

Abstract
Introduction
Treatment of patients with severe sepsis with agents antagonising the effects of C5a has been proposed based on beneficial effects in animal experiments and in vitro studies demonstrating upregulation of the C5a receptor (CD88) on granulocytes by endotoxin.
Materials and methods
CD88 expression on leukocytes from 12 patients with severe sepsis or septic shock was analysed by flow cytometer, and serum complement factors C3a and C5b-9 were measured by enzyme immunoassay techniques.
Results
The granulocyte CD88 expression on day 1 was lowered (36; range, 2–59) in comparison with controls (63; range, 25–88) (P < 0.001), despite complement activation, while the monocyte CD88 expression was unchanged. The receptor reduction correlated significantly to the APACHE II score (r2 = 0.35, P < 0.05). The recovery of CD88 expression was slow.
Discussion
In contrast to the findings in animals, it is concluded that granulocyte CD88 expression is reduced at the time when the diagnosis of severe sepsis or septic shock can clinically be made. The reason for this needs further investigation but it may be due to a previous complement activation or to cytokine effects.
Keywords: antiC5a treatment, complement receptor, leukocytes, septic shock
Introduction
The complement system is a part of the innate immune system and has several functions, such as clearance of immune complexes, opsonisation of pathogens, and direct lysis of invading pathogens by formation of the membrane attack complex [1]. During complement activation there is a generation of the biological peptides, C3a and C5a, referred to as anaphylatoxins. C3a is a chemotactic factor for human mast cells and eosinophils, and it induces the release of histamine and other vasoactive mediators. C5a has been found to have a wider range and higher grade of biological activity. C5a has a chemotactic effect on granulocytes, monocytes, and macrophages, all of which have receptors for C5a (CD88). In neutrophil granulocytes, C5a has also been shown to promote generation of superoxide anions and release of granule enzymes [2]. Furthermore, C5a has been shown to induce upregulation of adhesion molecules on neutrophils, and is thus also one of the factors responsible for neutrophil adhesion to endothelial cells [3].
To prevent complement-induced destruction of the autologous cells, the complement system is under strict control [4,5]. The autologous cell surfaces are protected from homologous complement by soluble and membrane-bound regulators [6]. Despite this protection, excessive or inappropriate complement activation has been associated with inflammatory responses in immune complex-dependent diseases and in adult respiratory distress syndrome [7,8]. Also, in patients with severe sepsis and septic shock, there is a marked activation of the complement system [9,10]. In all these conditions, C5a has been shown to be a mediator of pathophysiological significance [7,8,10] that has stimulated the development of specific anti-C5a strategies.
Recent studies in rats have demonstrated that anti-C5a treatment reduces both the mortality and the biological effects induced by endotoxin [11,12] or polymicrobial products in the caecal ligation puncture model [13]. Increased expression of the C5a receptor on the granulocyte has been proposed to be a component of the central mechanism, since it has been shown that endotoxin can upregulate the C5a receptor expression in vitro [14,15].
In human infection, CD88 expression of the granulocytes has only been investigated in HIV infection; and it was then found to be reduced in advanced disease [16]. To investigate whether this was also the case in human sepsis or whether there was an increase in C5a receptor expression similar to that found in the preclinical experiments, the C5a receptor expression on granulocytes and monocytes was prospectively studied in patients with severe sepsis or septic shock.
The present results demonstrate that the granulocyte CD88 expression was reduced, suggesting more complicated pathogenetic mechanisms than those found in animal experiments.
Materials and methods
Patients and controls
Patients who fulfilled clinical criteria supporting a presumptive diagnosis of severe sepsis or septic shock were prospectively enrolled in the study. The criteria for severe sepsis and septic shock were a modification of those defined by Bone et al. [17].
The patients had to fulfil all of the following four criteria. The acute disease was supposed to be caused by an infection (positive blood culture not required). Second, there had to be the presence of a systemic inflammatory response syndrome defined as two or more of the following criteria: temperature ≤ 35.6°C or ≥ 38.3°C; heart rate, ≥ 90 beats in the absence of a pacemaker; respiratory rate, ≥ 20 breaths/min or PaCO2 ≤ 4.3 kPa (32 mmHg); and white blood cell count, ≥ 12 × 109/l, ≤ 4 × 109/l, or >10% immature band forms.
