

Abstract
Expansion of sequencing efforts to include thousands of genomes is providing a fundamental resource for determining the genetic diversity that exists in a population. Now, high-throughput approaches are necessary to begin to understand the role these genotypic changes play in affecting phenotypic variation. Saccharomyces cerevisiae maintains its position as an excellent model system to determine the function of unknown variants with its exceptional genetic diversity, phenotypic diversity, and reliable genetic manipulation tools. Here we review strategies and techniques developed in yeast that scale classic approaches of assessing variant function. These approaches improve our ability to better map quantitative trait loci at a higher resolution, even for rare variants, and are already providing greater insight into the role that different types of mutations play in phenotypic variation and evolution not just in yeast but across taxa.
Keywords: quantitative traits, high-throughput screens, functional assay, genotype-to-phenotype, QTL mapping, MAVE, CRISPR, yeast genetics, population sequencing, variant effects, mutagenesis, saturation mutagenesis, massively parallel reporter assay, phenotyping, Saccharomyces cerevisiae
Introduction
Genetic diversity present across the budding yeast Saccharomyces cerevisiae population has produced the extraordinary phenotypic diversity that we see in this species today [1,2]. With a variety of wild ecological origins and over 12,000 years of domestication across the globe, S. cerevisiae isolates in different clades have distinct polymorphisms that facilitate adaptation to specific environments [3–5]. The overarching question that still remains after decades of genetics and genomics work is which of the genotypic differences and changes in genetic architecture between individuals give rise to phenotypic variation. A change in a single locus alone can be the cause for phenotypic differences and is known as a Mendelian trait. However, Mendelian traits are rare as most traits are complex and thus governed by multiple loci and gene-by-environment interactions. Understanding the genetic basis of complex traits has many implications for advancing therapeutics for disease, industrial applications, agricultural output for our changing climate, and our knowledge of evolution.
Curation, deep sequencing, and genomic analysis of 1,011 S. cerevisiae isolates collected from natural and domestic environments revealed the sheer number of variants present in the population [3]. Containing over 1.6 million single nucleotide polymorphisms, this collection highlights that foundational approaches like quantitative trait locus (QTL) mapping for understanding the effects of these SNPs would be prohibitively labor-intensive, time-consuming, and oftentimes impossible given the current limitations of these strategies [6]. Copy number variants (CNVs) further confound our understanding of the genetic architecture of traits; almost all open reading frames (ORFs) in the S. cerevisiae genome have a CNV in at least one of the 1,011 strains [3]. The huge diversity in populations compared to the little that is known about the effects of genotypic changes, with the added layer of intricacy that environmental factors play, necessitates high-throughput experiments that can confidently determine the impacts of variants of all types.
Even the huge number of variants as yet observed is dwarfed by the number of variants that could possibly exist. Obviously, this general problem is not limited to the yeast system: modern genetics is going to require high throughput approaches to understanding variation across taxa. Technological solutions developed in yeast can immediately be applied to other genomes either as a testbed for methods development to port to other systems, or by heterologously expressing genes in yeast.
While these challenges are daunting, the throughput of systems level approaches for determining and measuring variant function have increased considerably, particularly in S. cerevisiae, pointing to a path forward. The expansive genetic diversity of yeast, coupled with the extensive toolkit for dissecting genetic traits and engineering variants, provide an excellent foundation for understanding the impact of polymorphisms, learning fundamental principles underlying these effects, and ultimately predicting effects of potential mutations. Here we describe the latest technologies and strategies developed for addressing genotypic changes on a massive scale and what questions applications of these approaches can answer.
Approaches
Genome-wide Association Study (GWAS) and QTL mapping are the major approaches used to understand the effects of natural variants on complex traits [3,6,7]. Even though over one million samples have been incorporated in human GWAS analyses, the missing heritability problem, where mapped variants explain a small proportion of the total heritability [8], is still not resolved. Previously in S. cerevisiae, due to the high degree of mosaicism and presence of population structure, GWAS required additional statistical corrections to avoid false positives [9–13]. However, increases in sample size and diversity have improved the power of using GWAS for understanding the genetic basis of complex traits in yeast [3,7,11–14].
