, type-1 diabetes, renal fibrosis, and inflammation.[5] As a result, the development of Hpse inhibitors has received considerable attention and various approaches are being pursued such as modified heparins, sulfated oligosaccharides, small molecules and anti-Hpse antibodies.[6]
Hpse also regulates the lifecycle of many human viruses such as dengue virus (DENV), human papilloma virus (HPV), respiratory syncytial virus (RSV), adenovirus (ADNV), hepatitis C virus (HCV) and herpes simplex virus (HSV).[7] HSV is an enveloped double-stranded DNA virus that belongs to alpha-herpesvirus subfamily.[8] HSV type-1 is a major cause of corneal keratitis and encephalitis, which when left untreated can result in the loss of vision, neurological deficits, seizures or even death.[9] Although acyclovir and derivatives thereof can treat active herpes infections, these drugs do not establish permanent viral latency, allowing the virus to reactivate and cause clinical disease at a later point in time.[10] Therefore, there is a need to develop therapeutic strategies that target other key steps in the viral lifecycle such as entry and spread to other cells and tissues.
HSV-1 uses cell surface HSPGs of host cells as receptor.[11] Infection is initiated by attachment of viral glycoprotein B (gB) and gC to cell surface HSPGs. Next, gD, engages with one of its receptors, nectin-1, herpes virus entry mediator (HVEM) or 3-O-sulfated HS, resulting in a conformational change and exposure of a fusion-activating domain.[12] Activated gD binds to a heterodimer of the glycoproteins, gH and gL, resulting in cell penetration and capsid release. After replication, HSV-1 leaves host cells and spreads to other uninfected cells, which is facilitated by viral glycoprotein heterodimer, gE/gI redistribution to cell junctions. At these tight and adherent junctions, the virus utilizes gE/gI to attach to its receptors that support lateral spread from infected to uninfected cells. HSV can also produce fusion pores at these junctions to egress or be released into the ECM.[13]
Previously, we described that HSV-1 has a remarkable ability to modulate HS biosynthesis for optimal infection and viral spread.[14] During the initial stage of infection, cellular HS biosynthesis is upregulated enhancing viral attachment and cell entry. In later stages of infection, the expression of Hpse is upregulated ensuring that HSPG’s are cleaved resulting in the detachment of the virus.[7, 13b] We anticipated that inhibition of Hpse may prevent viral release and may represent a therapeutic strategy for HSV-1 and other viruses that rely on Hpse for infection.
In this study, we designed and synthesized differently sulfated di-, tetra-, hexa- and octasaccharides and evaluated their ability to inhibit Hpse activity by a colorimetric[15] and homogeneous time-resolved fluorescence (HTRF)[16] assay. It was found that increasing the chain length of the HS oligosaccharides results in more potent inhibition, and it revealed that 2-O-sulfation of IdoA is tolerated by the enzyme. Molecular modeling studies provided a rationale for the observed structure-activity relationship (SAR). Treatment of human corneal epithelial cells (HCEs) infected with HSV-1 with the hexa- and octasaccharide blocked viral induced shedding of HS which in turn resulted in a significant reduction of shedded virions. The compounds also inhibited migration and proliferation of immortalized HCEs thereby providing additional therapeutic properties.
Results and Discussion
Design and chemical synthesis of well-defined HS oligosaccharides as inhibitors of Hpse
Hpse is a retaining endoglycosidase that cleaves GlcA-GlcNS linkages of polymeric HS.[17] A crystal structure of Hpse in complex with a tetrasaccharide has provided a structural rationale for the catalytic mechanism and substrate recognition by this enzyme.[18] It has a large cleft in which residues Glu343 and Glu225 function as catalytic nucleophile and acid-base required for a retaining catalytic cleavage mechanism. Furthermore, the binding cleft is lined with basic amino acids (Lys159, Lys161, Lys231, Arg272, Arg273, Lys274 and Arg303) important for binding of negative charged HS chains. The sulfates of GlcNS(6S) at the −2 subsite and GlcNS6S at the +1 subsite make ionic interactions with side chains of basic amino acid in the binding cleft. Previous inhibition studies with differently sulfated trisaccharides had already indicated the importance of these sulfates for substrate recognition.[19] The co-crystal structure showed that IdoA at the −1 subsite, where glycosidic bond cleavage takes place, adopts a 2SO conformation. However, in this configuration the catalytic nucleophile and acid-base are not properly positioned for glycosidic bond cleavage making IdoA-containing compounds resistant to Hspe cleavage. The X-ray structures also indicated that GlcUA(2S) or IdoUA(2S) cannot be accommodated at the −1 subsite because of steric clashes between the 2-O-sulfate and Asn224.[18] We examined the binding cleft of Hpse (apo-structure PDB: 5E8M)[18], which is 30 Å in length, and anticipated that it can accommodate oligosaccharides as large as an octasaccharide. Therefore, we expected that IdoA containing HS-oligosaccharides of increasing chain length will provide increasingly potent inhibitors. It is known that glycol split heparins, in which vicinal diols have been oxidized by sodium periodate followed reduction of the resulting aldehydes by sodium borohydride to alcohols, can potently inhibit Hpse.[20] This oxidation affects mainly non-sulfated glucuronic and iduronic acid residues, and as a result glycol split heparin contains mainly IdoA2S moieties.[21] Thus, these observations indicate that Hpse can accommodate IdoA2S residues. Based on these findings, we were compelled to synthesize HS oligosaccharides 1-6 (Fig. 1D) and probe their inhibitory potential for Hpse. Compounds 1-4 were designed to probe the influence of C-2 sulfation of IdoA on inhibitory activity and compounds 5 and 6 to examine the importance of oligosaccharide length. Compounds 1-6 were prepared by a modular synthetic approach utilizing a set of disaccharide building blocks that mimic sulfated disaccharide moieties in HS.[22] At an appropriate stage of synthesis, levulinoyl (Lev) esters can selectively be removed to provide alcohols for sulfation.