The third criterion was the presence of one of the following parameters indicating organ dysfunction: acute alteration of mental status, defined as Glasgow Coma Scale <15, not confounded by sedative, hypnotic or other agents with central nervous system depressive effects; metabolic acidosis with pH < 7.30 or base deficit ≥ 5 mEq/l; hypoxia, in the absence of a pneumonia, defined asPaO2 < 9.3 kPa (70 mmHg) on air ventilation, acute reduction of PaO2 > 2 kPa (15 mmHg) on air ventilation, PaO2/FiO2 < 37.3 kPa (280 mmHg), or hypoxia requiring mechanical ventilation; coagulation abnormalities, defined as platelet count <100 × 109/l or <50% of a value measured within the previous 24 hours, an increase in International Normalised Ratio or partial thromboplastin time >50% above the normal value, or D-dimer concentration >0.5 mg/l; oliguria with urine output <30 ml/hour or <0.5 ml/kg/hour for ≥ 1 hour; and hypotension defined as persistent systolic blood pressure ≤ 90 mmHg or reduction >40 mmHg from a value measured within the previous 24 hours despite adequate fluid resuscitation.
Finally, an informed consent was necessary, or a presumed consent if the patient was not capable of making decisions because of altered mental health or sedation.
The exclusion criteria were age <18 years, rapidly progressing underlying disease, HIV/AIDS, cardiogenic shock as the primary underlying disease, haematologic underlying disease or cytotoxic therapy given within the previous week expecting neutropaenia.
The APACHE II score was calculated at the time of the first blood sampling and retrospectively at the onset of severe sepsis [18]. In patients sedated because of mechanical ventilation or for other reasons, the Glasgow coma score was recorded as normal [19].
To be able to perform the receptor and functional analyses, all blood samples were drawn in the morning. Samples were obtained on three occasions: on the day of inclusion (day 1), on either of days 2, 3 or 4, and on day 15. EDTA plasma samples for complement analyses were drawn on day 1.
The second sample was obtained on day 2 in five patients, on day 3 in five patients, and on day 4 in two patients. Since there were no statistical differences between these days, the results from these days are subsequently referred to as day 3.
Twenty healthy individuals with a median age of 46 years (range, 22–60 years) served as controls.
The study was performed with permission from the Ethics Committee, Faculty of Medicine, Uppsala University.
Methods
Preparation of leukocytes
Venous blood for receptor analyses on granulocytes and monocytes was collected in heparinised tubes (Venoject, Terumo Corporation, Belgium) and processed within 2 hours. Leukocytes were prepared according to a previously described method [20]. Briefly, 1 ml blood was mixed with 1 ml of 0.4% (w/v) paraformaldehyde in PBS and warmed to 37°C to fix the blood cells. The mixture was incubated for 4 min at 37°C. The blood sample was then incubated with 40 ml of 0.83% (w/v) NH4Cl in 0.01 M Tris-HCl buffer (0.01 mol/l Tris [hydroxymethyl]-aminomethane, pH7.4) for 15 min at 37°C to lyse the red blood cells. The cells were then centrifuged for 5 min at 350 × g at room temperature, and the supernatant and the red blood cell debris were removed. The remaining leukocytes were washed twice with PBS/citrate/human serum albumin. The cells were then diluted with PBS/citrate/human serum albumin and counted. The concentration of the granulocytes was adjusted to (1.7–2.5) × 106/ml.
Labelling of leukocytes with antibodies to cell surface antigens
To each tube were added 50 l cell suspension, optimally titrated FITC-labelled anti-CD88, clone W17/1 (Serotec, Raleigh, NC, USA) or FITC-labelled isotype control antibodies (Dakopatts A/S, Glostrup, Denmark), and phycoerythrin-labelled anti-CD14 (Dakopatts). This CD88 antibody has been demonstrated to bind to the same part of the receptor as C5a [21]. The samples were then incubated on ice for 30 min. After that, the cells were washed twice with ice-cold PBS/citrate. The cells were diluted with 200 l PBS/citrate/ human serum albumin and then kept on ice until analysis.
Flow cytometry
The samples were analysed on an EPICS-PROFILE II flow cytometer (Coulter Company Inc, Hialeah, FL, USA). The analysis used a fixed protocol, with the same settings maintained for forward scatter and side scatter throughout the study. The fluorescence was calibrated daily, to compensate for the variation of the signals from the flow cytometer, using standardised beads (Flow Set; Coulter Company Inc).