QTL mapping, on the other hand, controls environmental factors and leverages the segregation of parental genotypes to pinpoint causal genetic variants leading to measurable phenotypes of interest [22] (Figure 1, left panel). Phenotyping approaches for QTL mapping in yeast vary widely and include those that detect changes in molecular phenotypes such as gene expression or protein abundance [23–29], colony or cell morphology [20,30,31], flocculation patterns [20], growth rate [3,15,16,20], enzymatic function [32], compound production [33], translation termination efficiency [34], and many more [1,2].
Figure 1.
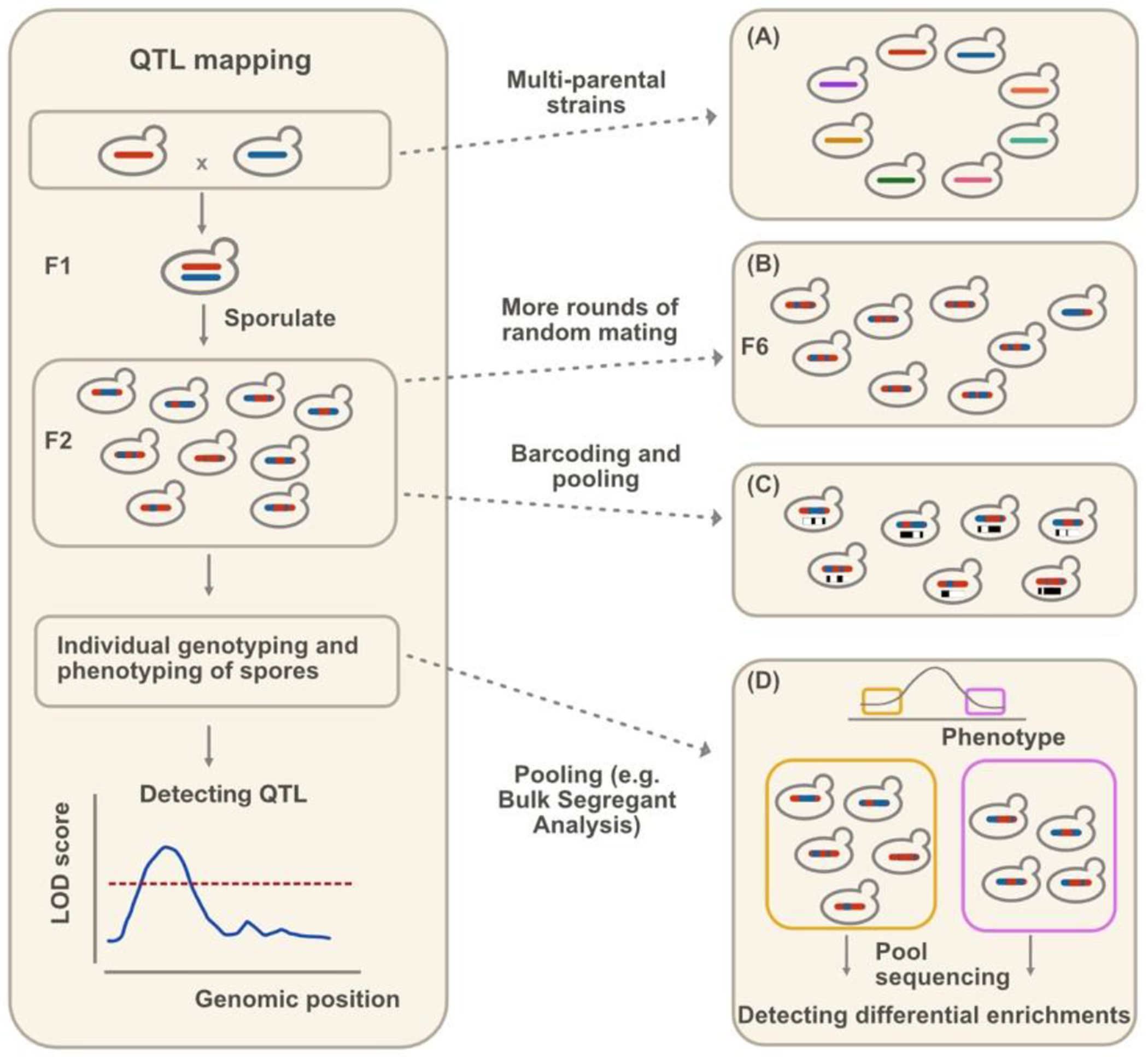
Traditional QTL mapping procedures (left panel) and recent advancements (right panel). The LOD (logarithm of odds) score indicates the probability that a QTL is present at that genomic position. QTL are identified by regions with LOD scores above a significant threshold. Recent advancements increase mapping precision by (A) using multiple parental strains [15], (B) more rounds of random mating [16,17], (C) high-throughput barcoding techniques [18,19], or (D) pooling assays [20,21]. In (D), the yellow and purple boxes represent yeast with extreme phenotypes, which are pooled and sequenced separately to detect alleles that are differentially enriched.