Figure 1.

Rational design of inhibitor of human heparanase (Hpse). (A) Surface representation of Hpse enzyme; (B) Details of the positively charged residues composing the substrate binding cleft; (C) Surface and stick representation of GlcNS-GlcA-GlcNS-GlcA-pNP bound to Hpse; (D) Chemical structure of target compounds 1-6.
The preparation of hexasaccharide 5 is described in Scheme 1. Thus, a triflic acid (TfOH) mediated glycosylation of glycosyl acceptor 7 with donor 8 yielded tetrassacharide 9 as only the α-anomer. The anomeric configuration was confirmed by the 3JH1,H2 coupling constant (3.9 Hz) and 13C chemical shifts of C-1 (97.7 ppm) (see Supplementary information). The fluorenylmethyloxycarbonate (Fmoc) group of 9 was removed by triethylamine (Et3N) in dichloromethane to give tetrasaccharide acceptor 10 that was glycosylated with disaccharide donor 11 to obtain hexassacharide 12 as only the α-anomer. The Lev esters were removed by treatment with hydrazine acetate and the resulting alcohols were sulfated by previously optimized conditions employing sulfur trioxide-triethylamine complex (SO3.NEt3) at elevated temperature (60°C) for a prolonged reaction time (16 h).[23] However, under these conditions, the Fmoc protection group was removed and the resulting alcohol get sulfated. To avoid oversulfation, the Fmoc of 12 was replaced by an acetyl ester to give 13 by removal of Fmoc by Et3N in dichloromethane followed by acetylation of the resulting hydroxyl by acetic anhydride in pyridine in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP). The Lev esters of 13 were removed by treatment with hydrazine acetate, and the resulting alcohols were selectively sulfated using SO3.NEt3 in dimethylformamide (DMF) at 60°C. Next, the methyl esters were saponified by first treatment with hydrogen peroxide (H2O2) and lithium hydroxide (LiOH) in tetrahydrofuran (THF) followed by saponification of the other esters using sodium hydroxide (NaOH) in methanol (MeOH) to give 14. Under Staudinger conditions using trimethylphosphine (PMe3) and NaOH, the azides of 14 were reduced to amines, which were N-sulfated employing sulfur trioxide-pyridine complex (SO3.Py) in MeOH in the presence of Et3N and NaOH to provide 15. Finally, the target hexasaccharide 5 was obtained by global deprotection of 15 involving two-step hydrogenation by first using palladium on carbon (Pd/C) in a mixture of tert-butanol/water (tBuOH/H2O) to cleave the protecting group of the anomeric linker, and then over palladium hydroxide on carbon (Pd(OH)2/C) to remove the benzyl and naphthylmethyl ethers. The compound was purified by size exclusion column chromatography over a Biogel P-2 and followed by Na+ exchange using Dowex [Na+] resin. HS oligosaccharides 1-4 and 6 were prepared in a similar manner as detailed in Supplementary information (Schemes S1-S3). The target compounds 1-6 were obtained in quantities ranging from 5 to 10 mg and fully characterized by high-resolution electrospray ionization-mass spectrometry (ESI-MS) and nuclear magnetic resonance (NMR). 1H and 13C resonances were fully assigned by 1D and 2D (1H-1H correlation spectroscopy [COSY], 1H-1H total COSY [TOCSY], and 1H-13C heteronuclear single quantum coherence spectroscopy [HSQC]) NMR experiments (see Supplementary information). The sites of sulfation were confirmed by downfield shifts of ring protons (∼0.5 ppm) and by downfield shift of ring carbons (∼4 ppm). Furthermore, complete O- and N-sulfation results in appearance of peaks corresponding to C-2GlcN, C-6GlcN and C-2IdoA as distinct clusters (see HSQC NMR spectra in Supplementary information).
Scheme 1.

Modular synthesis of hexasaccharide 5.
Evaluation of HS oligosaccharides as Hpse inhibitors
HS oligosaccharides 1-6 bear only IdoA moieties in their backbones and therefore it was expected that they will be resistant to hydrolysis by Hpse. To biochemically confirm this resistance, we employed a colorimetric assay based on detection of newly formed reducing ends upon cleavage of scissile bonds by Hpse employing the water soluble tetrazole reagent-1 (WST-1, Fig. 2A).[15] Thus, compound 1-6 (50 μM) and Fondaparinux (Fpx., positive control) were incubated at 37°C with Hpse (50 nM) and reactions were developed by treatment with WST-1 (at 60°C for 1 h). Notably, none of the oligosaccharides were cleaved by Hpse even after an incubation time of 16h (Fig. 2B).
Figure 2.