The granulocytes and monocytes were separated on the basis of their forward scatter and side scatter patterns, and the staining with anti-CD14 was used to check the identification of the monocytes. Gates were set around the granulocyte and monocyte populations, and the FITC fluorescence within the gates was measured. A minimum of 10,000 events in the granulocyte gate was counted. The granulocyte and monocyte expression of the C5a receptor (CD88) was measured as specific mean fluorescence intensity of the whole population of granulocytes and monocytes, and as the relative amount of CD88-positive granulocytes and monocytes. The specific mean fluorescence intensity of the granulocyte and monocyte C5a receptor expression was calculated by subtracting the background mean fluorescence intensity obtained with the negative isotype control mAb from the value obtained with the anti-CD88 mAb. The relative amounts of C5a receptor-positive granulocytes or monocytes were calculated as the relative numbers of granulocytes or monocytes, respectively, showing a higher fluorescence intensity when stained with the anti-CD88 mAb than with the negative control mAb.
The interassay variation was 25%, while the intra-assay variation was 7%. A patient sample was always analysed concomitantly with a sample obtained from a healthy control.
Analyses of complement components
The blood for complement analyses was collected in EDTA tubes (Becton Dickinson, Plymouth, UK) and kept on ice until centrifugation. The plasma was collected and stored at -70°C.
The concentration of C3a was analysed by an enzyme immunoassay that is a sandwich enzyme immunoassay employing the monoclonal antibody 4SD17.3 as the capture antibody. EDTA plasma was diluted 1/500 and analysed as described previously [22]. Bound C3a was detected with biotinylated rabbit anti-C3a followed by horseradish peroxidase-conjugated streptavidin (Amersham, Amersham, UK). Zymosan-activated serum [23], calibrated against a solution of purified C3a [24], served as standard and the values are given in nanograms per millilitre.
Soluble C5b-9 was analysed by a modified enzyme immunoassay, described by Mollnes et al. [25]. Plasma samples, diluted 1/5, were added to microtitre plates coated with anti-neoC9 mAb MCaE11. Soluble C5b-9 was detected by polyclonal anti-C5 antibodies diluted 1/500, followed by horseradish peroxidase-conjugated antirabbit immunoglobulin diluted 1/500 (both from Dako A/S, Denmark). Zymosan-activated serum defined as containing 40,000 arbitrary units per millilitre served as the standard. The interassay variation of the C3a and the C5b-9 analyses was 10%.
Leukocyte count
Leukocyte counts were made on a Coulter STKS (Beckman Coulter Inc, Hialeah, FL, USA).
Statistics
Statistical analysis was performed in a nonparametric manner using the Mann–Whitney U test for comparison of data between patients and controls and between patient groups. One healthy control was used at each analysis and a total of 20 control patients were employed. Some controls were therefore analysed on two or three occasions. For the Mann–Whitney analysis, only the value obtained at the first analysis was used in the calculations.
The Wilcoxon matched-pairs test was employed for comparison of individual data within the patient group. The Spearman rank-order correlation coefficient was calculated for correlation analyses.
A difference between two groups was considered significant when P < 0.05. The software STATISTICA (StatSoft Inc., Tulsa, OK, USA) was used in the statistical calculations.
Results
Twelve patients (seven males and five females), with a median age of 58 years (range, 21–85 years), admitted to the intensive care unit (ICU) fulfilled the criteria and were entered into the study. Patient characteristics are presented in Table 1. Three patients were admitted to the ICU with a severe infection as the principal cause. Six patients had probable postoperative infections, one patient had a postburn infection, one patient had a pancreatic abscess, and one patient developed pneumonia after having been admitted to the ICU because of severe hypercalcaemia and malnutrition.
Table 1.