High-throughput genome-wide identification of genes contributing to trait differences
Pooled approaches—such as bulk segregant analysis, which sequences pooled individuals with extreme phenotypes [21], and barcoding multiple parental strains [18] or individual segregants [19] in order to trace lineages of pooled segregants throughout a screening—improve the throughput of QTL mapping (Figure 1, right panel). Although these approaches can be enriched for false positives due to beneficial mutations, genomic instability, and diploidization events that may arise during the growth phase [20], advancements in automated workflows now allow phenotyping of a remarkable number of individual segregants. The largest QTL mapping study in yeast to date phenotyped an astounding 100,000 F2 barcoded segregants [18]. Collectively, QTL mapping studies have measured over 100 complex traits [15–18,20,21]. Furthermore, deliberate selection of parent strains can survey most of the natural variation in the S. cerevisiae population, with 82% of biallelic SNPs captured by just 16 parental natural isolates [15]. Progeny of these 16 isolates, as well as hybrids from a diallel cross with 55 natural isolates, are also enriched for rare variants [15,35]. Screening of these crosses confirmed that variants of large effects are usually rare in the total population, consistent with negative selection on these alleles [15,18,35].
Ascertainment biases in QTL mapping still exist, resulting in variants of large effects being overrepresented in functional studies and variants of small effects being overshadowed. Identification of polymorphic differences between two strains and individually introducing the variants from one strain to another exposes the functional effects of those variants in an unbiased manner. Several advances made in CRISPR/Cas9 precise editing techniques have been used to introduce SNPs identified between two strains in a high-throughput manner (Figure 2B) [36–39]. Such studies leverage the use of retrons, base editing, and protein fusions to increase the efficiency of homology-directed repair (HDR) over nonhomologous end joining (NHEJ) in yeast [36–38]. These pooled variant studies have been able to interrogate the impacts of over 16,000 SNPs (in some cases over 30,000 SNPs) in one experiment, although variants of small effect tend to have low reproducibility and high false discovery rates [36,37,39]. Once generated, libraries can be screened in various conditions to measure the interaction between each variant and the environment. Overcoming other limitations such as decreased editing accuracy with increased distance to PAM (protospacer-adjacent motifs, or the cut-site motif recognized by Cas9) sites or high oligonucleotide error rates will improve the power and accuracy of these assays.
Figure 2.