Biochemical and biophysical evaluation of compound 1-6. (A) Schematic diagram for WST-1 assay; (B) Hydrolysis assays; (C) Schematic diagram for TR-FRET assay; (D) IC50 determination using TR-FRET assays; (E) Summary table of IC50; (F) Inhibitor screening using WST-1 assay; (G) Kinetic measurements to determine the inhibition constant (Ki) value of compound 6 (4.4 μM); (H) Summary table of Ki value, inhibition mode and coefficient of determination (R2); (I) Structure of biotinylated octamer (16); (J) Surface plasmon resonance (SPR) sensorgram of Hpse binding with 16. Data are presented as mean ± SD (n=3), representative experiments are shown which have been repeated at least three times.
The inhibitory potential of the synthetic compounds was evaluated by a homogenous time-resolved fluorescence assay (HTRF®, CisBio/PerkinElmer).[16] This assay is based on Förster resonance energy transfer (FRET) between two fluorophores; a donor (europium cryptate, K) and an acceptor (allophycocyanin, XL665). The time-resolved (TR) component eliminates short-lived background fluorescence which in combination with large Stokes shift makes the assays highly sensitive.[24] The HTRF® heparanase assay utilizes a bifunctional HS substrate (biotin-HS-K) labeled with europium cryptate and biotin. The biotin moiety of the HS substrate interacts with streptavidin-XL665 to form a FRET donor-acceptor complex which upon cleavage by Hpse is lost (Fig. 2C). First, we incubated biotin-HS-K either with Hpse or buffer control at 37°C for 1 h, followed by the addition of XL665 at room temperature for 20 min. As anticipated, in the presence of Hpse the substrate was hydrolysed and no FRET signal was observed, whereas the control reaction, in which the substrate is still intact, gave a strong signal (Fig. S2). Next, a wide range of concentrations of compounds 1-6 were employed in the HTRF® assay and half-maximal inhibitory concentrations (IC50 values) were determined by nonlinear regression of log(inhibitor) vs. response-variable slope (Fig. 2D and Fig. S3). It was found that compounds having a 2-OS moiety were somewhat more potent inhibitors than the corresponding oligosaccharides lacking this moiety (1 vs. 2 and 3 vs. 4). Furthermore, by increasing the chain length, the inhibitory potential increased substantially (1 vs. 3 vs. 5 vs. 6), and compound 6 exhibited an IC50 value of 1.1 μM (Fig. 2F). To examine whether a further increase in HS oligosaccharide chain length will result in subsequent improvement in inhibitory activity, isolated heparin oligomers (Iduron, UK) with degree of polymerization (DP) of 10, 12 and 14 were investigated (Fig. S4A). The DP-12 and −14 oligomers gave IC50 value of 0.38 and 0.14 μM, respectively (Fig. S4B-D), indicating that by using longer HS oligomers the activity can be further improved.
Enzyme kinetic studies can provide further insight in mode of inhibition of the synthetic compounds. To reliably characterize inhibition kinetics, full length HS is an unsuitable substrate because it is heterogeneous and provide multiple points of cleavage. Furthermore, fragments produced by the action of Hpse may invoke inhibition. In contrast, the pentasaccharide Fpx., which is clinically employed as an anti-coagulating agent, has a GlcA moiety that can be cleaved by Hpse introducing a reducing end that can be detected by WST-1.[15] Therefore, oligosaccharides 1-6 were examined as inhibitors of Hpse mediated hydrolysis of Fpx. at an initial concentration of 50 μM (Fig. 2F).
Hexasaccharide 5 and octasaccharide 6 showed promising inhibitory activities, while disaccharides 1 and 2 and tetrasaccharides 3 and 4 showed no detectable activity at the screening concentration. Next, inhibitory constants (Ki) of compounds 5 and 6 were determined by incubating various concentration of Fpx. (10 μM to 200 μM) with Hpse in the presence of inhibitors (5 or 6, 2 to 50 μM) and fitting the data to enzyme kinetics-inhibition equations built in Prism software. Good fits for competitive inhibition were obtained giving a Ki of 40.9 μM (R2 = 0.929) for 5 and 4.4 μM (R2 = 0.929) for 6 (Fig. 2G-H and Fig. S1).
Next, the binding affinity of the most potent inhibitor (6) for Hpse was evaluated by surface plasmon resonance (SPR). Thus, compounds 6 was biotinylated by coupling the anomeric aminopentyl linker with sulfo-N-hydroxysuccinamide-long chain (LC)-biotin to afford Octamer-LC-biotin (16, Fig. 2I), which was immobilized on the streptavidin-functionalized CM5 sensor chip. Binding experiments were performed by employing different concentrations of the Hpse as analytes (Fig. 2J, representative sensorgram of three independent runs is presented) and equilibrium dissociation constant (KD) value of 58 nM was determined using a 1:1 Langmuir binding model (For detailed fitting data see Fig. S5).