Patient characteristics
| Patient | Sex, age (years) | Underlying disease | Diagnosis of infection | Significant culture findings | APACHE II score, debut/inclusion | Day 28 mortality |
| 1 | Male, 56 | Renal transplantation | Streptococcal sepsis | β-Haemolysing streptococci group A in wound culture | 27/26 | Alive |
| 2 | Female, 85 | Hypertension | Bilateral pneumonia | No positive cultures | 21/20 | Alive |
| 3 | Female, 53 | Arteriosclerosis, infected aortic graft | Postoperative sepsis | Coagulase-negative staphylococci in graft culture | 15/16 | Dead |
| 4 | Male, 81 | Cholelithiasis | Klebsiella sepsis, cholecystitis | Klebsiella pneumophila in blood culture | 22/16 | Alive |
| 5 | Male, 21 | Pancreatitis | Pancreatic abscess | No positive cultures | 12/13 | Alive |
| 6 | Male, 74 | Aortic aneurysm | Postoperative abdominal infection | Proteus mirabilis, Citrobacter freudi, Candida albicans and Candida glabrata in abdominal fluid and wound cultures | 20/18 | Dead |
| 7 | Male, 55 | Stroke, alcohol abuse, angina pectoris | Burn wound infection | Enterobacter cloacae and Klebsiella pneumophila in wound cultures | 16/18 | Alive |
| 8 | Male, 61 | Alcohol abuse, malignancy, hypercalcemia | Pneumonia | No positive cultures | 18/11 | Alive |
| 9 | Female, 78 | Angina pectoris, necrotising pancreatitis | Postoperative sepsis | Candida albicans in tracheal culture | 15/22 | Alive |
| 10 | Female, 51 | Diabetes, arteriosclerosis, myocardial infarctions, infarction in cerebellum, embolus in arteria poplitea and compartment syndrome | Necrotising infection of the calf muscles | Enterococcus faecalis in blood culture | 28/18 | Alive |
| 11 | Male, 74 | Sigmoideum cancer | Postoperative abdominal infection? | No positive cultures | 13/13 | Alive |
| 12 | Female, 73 | Idiopathic thrombocytopaenia, arteriosclerosis, aortic graft | Postoperative sepsis | Staphylococcus aureus in wound culture | 12/14 | Alive |
Infection was verified in all but one patient. In three patients treated with broadspectrum antibiotics for several days before sampling, cultures were negative or they demonstrated bacteria or fungi that were interpreted as colonisation. Causes of the deterioration other than infection could be ruled out in these patients. Although an infection cannot be excluded in patient 11, a retroperitoneal haematoma might have explained the postoperative deterioration at the time of enrolment. Culture findings are presented in Table 1.
Because patient 11 fulfilled the prospectively designed inclusion criteria for severe sepsis and would have been enrolled in a clinical trial using these criteria, this patient was included in the further analyses. Patient 3 died in septic shock after 5 days, and patient 6 died after 28 days. Patient 10 was referred to another hospital on day 5. The ICU mortality, the 28-day mortality, and the hospital mortality were 8, 17, and 33%, respectively. The median length of the ICU stay was 15 days.
The first blood samples were obtained within 24 hours in eight patients after having fulfilled the criteria for severe sepsis, and in another four patients within 36 hours. The median APACHE II score at onset of severe sepsis was 17 (range, 12–28). At the time of the first blood sampling, the APACHE II score had improved in three patients, had worsened in four patients, and was more or less unchanged in the other patients, the median APACHE II score still being 17 (range, 13–26). All patients were febrile at this time and demonstrated serum C-reactive protein (CRP) concentration >100 mg/l, which was on the rise in all but three patients.
The median CD88 expression on the granulocytes in the healthy control group was 63 (range, 25–88). At the time of first sampling on day 1, the granulocyte CD88 expression in the patients with severe sepsis was significantly lower at 36 (range, 2–59; P < 0.001) (Fig. 1).
Figure 1.
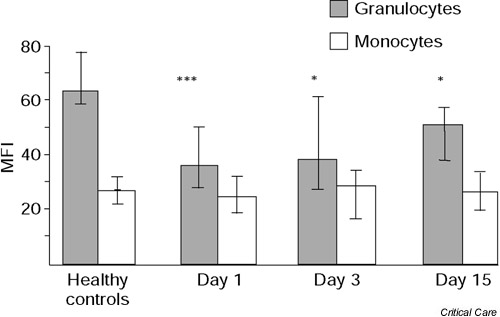
Granulocyte and monocyte expression of CD88 in patients with severe sepsis on day 1, day 3, and day 15. Values are expressed as the median, and the 25th and 75th percentiles. *P < 0.05, ***P < 0.001 in the comparison with the healthy controls (Mann–Whitney U test). MFI, mean fluorescence intensity.
In comparison with the value obtained from the healthy individuals analysed on the same occasion, CD88 expression was lower in all patients except patient 11. There was some increase in CD88 expression on day 3 (37; range, 6–101), and the increase was significant after 2 weeks in comparison with day 1 (51; range, 20–105) (P < 0.05). Both on day 3 and on day 15, however, the granulocyte CD88 expression was still significantly reduced in comparison with that of the healthy controls (P < 0.05). The recovery followed an individual course, since granulocyte CD88 values on day 15 were highly correlated with those on day 1 (r2 = 0.64, P < 0.01). The median granulocyte CD88 expression in patients who remained in the ICU on day 15 had been 35 (range, 2.5–59) on day 1, compared with 44 (range, 26–57) on day 1 in those who were discharged from the ICU on day 15 (P < 0.05). Granulocyte CD88 expression on day 1 was 18.9 in the patient who died in the ICU on day 5. This expression was further reduced to 12.6 on the day before death.