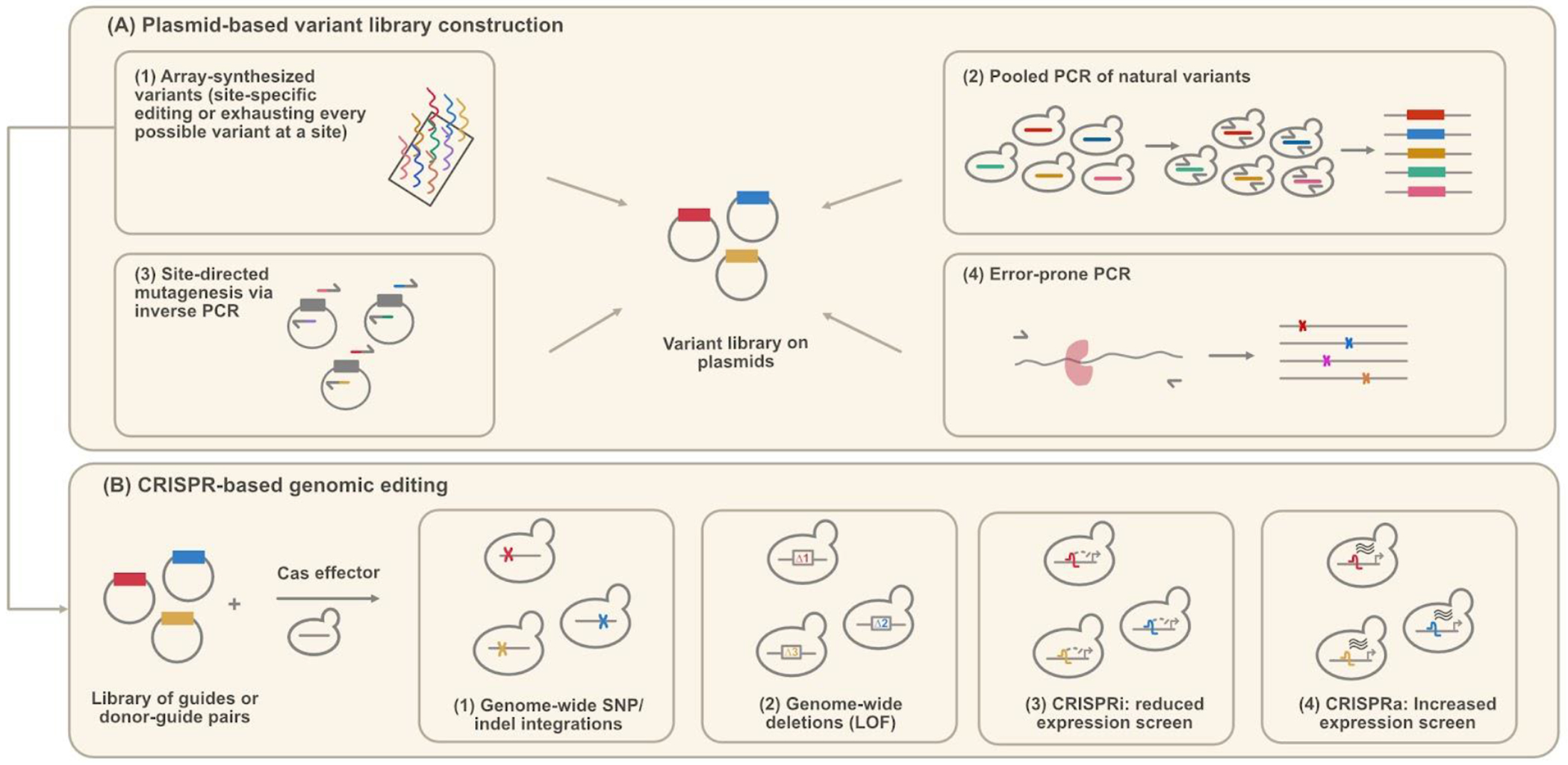
Summary of yeast library construction and genomic editing methods. (A) Plasmid-based variant library construction. The effects of these variants can be determined by transforming yeast cells with this plasmid library. Commonly-used library construction methods include (1) array-synthesized variants [37,46,47,54,55], (2) pooled PCR of natural variants [48], (3) site-directed mutagenesis [46,52,53], and (4) error-prone PCR [50,51]. (B) CRISPR-based variant construction. Yeast cells are transformed with a plasmid library of guide or donor-guide pairs so that genetic changes are incorporated into the genome. These methods include (1) Genome-wide SNP/indel editing [36–39], (2) Genome-wide deletions [41], (3) CRISPRi to decrease target expression [41–43], and (4) CRISPRa to increase target expression [41].
Precise editing approaches have been useful for identifying expression QTLs (eQTLs), or cis-regulatory variants that alter expression and affect phenotype, making it possible to pinpoint causal cis- and trans-acting mutations in S. cerevisiae [23,27,40]. Similarly, genome-wide perturbations that upregulate or downregulate nearly all genes in one assay can identify eQTLs as well, although not always at nucleotide resolution [41–44]. Scaled eQTL studies can now simultaneously measure expression and protein abundance with greater statistical power, providing a better understanding of how promoters and post-transcriptional processes may affect complex traits [23,27,40]. In addition to changes in expression and protein abundance patterns, complete loss of function (LOF) by full gene deletions, frameshift mutations, temperature sensitive mutations, or introduction of premature stop codons can be investigated in a high-throughput manner to determine the impact of different types of LOF mutations [38,45].