Molecular modelling to rationalize inhibition data
To rationalise the observed differences in inhibition potency, we performed docking studies of compounds 1, 3, 5, and 6 with Hpse.[25] The 3D structures of the oligosaccharides were built using the GLYCAM carbohydrate builder web tool (http://glycam.org) and energy minimized by GROMACS.[26] The resulting structures were superimposed to the tetrasaccharide ΔDP-4: ΔHexUA2S-GlcNS6S-IdoA-GlcNS6S observed in a co-crystal with Hpse (PDB: 5E9C)[18] using the PyMol alignment tool.[27] In particular, the first internal IdoA2S residue from the non-reducing end, was superimposed onto the IdoA ring of the ΔDP-4 which occupies the catalytic site of the enzyme (−1 site). After ligand replacement, the resulting structures were energy minimized and analysed for intermolecular interactions. It showed that the oligosaccharides can establish several intermolecular interactions with Hpse in a size-dependent manner. We observed engagement of the positively charged amino acids within the cleft of Hpse by sulfates of the oligosaccharides leading to shape complementarity. Octasaccharide 6 established 26 intermolecular interactions, of which 14 are hydrogen bonds and 12 are electrostatic (Fig. 3A-C and Table S1). Similarly, the hexasaccharide 5 established 24 polar intermolecular interactions, 13 of which are hydrogen bonds and 11 are electrostatic (Fig. S9 and Table S2). The tetrasaccharide 3 established 21 intermolecular interactions of which 11 are hydrogen bonds and 10 are electrostatic (Fig. S10 and Table S3), while the disaccharide 1 made 16 interactions of which 11 are hydrogen bonds and only five are electrostatic (Fig. S11 and Table S4). The difference in the number of intermolecular interactions correlates with their IC50 values. While the intermolecular interactions at the catalytic sites −1 and +1 are preserved for all studied oligosaccharides, the main difference lies in the contribution of the residues upstream and downstream of the cleft and in the case of larger hexa- and octasaccharides additional contacts could be made whereas this is not the case for the di- and tetrasaccharide.
Figure 3.

Docked structure of the Hpse in complex with 6. (A) Surface and stick representation, electrostatic potential surface, coloured by charge density; (B) Stick representation of the protein residues interacting with 6; (C) Schematic representation of the intermolecular interactions, electrostatic interactions; (D) Differences in IdoA and (E) IdoA2S binding at the Hpse catalytic site.
To investigate how the enzyme accommodates IdoA2S at the catalytic site −1, the 3D structure of IdoA2S-GlcNS6S-OMe was built and superimposed on the ΔDP-4 bound to Hpse (PDB: 5E9C). As expected, the presence of the sulfate group at the C-2 of the IdoA generated steric clashes with some residues within the catalytic pocket (Glu343, His296, and Asn224). After energy minimization, the resulting structure showed a rearrangement of several side chains of the residues surrounding the sulfate group (Fig. 3D-E and Fig. S6). As result, a larger cavity was formed to host the sulfate. In this cavity, Thr97, Asn224, His296, and Gln383 established hydrogen bonds with the sulfate, while residues Arg93 and Lys232 participated by electrostatic interactions through their positively charged side chains (Fig. 3E).
A comparison with the above-mentioned crystal structure of Hpse showed that the hydroxyl group at C-2 of IdoA forms hydrogen bonds with Asn224 and Glu343 (Fig. 3D). Glu343 has been identified as one of the two nucleophilic catalytic residues together with Glu225. Interestingly, the replacement of the hydroxyl by a sulfate moves back the negatively charged side chain of the Glu343 due to the repulsive electrostatic interaction. The larger distance between the carboxylate of the Glu343 and the anomeric carbon of the IdoA ring of the substrate may contribute to the resistance of IdoA2S containing oligosaccharides to Hpse-mediated hydrolysis.
Hpse inhibitors block viral egress by inhibiting shedding of cell surface HS during HSV-1 infection
Previously, we described that in response to HSV-1 infection, the expression of Hpse is upregulated, which results in HS shedding allowing outgoing virions to escape and spread to other cells.[14, 28] We hypothesized that inhibition of Hpse by compounds 5 and 6 will prevent virus induced HS degradation which in turn should block viral release. Thus, we pre-treated (−2 hours post-infection, hpi), neutralized (0 hpi) or therapeutically (2–12 hpi) administered compounds 5 or 6 to HCEs[29] infected with HSV-1 strain 17GFP, and examined the plaque count of viruses that remained associated with cells or released into the media.[30] The administration of compounds 5 and 6 at different time points during the infection experiments allowed for the identification of specific stages in the viral life cycle that are affected. The introduction of the compounds after infection, particularly at late time points (8 and 12 hpi), is expected to primarily influence viral release. On the other hand, pretreatment of the cells (−2 hpi) or simultaneous administration of the compound (0 hpi) along with the virus have a more significant impact on viral attachment and entry. At 24 hpi, cell lysates containing intracellular virus and supernatants containing extracellular egressed virus were collected and overlaid on Vero cells to perform plaque assays to count infectious particles (Fig. 4A-B). As expected, there were substantial reductions in extracellular virus in the samples that were treated pre- or post-infection with 5 and 6. Importantly, a large reduction was observed even when the inhibitors were administrated at 8 or 12 hpi supporting the notion the HS-oligosaccharides suppress viral release. To quantify the potency of the compounds in restricting viral spread, a fluorescence-based inhibition assays was performed using green fluorescent protein (GFP)-reporter virus at low multiplicity of infection (MOI, number of virions added per cell).[31] In this assay, viral spread can be quantified between control groups by measuring the percentage of GFP positive cells. Thus, HCEs were infected with 17 GFP virus at 0.1 MOI and at 2 hpi different concentrations of compounds 5, 6, and UFH were added. After 24 h, the ratio of infected cells (GFP-positive) vs. total cells (DAPI nuclear stain) was assessed to determine IC50 values (Fig. S12). In the absence of HS oligosaccharides or heparin, all cells were GFP positive demonstrating that viral spread had occurred. Both 5 and 6 inhibited HSV-1 spread and gave an IC50 value of 55 μg/mL and 42 μg/mL, respectively. Unfractionated heparin (UFH) is more potent inhibitor with an IC50 value of 1 μg/mL.