Monocyte CD88 expression was considerably lower than granulocyte CD88 expression, both in healthy controls (P < 0.01) and in the patients (P < 0.05). In contrast to the granulocyte CD88 expression, the monocyte CD88 expression was totally unchanged over the time of observation and there was no difference between patients and the controls (Fig. 1).
The leukocyte counts on the different days are presented in Table 2. There was no correlation between granulocyte count and CD88 expression on day 1 (r2 = 0.05) or on day 3 (r2 = 0.03), indicating that the reduction in C5a receptor expression during acute infection cannot be explained by a variation in the number of leukocytes. On day 15, however, there was a small but significant negative correlation with lower values in patients with leukocytosis (r2 = 0.24; P < 0.05).
Table 2.
Leukocyte cell count on day 1, on day 3 and on day 15
| Day 1 | Day 3 | Day 15 | |
| Total leukocyte cell count (× 109/l) | 16.2 (8.1–34.6) | 18.3 (6.3–34.6) | 11.2 (7.2–18.9) |
| Polynuclear cells (× 109/l) | 13.6 (5.8–33.5) | 15.6 (3.7–31.4) | 8.0 (4.0–16.6) |
| Mononuclear cells (× 109/l) | 1.7 (0.7–15.5) | 2.1 (0.5–6.0) | 2.2 (1.4–5.5) |
Data presented as median (range).
The granulocyte expression of CD88 on day 1 correlated negatively to the severity of disease as measured by the APACHE II score at inclusion (r2 = 0.35, P < 0.05) (Fig. 2). This correlation was influenced neither by the changes in APACHE II score from the onset of severe sepsis nor by the duration of severe sepsis. Overall, there was no significant correlation either to the varying duration of the severe sepsis at inclusion or to the duration of acute illness. There was no correlation between APACHE II score and monocyte CD88 expression (r2 = 0.00).
Figure 2.
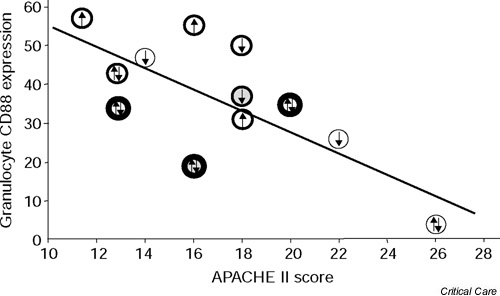
Correlation between the granulocyte CD88 expression and the APACHE II score on day 1. Upward arrows indicate an increase from onset of the severe sepsis in APACHE II score >2, and downward arrows indicate a decrease from onset of the severe sepsis in APACHE II score >2. Double-directed arrows represent changes <2.○, a patient with a duration of sepsis of 12 hours; ??, a patient with a duration of 13–24 hours; and ??, a patient with a duration >24 hours. Patients who died within 28 days are marked with shaded symbols. A regression line has been calculated using the method of least squares.
Granulocyte CD88 expression on day 1 did not correlate to the inflammatory process as indicated by the level of serum CRP. Later during the clinical course, however, these parameters correlated significantly (r2 = 0.42, P < 0.05 on day 2; r2 = 0.50, P < 0.05 on day 15). There was no significant correlation between the monocyte CD88 expression and the concentration of serum CRP on any of the days.
C5b-9 levels were increased in all patients and C3a concentrations were increased in all but one patient on day 1 (Table 3). There was no correlation between C5b-9 or C3a levels and neutrophil or monocyte CD88 expression.
Table 3.
Concentrations of C3a and C5b-9 on day 1
| Number of patients with values above upper normal limit | Median | Range | Normal range | |
| C3a | 10/11 | 1106 ng/ml | 259–1529 ng/ml | 92–268 ng/ml |
| C5b-9 | 11/11 | 147 AU/ml | 64–562 AU/ml | 12–56 AU/ml |
AU, arbitrary units
Discussion
To avoid clinical trials with little chance of proving efficacy of different anti-C5a strategies, it was of interest to study whether the increased expression of the C5a receptor found in the animal experiments was found also in human sepsis. Theoretically, it would also have been of interest to study the C5a concentration. However, C5a concentration is not a good parameter to follow complement activation.
C5a binds rapidly to the C5a receptor and, in contrast to C3a, C5a is cleared from plasma within minutes [26,27]. In a clinical trial including patients with sepsis and complement activation, as assessed by increased C3a concentrations and C3a/C3 ratios, the C5a values found were identical to those in healthy controls [10]. Besides being an important parameter of complement activation of its own, the concentration of C5b-9 analysed in this study is an indirect measurement of the C5a production, since C5 at activation is split into C5a and C5b.