High-throughput interrogation of variant effects at a causal locus
When genes of interest are identified by any of these mapping methods, the question arises of how natural genetic variation or potential new mutations in these genes impact function. Multiplexed Assays of Variant Effects, or MAVEs, can interrogate the effects of substitutions and cis-regulatory mutations in one locus by creating large libraries of variants and measuring en masse how they affect fitness, protein interactions, expression, splicing, or enzymatic function [49]. MAVEs encompass a variety of methods such as Massively Parallel Reporter Assays (MPRAs), Deep Mutational Scanning (DMS), and Saturation Genome Editing (SGE). Variants for MAVEs can be generated through a variety of methods (Figure 2A): Error-prone PCR is a cost-effective approach that introduces random mutations into a region of interest and can explore both single and combinatorial effects of mutations, but due to its random nature, specific mutations and variants of interest are not guaranteed to be generated [50,51]. Inverse PCR allows for site-directed mutagenesis and has now been scaled for MAVEs, but it remains quite labor intensive [46,52,53]. Oligonucleotide synthesis also allows for deep interrogation into single substitutions and can include more complex variants, or variants with more than one mutation, but has a higher cost and error rate [47,54,55]. Saturation mutagenesis can now be performed endogenously with CRISPR/Cas9 editing, namely by designing synonymous mutations in the donor DNA strand or introducing a heterologous intermediate sequence to prevent unintentional recognition and cleavage due to strand complementarity to the guide RNA [37,56] (Figure 2B). Similarly, improvements in the use of single-stranded oligodeoxynucleotides (ssODN) in yeast permit the scaling of precise, multisite editing without double-stranded breaks [57]. Sequencing full variants or short DNA tags that act as barcodes for each variant allows the tracking of these variants throughout an assay, which can be used to infer variant function (Figure 3).
Figure 3.
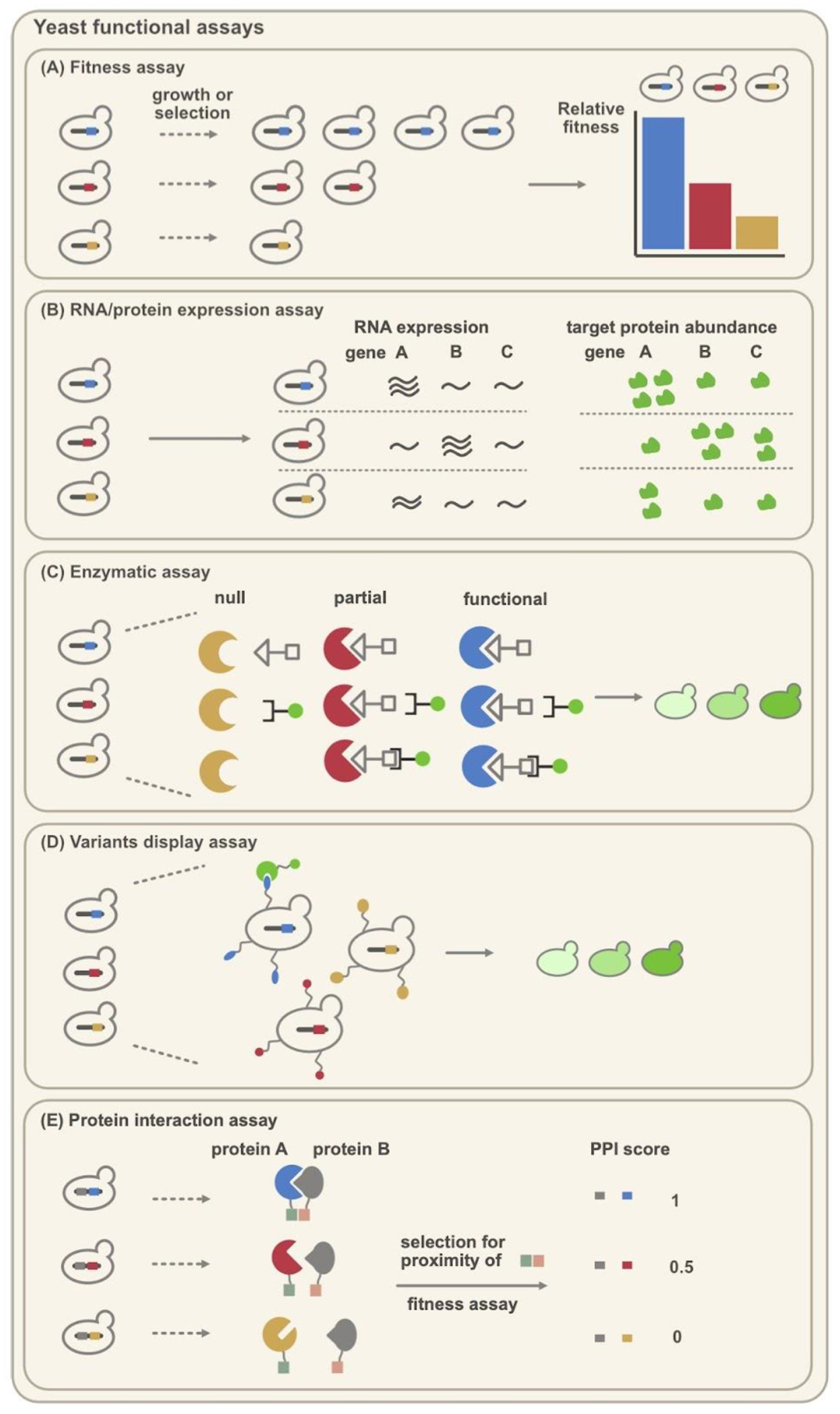