Figure 4.

HPSE inhibitors 5 and 6 decrease viral egress from HSV-1 infected HCEs. (A) Plaque assay analysis comparing extracellular and intracellular virus; (B) Extracellular virus collected from the cell supernatants for all time points were titrated and plaque counts are shown; (C) Addition of 5 and 6 sequesters cell surface HS during HSV-1 infection; (D) Quantification of fig. 4C; (E) Flow cytometry evaluation of HS shedding in infected HCEs; (F) Quantification of flow cytometry analysis conducted in fig. 4E. Data are presented as mean ± SEM (n=3), representative experiments are shown which have been repeated at least three times.
To evaluate possible inhibition of viral attachment and entry into HCEs by compounds 5, 6 and UFH, a recombinant virus (KOS gL86) was employed,[32] which expresses β-galactosidase derived from the lacZ gene that has been substituted for the gL gene that encodes a fusion glycoprotein. HCEs were concurrently exposed to the KOS gL86 virus and compound 5, 6 and UFH. The infection was allowed to continue for 6 h, facilitating β-galactosidase production, thereby serving as an indicator of viral entry. The levels of β-galactosidase activity were quantified using ortho-nitrophenyl β-D-galactopyranoside (ONPG) as a substrate. It was observed that all compounds robustly inhibited β-galactosidase activity, indicating that they also act as entry inhibitors. UFH impeded HSV-1 entry into HCEs with an IC50 value of 2 μg/mL, while compounds 5 and 6 displayed IC50 values of 27 μg/mL and 22 μg/mL, respectively (Fig. S13). Collectively, these results indicate that compounds 5 and 6 have a dual mode of action and can inhibit viral entry as well as release.
We performed additional studies to confirm that the reduction in egressed virus is due to blocking of virus-induced cell surface HS shedding. Thus, 2 h after mock or infection with HSV-1, HCEs were treated with compounds 5 or 6, and HS expression was examined 24 h post-infection by fluorescence microscopy using an anti-HS antibody (10E4 epitope)[14] and a secondary antibody labeled with AlexaFluor-546. Cells were stained with the antibodies prior to fixation to ensure only cell surface HS was detected (Fig. 4D). As expected, HSV-1 infection of HCEs resulted in the significant loss of cell surface HS. However, mean intensity density analysis of fluorescence intensities of HCEs treated with 5 and 6 showed substantially higher levels of cell surface HS, which was like that of non-infected cells (Fig. 4E).
These results indicate that compounds 5 and 6 prevent removal of HS from the cell surface during HSV-1 infection. Additional flow cytometry analysis was performed to quantify and compare cell surface HS expression more accurately. As can been seen in Fig. 4E-F, the number of cells that were positive for cell surface HS was much greater in samples treated with 5 and 6 during HSV-1 infection, further confirming that inhibition of Hpse prevents virus induced HS-shedding. It is of interest to note that there was no difference in non-infected samples that were treated with 5 or 6 indicating that the compounds do not interfere with baseline surface HS turnover.
Inhibition of cell migration and proliferation
In addition to being a pro-viral and pro-inflammatory factor, Hpse activity has been linked to various pro-survival activities such as cell migration, and proliferation.[33] It has been shown that exogenous administration of Hpse stimulates phosphatidylinositol 3-kinase (PI3K)-dependent endothelial cell migration and proliferation.[34] On the other hand, inhibition of Hpse suppresses cell migration and proliferation of non-infected cells.[35] Previously, we have shown that HSV-1 infected murine corneas that overexpress a constitutively active form of Hpse in the epithelium have reduced wound healing capacity.[36] We hypothesized that compounds 5 and 6 will improve HCEs wound healing ability by suppressing viral-induced Hpse activity.
Monolayer scratch assays are a cost effective and time efficient approach to determine the migratory potential of the cells in vitro.[37] Therefore, confluent monolayers of HCEs were disjointed by a single ~100 μm scratch followed by infection with mock or HSV-1 and the addition of 5, 6 or vehicle control. Cells from the edge of the scratch would migrate to the center and inhibition of Hpse was expected to slow this activity. The migration site was monitored for a period of 24 h by taking images at hourly intervals (Fig. 5A, non-infected and Fig. 5C, HSV-1 infected). The migration assay images were analyzed and the % of uncovered area was calculated in each frame of the image (Fig. 5B and 5D). Compounds 5 and 6 caused a significant decrease in migration only allowing ~75% and 50% coverage of the migration site, respectively in non-infected HCEs.
Figure 5.

Cell migration and cell proliferation is restricted by 5 and 6. (A) Image of the scratch site to monitor extent of migration of cells; (B) Quantification of uncovered area from fig. 5A; (C) Image of the scratch site to monitor HSV-1 infected cells; (D) Quantification of % uncovered area for images taken in fig. 5C; (E) Cell proliferation assays via flow cytometry; (F) Quantification of fig. 5E. Data are presented as mean ± SEM (n=3), representative experiments are shown which have been repeated at least three times.