In this study patients with severe sepsis or septic shock, the increased levels of C3a and C5b-C9 indicate complement activation in agreement with the animal experiments and a previous clinical study [10]. In contrast to the findings in the animal experiments [14,15], however, CD88 expression on the granulocytes was markedly reduced. In the acute phase, this was probably not a compensatory effect due to the leukocytosis because the correlation between granulocyte count and expression of CD88 was slightly positive on day 1. There was a slow recovery and, after 2 weeks, the granulocyte CD88 expression was still lower than in healthy controls.
Although there are signs of complement activation, the present results demonstrate that granulocyte CD88 expression is reduced at the time when the diagnosis of severe sepsis or septic shock can clinically be made. Since the analyses of the CD88 expression had to be performed within 2 hours after blood sampling, because of the changes in cell surface receptor expression during longer storage periods, all the analyses had to be performed during daytime working hours. There was therefore a delay after the onset of severe sepsis or septic shock. However, as can be seen in Fig. 2, the duration of the severe sepsis did not seem to affect the results. Statistically, there was no correlation between duration of sepsis and the granulocyte CD88 expression.
It may also be speculated that deteriorating patients, being in a more uncontrolled phase, perhaps would have higher values than patients demonstrating signs of improvement. The present data do not suggest that this factor was of great importance (Fig. 2). Although a minor effect of these factors cannot be excluded, it must be emphasised that the CD88 expression was lower in all patients but one than in the concomitantly analysed healthy controls, irrespective of the duration of sepsis or whether the sepsis was improving. Furthermore, the patient in whom the granulocyte CD88 expression was within the range of the controls was the one in whom infection could not be verified.
Moreover, it may be argued that the patients included in this study were not ill enough. However, the reduction in granulocyte CD88 expression was more pronounced in the more severe cases, as indicated by the negative correlation to the APACHE II score and by the fact that patients who were not discharged from the ICU 2 weeks later had shown lower values on day 1.
In the animal experiments in which anti-C5a strategies were beneficial, the treatment was given concomitantly or before the endotoxin injection [11,12] or the establishment of the infection [13]. In humans, a transient increase in C5a binding to granulocytes has been demonstrated 3 hours after low-dose endotoxin administration [14]. After that there was a successive decrease, and after 24 hours the mean value was below that at baseline.
It has been shown in in vitro experiments that, after binding of C5a, the receptor complex is rapidly internalised, thus rendering the cells resistant to subsequent challenges with C5a [28]. Thus, it cannot be excluded that, in our patients with sepsis and signs of complement activation, there might have been an initial transient upregulation of the granulocyte C5a receptor. At the time of diagnosis, however, this expression has become reduced, possibly as a consequence of a previous stimulation. If this is the scenario, it might be hypothesised that anti-C5a treatment may be effective if given before or concomitantly with infectious stimuli, but without effect if given later in the septic course. To our knowledge, there are so far no results published from animal experiments in which the anti-C5a treatment has been postponed until signs of severe sepsis have become evident.
Another reason for the discrepancy may be that endotoxin or polymicrobial sepsis by the caecal ligation puncture model caused the septic reaction in the experimental models, while infections in our heterogeneous patient group with clinical sepsis were caused both by Gram-positive and Gram-negative bacteria. However, our limited data do not indicate a difference between Gram-positive and Gram-negative infections. To our knowledge, there are no animal experiments investigating the C5a receptor or anti-C5a treatment in Gram-positive infections.
It must also be emphasised that a low granulocyte C5a receptor expression does not theoretically exclude an ongoing granulocyte activation by C5a. One possibility would be that the reduced receptor level detected by the CD88 antibody may be caused by receptor-bound C5a, and another that the receptor of the internalised complex is rapidly transported back to the cell surface to bind more C5a. Studies in humans and animals, however, have all shown a rapid internalisation of the ligand–receptor complex after binding, and thereafter there is a desensitisation to subsequent challenges with C5a [13,29]. There are no data indicating an increase in recycling of the receptor in sepsis or in any other condition.
Other reasons for the reduction in granulocyte C5a receptor expression may be the inflammatory response or the infection per se. The correlation to the CRP response might suggest that there is a relation to the inflammatory response. This is best studied in in vitro experiments and in a new prospective clinical investigation in which there is parallel inclusion of systemic inflammatory response syndrome patients with and without infection. Another theoretical possibility might be that immature granulocytes recently released from the bone marrow may have a lower CD88 expression. With the rapid turnover that occurs during severe sepsis and septic shock, such cells would account for a substantial part of the granulocyte cell population. There are presently no data to support a lower CD88 expression on younger cells, but the negative correlation between granulocyte count and CD88 expression on day 15 may be consistent with such a mechanism.