Summary of high-throughput functional assays in yeast. The starting library can be developed using either plasmid, integrative, or CRISPR-based methods from Figure 2. These functional assays include but are not limited to (A) Fitness assay [48], (B) RNA or protein expression assay [23,27–29], (C) Enzymatic assay [46], (D) Variants display assay [64], and (E) Protein interaction assay [65–67]. Note that for simplicity, we only represent one variant library in the protein interaction assay panel (E). In reality, two sets of protein variant libraries can be used to explore protein-protein interactions [65,67].
MAVEs in yeast have been useful for predicting how new mutations alter phenotype, not just for yeast genes, but for genes from other systems as well (Figure 3). Remarkably, many human genes complement their orthologous S. cerevisiae gene knockouts, sequence homology can be taken advantage of for those that don’t, and even genes without homologs can be functional when heterologously expressed in yeast [46,47,58–64].
This style of high-throughput, pooled phenotype testing can also be applied to more complex allele libraries [48,65,67,68]. For example, we have generated and phenotyped a library of all natural alleles of one gene, including multiple SNPs and insertion/deletions in one haplotype[48]. This MAVE approach for understanding gene function on a species-wide scale reveals not only the natural fitness distribution of variants in this gene, but informs its evolutionary history as well. Decreases in cost along with increases in throughput and accuracy of long-read sequencing enable the genotyping of large libraries containing complex variants that are longer than what can be fully captured from high-throughput sequencing [68].
Finally, variant libraries need not be based on natural sequences at all. Generation of randomized sequences has been useful for understanding how translation and transcription factor-binding sites are utilized in promoter regions, with the effects of up to 100 million sequences determined in one experiment [69–71]. The increasing number of studies focusing on exploring this space enhances our understanding of variant effects and is fundamental to computational programs more accurately predicting function [29,72–76].