Previously, we demonstrated that the expression and activity of Hpse increases upon HSV-1 infection.[36] The elevated Hpse activity corresponds with accelerated cell migration, which is also evident from the results presented here. However, starting at 12 hpi, the cells start to circularize and undergo cell death leading to an increase in uncovered area. As illustrated in Fig. 5B, the non-treated cells showed diminished wound healing capabilities primarily due to extensive infection and cell death. On the other hand, cells treated with 5 and 6 exhibited a significant increase in migration, indicating improved wound healing abilities that can be attributed to the antiviral activity of 5 and 6, as described above.
To examine whether the addition of Hpse inhibitors such as 5 and 6 cause a decrease in cell proliferation, we monitored cell proliferation of HCEs using CytoPainter assay. This assay is similar to a traditional Carboxyfluorescein succinimidyl ester (CFSE) assay where cells are initially dyed with the CFSE dye, and the mean fluorescence intensity depletes during every cell division cycle.[38] We performed a flow cytometric analysis of cells treated with 5 and 6 to determine the extent of cells retaining original fluorescence intensity (Fig. 5E-F). Quantitative analysis of the flow cytometry showed that 5 and 6 treated populations had significantly higher cells retaining the original fluorescence intensity when compared to vehicle control treated population. These results indicate that 5 and 6 inhibit cell-migration and proliferation in HCEs.
Evaluation of anticoagulation and cytotoxic effects
The use of heparin as a therapeutic is complicated by structural heterogeneity and the risk of causing bleeding and thrombocytopenia.[39] The hexa- and octa-saccharide described here are devoid of 3-O-sulfated GlcNS which is critical for the anticoagulation activity of heparin. To confirm this lack of activity, we measured AT-III mediated anti-Factor-IIa and anti-Factor-Xa activities of compounds 5, 6, and UFH using a colorimetric assay (Biophen Anti-IIa and Anti-Xa kits, two-stage chromogenic assays) (Figs. S15-S17). As anticipated, UFH gave an IC50 value of 0.1 μg/mL for Factor-IIa (Fig. S16) and 0.1 μg/mL for Factor-Xa (Fig. S17). Compounds 5 and 6 did not show any inhibition of Factor-IIa and inhibited Factor-Xa with high IC50 values of 85 and 52 μg/mL, respectively (Figs. S16 and S17).
The cytotoxicity of compounds 5, 6, and UFH was evaluated on HCE cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. It was found that compound 5 (8–1000 μM, 24 h), 6 (8–1000 μM, 24 h), and UFH 5 (0.8–100 μg/mL, 24 h) did not affect the viability of HCEs and exhibits no toxicity (Fig. S14).
Conclusion
We have designed and prepared a range of HS oligosaccharides to examine the importance of chain length and 2-O-sulfation of IdoA for Hpse inhibitory activity. In two different assay formats, it was found that by increasing the chain length of IdoA containing oligosaccharides, the inhibitory activity substantially improved. An octasaccharide composed of IdoA2S-GlcNS6S repeating units competitively inhibited the cleavage of Fpx. with Ki of 4.4 μM. Docking studies showed that the binding cleft of Hpse can accomodate an octasaccharide and that each monosaccharide residue can establish electrostatic interactions and/or hydrogen bonds with the protein. The inhibition studies also showed that HS oligosaccharides having 2-O-sulfate moieties can be accommodated by Hpse. The computational studies indicated that in this binding mode, catalytic residues at the −1 subsite are displaced which may contribute to resistance to hydrolysis. It is known that heparin, which is rich in IdoA2S-GlcNS6S moieties, can inhibit Hpse.[4-6] The studies presented here support that IdoA2S containing residues within heparin contribute to its inhibitory activity. HS has much lower levels of sulfation and structural motifs such as that of hexasaccharide 5 and octasaccharide 6 are very rare in HS. Thus, cell surface HS may have a much lower propensity to inhibit Hpse than heparin. The use of heparin as a therapeutic is complicated by structural heterogeneity and the risk of causing bleeding and thrombocytopenia. The hexa- and octasaccharides described here are devoid of 3-O-sulfated GlcNS which is critical for the anticoagulation activity of heparin and as expected, did have no- or very reduced activity for Factor-IIa and Factor-Xa, respectively.
Previous studies have shown that the modification of sulfated oligosaccharides with a lipophilic aglycon can improve Hpse inhibitors potency.[40] Multivalent display of sulfated oligosaccharides is another approach to improve the inhibitory activity of sulfated oligosaccharide.[41] Also, HS-configured cyclophellitol pseudo-disaccharides have been reported to be highly active mechanism-based Hpse inhibitors.[42] A future goal is to examine if the inhibitory potential of compounds such as 5 and 6 can be improved by such approaches.
HSV-1 employs HS as receptor for cell attachment and entry. Previously, we showed that during late-stage infection, the virus induces the upregulation of Hpse to remove cell surface HS allowing detachment of newly formed virions from cells.[7, 13b] We also showed that genetic knockdown of Hpse in vivo results in significant reduction of viral shedding. Therefore, we anticipated that pharmacological inhibition of Hpse by compounds such as 5 and 6 may also prevent viral release thereby exerting a therapeutic effect. To test this hypothesis, detailed kinetic infection studies were performed in which compounds 5 and 6 were administered pre- or post HSV-1 challenge followed by examination of the number of virions associated with cells and released into the media. Treatment of HSV-1 infected cells as late as 8 and 12 hpi with the HS oligosaccharides resulted in a very significant reduction in released virions. Moreover, the HS-oligosaccharide prevented HSV-1 induced shedding of cell surface HS from immortalized HCE cells. Combined these observations support the notion that inhibition of Hpse by compounds such as 5 and 6 can reduce viral release and spread.