In contrast to the changes in the granulocyte CD88 expression, there were no changes in the monocyte expression. The reason for this discrepancy is not known but may be caused by the differential effects of proinflammatory or anti-inflammatory substances on granulocytes and monocytes, by differences in the affinity to C5a, or by a bone marrow effect in combination with varying half-lives.
Conclusion
A reduction in granulocyte C5a receptor expression has been demonstrated in patients at the time when the diagnosis of severe sepsis or septic shock can be clinically established. The number of patients in the present study is limited. However, the strongly significant reduction in granulocyte CD88 expression and its correlation with disease severity in included patients clearly indicate that severe sepsis in clinical practice reflects other mechanisms than those seen in the animal experiments and that inclusion of further patients would not reasonably have led to an alternative conclusion. The reduction in granulocyte C5a expression may perhaps implicate a risk that the clinical response to anti-C5a treatment might be more limited than that seen in animal experiments. However, this needs further experimental and clinical investigations.
Key messages
· The C5a receptor expression on granulocytes from septic patients was reduced compared with healthy controls
· The reduction of C5a receptor expression correlated to the APACHE II score
· The granulocyte C5a receptor expression on day 1 was higher in patients who were discharged from the intensive care unit in contrast to those still being in the intensive care unit on day 15
· The recovery of granulocyte C5a receptor expression was slow
· The monocyte C5a receptor expression was unchanged
Competing interests
None declared.
Abbreviations
APACHE II = Acute Physiology and Chronic Health Evaluation II; CRP = C-reactive protein; FiO2 = fraction of oxygen in inspired air; FITC = fluorescein isothiocyanate; ICU = intensive care unit; mAb = monoclonal antibody; PaCO2 = arterial partial pressure of carbon dioxide; PaO2 = arterial partial pressure of oxygen; PBS = phosphate-buffered saline.
Acknowledgments
Acknowledgements
The authors would like to thank their research nurse Elisabeth Pettersson and their biomedical assistants Riitta Laukkanen-Mållberg, Lena Gröndahl and Anita Nyberg Axelsson for their excellent assistance in this project. This work was supported in part by the Medical Research Council, Sweden.
References
- Speth C, Würzner R, Stoiber H, Dierich MP. The complement system: pathophysiology and clinical relevance. Wiener Klin Wochensch. 1999;111:378–391. [PubMed] [Google Scholar]
- Webster RO, Zanolari B, Henson PM. Neutrophil chemotaxis in response to surface bound C5a. Exp Cell Res. 1981;129:55–62. doi: 10.1016/0014-4827(80)90330-4. [DOI] [PubMed] [Google Scholar]
- Foreman KE, Glovsky MM, Warner RL, Horvath SJ, Ward PA. Comparative effects of C3a and C5a on adhesion molecule expression on neutrophils and endothelial cells. Inflammation. 1996;20:1–9. doi: 10.1007/BF01487740. [DOI] [PubMed] [Google Scholar]
- Lindahl G, Sjöbring U, Johnsson E. Human complement regulators: a major target for pathogenic microorganisms. Curr Opin Immunol. 2000;12:44–51. doi: 10.1016/s0952-7915(99)00049-7. [DOI] [PubMed] [Google Scholar]
- Morgan PB. Regulation of the complement membrane attack pathway. Crit Rev Immunol. 1999;19:173–198. [PubMed] [Google Scholar]
- Lachmann PJ. The control of homologous lysis. Inmmunol Today. 1991;12:312–315. doi: 10.1016/0167-5699(91)90005-E. [DOI] [PubMed] [Google Scholar]
- Stevens JH, O'Hanley P, Shapiro JM, Mihm FG, Satoh PS, Collins JA, Raffin TA. Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest. 1986;77:1812–1816. doi: 10.1172/JCI112506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5a monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA. 1995;92:8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedegård G, Cui L, Hugli TE. Endotoxin-induced shock in the rat. Am J Pathol. 1989;135:489–497. [PMC free article] [PubMed] [Google Scholar]