Conclusions
Advancements in approaches for identifying QTLs as well as the underlying causal genes and nucleotides have revealed an extraordinary amount of information about complex traits. High throughput methods such as those described here facilitate moving beyond individual example cases and allow for general patterns to surface that begin to reveal the categories of complex traits and what is required for a comprehensive understanding of their genetic basis. Complex traits vary in patterns themselves and can involve multiple common variants, multiple rare variants, or a combination of both [15,18], justifying the need for independent interrogations of variants under multiple environments. For some traits, variants clustered in one locus can have effects in the same direction [36,77]; for others, however, variants can have canceling effects that result in neither variant being detected in a QTL [23]. These variants can be coding or noncoding, and the effect sizes of these mutations are relatively similar [16,36]. Effects of variants can be additive, although greater sample sizes of segregants revealed that the resulting phenotypes are more indicative of epistatic interactions [15,18,19,35]. Identifying epistatic interactions is still challenging, and most high-throughput approaches for doing so have been through whole gene deletions or engineered mutant alleles [67,78–81] or by investigating genetic network changes as a result of single gene perturbations [29,82]. Thus, the effects of most pairwise and complex SNP interactions at the genomic scale remain to be determined. Impacts of higher-order interactions are still rather elusive, although a recent study shows that this can be investigated using a hierarchical gene drive system [83].
The end goal of these studies is to understand the principles behind how genotypic changes alter phenotype. Saturation-level analysis can be achieved by taking a gene-centric approach to understand how variation in a locus affects phenotype. Good candidates for such analysis are the causal genes identified by QTL studies. Certain genes (such as MKT1, HAP1, IRA2) continually resurface in QTL maps, indicating a high degree of pleiotropy [16,18,20,84–89]. Yet, how regulatory and substitution changes alter their function under these various environments and across many more alleles and genetic backgrounds is still largely unknown. Measuring the effects of genes using standing variation can reveal patterns of evolution and signatures of selection [48]. Insertion/deletion mutations are commonly seen in natural variants as well; although indel effects are poorly understood, they can now be investigated using MAVE approaches [90, 91]. Moreover, coupling growth phenotypes with molecular phenotypes like gene expression or protein abundance can lead to mechanistic understanding. They also illustrate a high-throughput way for studying distribution of fitness effects of genes, which is an important and long-standing question in understanding evolution.
Even with the high-throughput approaches developed to date, many challenges and prospects in identifying causal variants persist. In order to measure variant effects on complex traits, the traits must have phenotypes that can be measured accurately in high throughput. Additionally, knowing only one of the multiple traits affected by a pleiotropic gene may confound interpretations of how variation affects fitness. The impact of intergenic regions remains comparatively understudied as well. Increases in whole-genome sequencing have also revealed that causal copy number variants explain a larger fraction of phenotypic variance when compared to SNPs [3]. Thus, future studies will need to move beyond nucleotide-level variants; increasing the throughput for studying the effects of mutations such as copy number variation, translocation, and aneuploidies will provide a more exhaustive view of how genotype affects phenotype. Finally, the success of heterologously expressing human genes in yeast to understand variant function is evidence that this versatile model organism can test gene function across other organisms as well. Applications of these high-throughput methods across taxa will inform the evolutionary history of selection, adaptation, and drift in spanning diverse populations.
Acknowledgements
We thank Joseph Schacherer and Aimée Dudley for critical reading of this manuscript. PJ was supported by the PEW Charitable Trust awarded to Kelley Harris and Burroughs Wellcome Fund Career Award at the Scientific Interface awarded to Kelley Harris. CCY was supported by the National Science Foundation Graduate Research Fellowship [award number DGE-1762114]. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health [grant number R01 GM101091] and by the National Human Genome Research Institute of the National Institutes of Health [grant number R01 HG010378].
Footnotes
Declarations of interest: none.