It is possible that administration of the HS oligosaccharides before or at the same time of HSV-1 challenge may also inhibit cell attachment and entry. Therefore, viral entry studies were performed using the recombinant virus, KOS gL86, in which the gene for a fusion glycoprotein (gL) is replaced by the lacZ gene that encodes a β-galactosidase.[43] It was found that the HS oligosaccharides inhibit β-galactosidase activity indicating that they have a dual mode of action by impeding viral entry as well as release. The greatest reduction in released virus was observed when the compounds were administered pre- or at the time of infection (Fig. 4B), which agrees with a dual mode of action.
The biological activity of the HS oligosaccharides was further demonstrated by their ability to inhibit the migration and proliferation of HCEs. Hpse plays an important role in the progression of ocular herpes pathogenesis by promoting the formation of new blood vessels (angiogenesis), which allow immune cells to migrate into the corneal tissue causing corneal opacity and inflammation resulting in herpes keratitis, a major contributor to infectious blindness. Inhibition of Hpse in corneal cells is important for wound healing and modulation of ocular inflammation. Collectively, these observations demonstrate that Hpse inhibitors can prevent viral release and subsequent spread to other cells and tissues.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (P41GM103390 and HLBI R01HL151617 to G.-J.B.; R01EY029426, R01EY024710, and P30EY001792 to D.S.). We thank Dr. Apoorva Joshi and Dr. Vito Thijssen for technical assistance. We thank Dr. Gideon J. Davies (University of York, York, UK) for providing human heparanase.
Footnotes
Conflict of Interest
The authors declare no competing financial interest.
References
- [1].a) Parish CR, Freeman C, Hulett MD, Biochem. Biophys. Acta 2001, 1471, M99–M108; [DOI] [PubMed] [Google Scholar]; b) Vlodavsky I, Friedmann Y, J. Clin. Invest 2001, 108, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sarrazin S, Lamanna WC, Esko JD, Cold Spring Harbor Perspect. Biol 2011, 3, a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Pikas DS, Li J-P, Vlodavsky I, Lindahl U, J. Biol. Chem 1998, 273, 18770–18777; [DOI] [PubMed] [Google Scholar]; b) Okada Y, Yamada S, Toyoshima M, Dong J, Nakajima M, Sugahara K, J. Biol. Chem 2002, 277, 42488–42495; [DOI] [PubMed] [Google Scholar]; c) Peterson SB, Liu J, J. Biol. Chem 2010, 285, 14504–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rivara S, Milazzo FM, Giannini G, Future Med. Chem 2016, 8, 647–680. [DOI] [PubMed] [Google Scholar]
- [5].Vlodavsky I, Ilan N, Sanderson RD, Adv. Exp. Med. Biol 2020, 1221, 3–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) McKenzie EA, Br. J. Pharmacol 2007, 151, 1–14; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohan CD, Hari S, Preetham HD, Rangappa S, Barash U, Ilan N, Nayak SC, Gupta VK, Basappa I. Vlodavsky KS Rangappa, iScience 2019, 15, 360–390; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Coombe DR, Gandhi NS, Front. Oncol 2019, 9, 1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thakkar N, Yadavalli T, Jaishankar D, Shukla D, Pathogens 2017, 6, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Whitley RJ, Roizman B, Lancet 2001, 357, 1513–1518. [DOI] [PubMed] [Google Scholar]
- [9].Koujah L, Suryawanshi RK, Shukla D, Cell. Mol. Life Sci 2019, 76, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kukhanova MK, Korovina AN, Kochetkov SN, Biochemistry (Mosc) 2014, 79, 1635–1652. [DOI] [PubMed] [Google Scholar]
- [11].Shukla D, Spear PG, J. Clin. Invest 2001, 108, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Spear PG, Eisenberg RJ, Cohen GH, Virology 2000, 275, 1–8; [DOI] [PubMed] [Google Scholar]; b) Spear PG, Cell. Microbiol 2004, 6, 401–410. [DOI] [PubMed] [Google Scholar]
- [13].a) Tiwari V, Tarbutton MS, Shukla D, Molecules 2015, 20, 2707–2727; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Banerjee A, Kulkarni S, Mukherjee A, Front. Microbiol 2020, 11, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, Shukla D, Nat. Commun 2015, 6, 6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hammond E, Li CP, Ferro V, Anal. Biochem 2010, 396, 112–116. [DOI] [PubMed] [Google Scholar]
- [16].Roy S, El Hadri A, Richard S, Denis F, Holte K, Duffner J, Yu F, Galcheva-Gargova Z, Capila I, Schultes B, Petitou M, Kaundinya GV, J. Med. Chem 2014, 57, 4511–4520. [DOI] [PubMed] [Google Scholar]
- [17].Wilson JC, Laloo AE, Singh S, Ferro V, Biochem. Biophys. Res. Commun 2014, 443, 185–188. [DOI] [PubMed] [Google Scholar]
- [18].Wu L, Viola CM, Brzozowski AM, Davies GJ, Nat. Struct. Mol. Biol 2015, 22, 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu S, Li J, Loka RS, Song Z, Vlodavsky I, Zhang K, Nguyen HM, J. Med. Chem 2020, 63, 4227–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I, J. Biol. Chem 2005, 280, 12103–12113. [DOI] [PubMed] [Google Scholar]
- [21].Alekseeva A, Casu B, Cassinelli G, Guerrini M, Torri G, Naggi A, Anal. Bioanal. Chem 2014, 406, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Arungundram S, Al-Mafraji K, Asong J, Leach FE, Amster IJ, Venot A, Turnbull JE, Boons G-J, J. Am. Chem. Soc 2009, 131, 17394–17405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chopra P, Joshi A, Wu J, Lu W, Yadavalli T, Wolfert MA, Shukla D, Zaia J, Boons G-J, Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2012935118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Holzwarth AR, in Methods in Enzymology, Vol. 246, Academic Press, 1995, pp. 334–362. [DOI] [PubMed] [Google Scholar]
- [25].a) Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, J. Comput. Chem 2009, 30, 2785–2791; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Trott O, Olson AJ, J. Comput. Chem 2010, 31, 455–461; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nivedha AK, Thieker DF, Hu H, Woods RJ, J. Chem. Theory Comput 2016, 12, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E, SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- [27].Schrödinger L, DeLano W, http://www.pymol.org/pymol, 2020.