- Stöve S, Welte T, Wagner TOF, Kola A, Klos A, Bautsch W, Köhl J. Circulating complement proteins in patients with sepsis or systemic inflammatory response syndrome. Clin Diagnostic Lab Immunol. 1996;3:175–183. doi: 10.1128/cdli.3.2.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno M, Nishikawa K, Okada N, Matsuo S, Ito K, Okada H. Inhibition of a membrane complement regulatory protein by a monoclonal antibody induces acute lethal shock in rats primed with lipopolysackaride. J Immunol. 1999;162:5477–5482. [PubMed] [Google Scholar]
- Strachan AJ, Woodruff TM, Haaima G, Fairlie DP, Taylor SM. A new small molecule C5a receptor antagonist inhibits the reverse-passive Arthurs reaction and endotoxic shock in rats. J Immunol. 2000;164:6560–6565. doi: 10.4049/jimmunol.164.12.6560. [DOI] [PubMed] [Google Scholar]
- Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Friedl HP, Ward PA. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- Fukuoka Y, Ember JA, Hugli TE. Cloning and characterization of rat C3a receptor: differential expression of rat C3a and C5a receptors by LPS stimulation. Biochem Biophys Res Commun. 1998;242:663–668. doi: 10.1006/bbrc.1997.8034. [DOI] [PubMed] [Google Scholar]
- Granowitz EV, Porat R, Gelfand JA, Wolff SM, Dinarello CA. Administration of low-dose endotoxin to healthy humans increases C5a binding to circulating neutrophils. J Infect Dis. 1994;169:480–482. doi: 10.1093/infdis/169.2.480. [DOI] [PubMed] [Google Scholar]
- Hart SP, Ross JA, Haslett C, Dransfield I. Molecular characterization of the surface of apoptotic neutrophils: implication for functional downregulation and recognition by phagocytes. Cell Death Differ. 2000;7:493–503. doi: 10.1038/sj/cdd/4400680. [DOI] [PubMed] [Google Scholar]
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RMH, Sibbald WJ. ACCP/SCCM Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–829. [PubMed] [Google Scholar]
- Knaus WA. Measuring the Glascow coma score scale in the intensive care unit: potentials and pitfalls. Intensive Care World. 1994;11 [Google Scholar]
- Hamblin A, Taylor M, Bernhagen J, Shakoor Z, Mayall S, Noble G, McCarthy D. A method of preparing blood leukocytes for flow cytometry which prevents upregulation of leukocyte integrins. J Immunol Methods. 1992;146:219–228. doi: 10.1016/0022-1759(92)90231-h. [DOI] [PubMed] [Google Scholar]
- Opperman M, Raedt U, Hebell T, Schmidt B, Zimmerman B, Götze O. Probing the human receptor for C5a anaphylatoxin with site-directed antibodies. J Immunol. 1993;151:3785–3794. [PubMed] [Google Scholar]
- Nilsson Ekdahl K, Nilsson B, Pekna M, Nilsson UR. Generation of iC3 on the interphase between blood and gas. Scand J Immunol. 1992;35:85–91. doi: 10.1111/j.1365-3083.1992.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Mayer MM. Complement and Complement Fixation Experimental Immunochemistry. Thomas Springfield: Illinois; 1961.
- Bokisch VA, Müller-Eberhard HJ, Cochrane CG. Isolation of a fragment (C3a) of the third component of human complement containing anaphylatoxin and chemotactic acctivity and description of an anaphylatoxin inactivator of human serum. J Exp Med. 1969;129:1109–1130. doi: 10.1084/jem.129.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollnes T, Lea T, Froland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against neoantigen on the complex. Scand J Immunol. 1985;22:703–710. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- Chenoweth DE, Goodman MD. The C5a receptor of neutrophils and macrophages. Agents Actions. 1983;12:252–273. doi: 10.1007/978-3-0348-9352-7_15. [DOI] [PubMed] [Google Scholar]
- Opperman M, Götze O. Plasma clearance of the human C5a anaphylatoxin by binding to leukocyte C5a receptors. Immunology. 1994;82:516–521. [PMC free article] [PubMed] [Google Scholar]
- van Epps DE, Simpson S, Bender JG, Chenoweth DE. Regulation of C5a and formyl peptide receptor expression on human polymorphonuclear leukocytes. J Immunol. 1990;144:1062–1068. [PubMed] [Google Scholar]
- Naik N, Giannini E, Brouchon L, Boulay F. Internalization and recycling of the C5a anaphylatoxin receptor: evidence that the agonist-mediated internalization is modulated by phosphorylation of the C-terminal domain. J Cell Sci. 1997;110:2381–2390. doi: 10.1242/jcs.110.19.2381. [DOI] [PubMed] [Google Scholar]