- [28].Hopkins J, Yadavalli T, Agelidis AM, Shukla D, J. Virol 2018, 92, e01179–01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shah A, Farooq AV, Tiwari V, Kim MJ, Shukla D, Mol. Vis 2010, 16, 2476–2486. [PMC free article] [PubMed] [Google Scholar]
- [30].a) Yadavalli T, Ames J, Agelidis A, Suryawanshi R, Jaishankar D, Hopkins J, Thakkar N, Koujah L, Shukla D, Sci. Adv 2019, 5, eaax0780; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yadavalli T, Suryawanshi R, Ali M, Iqbal A, Koganti R, Ames J, Aakalu VK, Shukla D, Ocul. Surf 2020, 18, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 5844–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Montgomery RI, Warner MS, Lum BJ, Spear PG, Cell 1996, 87, 427–436. [DOI] [PubMed] [Google Scholar]
- [33].a) Ilan N, Elkin M, Vlodavsky I, Int. J. Biochem. Cell Biol 2006, 38, 2018–2039; [DOI] [PubMed] [Google Scholar]; b) Vlodavsky I, Goldshmidt O, Zcharia E, Atzmon R, Rangini-Guatta Z, Elkin M, Peretz T, Friedmann Y, Semin. Cancer Biol 2002, 12, 121–129. [DOI] [PubMed] [Google Scholar]
- [34].a) Riaz A, Ilan N, Vlodavsky I, Li J-P, Johansson S, J. Biol. Chem 2013, 288, 12366–12375; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Che G, Wang Y, Zhou B, Gao L, Wang T, Yuan F, Zhang L, Dis. Markers 2018, 2018, 7413027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a) Lider O, Mekori YA, Miller T, Bar-Tana R, Vlodavsky I, Baharav E, Cohen IR, Naparstek Y, Eur. J. Immunol 1990, 20, 493–499; [DOI] [PubMed] [Google Scholar]; b) Courtney SM, Hay PA, Buck RT, Colville CS, Phillips DJ, Scopes DIC, Pollard FC, Page MJ, Bennett JM, Hircock ML, McKenzie EA, Bhaman M, Felix R, Stubberfield CR, Turner PR, Bioorg. Med. Chem. Lett 2005, 15, 2295–2299. [DOI] [PubMed] [Google Scholar]
- [36].Agelidis AM, Hadigal SR, Jaishankar D, Shukla D, Cell Rep. 2017, 20, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Y, Chia GS, Tham CY, Jha S, J. Vis. Exp 2017, 56248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Quah BJ, Parish CR, J. Vis. Exp 2010, 2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Onishi A, Ange KS, Dordick JS, Linhardt RJ, Front. Biosci. (Landmark Ed) 2016, 21, 1372–1392. [DOI] [PubMed] [Google Scholar]
- [40].Ferro V, Liu L, Johnstone KD, Wimmer N, Karoli T, Handley P, Rowley J, Dredge K, Li CP, Hammond E, Davis K, Sarimaa L, Harenberg J, Bytheway I, J. Med. Chem 2012, 55, 3804–3813. [DOI] [PubMed] [Google Scholar]
- [41].a) Loka RS, Yu F, Sletten ET, Nguyen HM, Chem. Comm 2017, 53, 9163–9166; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zubkova OV, Ahmed YA, Guimond SE, Noble S-L, Miller JH, Alfred Smith RA, Nurcombe V, Tyler PC, Weissmann M, Vlodavsky I, Turnbull JE, ACS Chem. Biol 2018, 13, 3236–3242. [DOI] [PubMed] [Google Scholar]
- [42].de Boer C, Armstrong Z, Lit VAJ, Barash U, Ruijgrok G, Boyango I, Weitzenberg MM, Schröder SP, Sarris AJC, Meeuwenoord NJ, Bule P, Kayal Y, Ilan N, Codée JDC, Vlodavsky I, Overkleeft HS, Davies GJ, Wu L, Proc. Natl. Acad. Sci. U.S.A 2022, 119, e2203167119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG, Cell 1999, 99, 13–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.