References
- 1.Fay JC: The molecular basis of phenotypic variation in yeast. Curr Opin Genet Dev 2013, 23:672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peltier E, Friedrich A, Schacherer J, Marullo P: Quantitative Trait Nucleotides Impacting the Technological Performances of Industrial Saccharomyces cerevisiae Strains. Front Genet 2019, 10:683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, et al. : Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai F-Y, Han D-Y, Duan S-F, Wang Q-M: The Ecology and Evolution of the Baker’s Yeast Saccharomyces cerevisiae. Genes 2022, 13:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallone B, Steensels J, Prahl T, Soriaga L, Saels V, Herrera-Malaver B, Merlevede A, Roncoroni M, Voordeckers K, Miraglia L, et al. : Domestication and Divergence of Saccharomyces cerevisiae Beer Yeasts. Cell 2016, 166:1397–1410.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackay TFC, Stone EA, Ayroles JF: The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 2009, 10:565–577. [DOI] [PubMed] [Google Scholar]
- 7.Sardi M, Paithane V, Place M, Robinson DE, Hose J, Wohlbach DJ, Gasch AP: Genome-wide association across Saccharomyces cerevisiae strains reveals substantial variation in underlying gene requirements for toxin tolerance. PLoS Genet 2018, 14:e1007217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. : Finding the missing heritability of complex diseases. Nature 2009, 461:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diao L, Chen KC: Local ancestry corrects for population structure in Saccharomyces cerevisiae genome-wide association studies. Genetics 2012, 192:1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connelly CF, Akey JM: On the prospects of whole-genome association mapping in Saccharomyces cerevisiae. Genetics 2012, 191:1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strope PK, Skelly DA, Kozmin SG, Mahadevan G, Stone EA, Magwene PM, Dietrich FS, McCusker JH: The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Research 2015, 25:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galardini M, Busby BP, Vieitez C, Dunham AS, Typas A, Beltrao P: The impact of the genetic background on gene deletion phenotypes in Saccharomyces cerevisiae. Mol Syst Biol 2019, 15:e8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peter J, Friedrich A, Liti G, Schacherer J: Extensive simulations assess the performance of genome-wide association mapping in various Saccharomyces cerevisiae subpopulations. Philos Trans R Soc Lond B Biol Sci 2022, doi: 10.1098/rstb.2020.0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maclean CJ, Metzger BPH, Yang J-R, Ho W-C, Moyers B, Zhang J: Deciphering the Genic Basis of Yeast Fitness Variation by Simultaneous Forward and Reverse Genetics. Mol Biol Evol 2017, 34:2486–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.*.Bloom JS, Boocock J, Treusch S, Sadhu MJ, Day L, Oates-Barker H, Kruglyak L: Rare variants contribute disproportionately to quantitative trait variation in yeast. eLife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study used 16 natural isolates as the multi-parental genotypes and performed QTL analysis on 13,950 recombinant haploid yeast segregants, which captured the majority of common variants in S. cerevisiae. They examined the growth of each segregant under 38 diverse environments and found genetic effects account for over half of the total genetic variation and rare variants attribute disproportionally more to the variation.
- 16.Jakobson CM, Jarosz DF: Molecular Origins of Complex Heritability in Natural Genotype-to-Phenotype Relationships. Cell Syst 2019, 8:363–379.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas R, Horev G, Lipkin E, Kesten I, Portnoy M, Buhnik-Rosenblau K, Soller M, Kashi Y: Mapping Ethanol Tolerance in Budding Yeast Reveals High Genetic Variation in a Wild Isolate. Front Genet 2019, 10:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.*.Nguyen Ba AN, Lawrence KR, Rego-Costa A, Gopalakrishnan S, Temko D, Michor F, Desai MM: Barcoded Bulk QTL mapping reveals highly polygenic and epistatic architecture of complex traits in yeast. Elife 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study developed a high-throughput barcoded bulk quantitative trait mapping which genotyped and phenotyped 100,000 offspring from yeast crosses, becoming the largest high-throughput QTL analysis in yeast to date. They applied this method to map eighteen complex traits, and found many small effect loci contributing to trait variation. They also found epistasis is playing an important role.
- 19.*.Matsui T, Mullis MN, Roy KR, Hale JJ, Schell R, Levy SF, Ehrenreich IM: The interplay of additivity, dominance, and epistasis on fitness in a diploid yeast cross. Nat Commun 2022, 13:1463. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study systematically explored the effect of dominance and epistasis in quantitative trait variation through ~200,000 barcoded diploids in different environments. They found similar degrees of additive effects compared to previous smaller datasets, but recover ~40% more genetic interactions in each environment.
- 20.Wilkening S, Lin G, Fritsch ES, Tekkedil MM, Anders S, Kuehn R, Nguyen M, Aiyar RS, Proctor M, Sakhanenko NA, et al. : An evaluation of high-throughput approaches to QTL mapping in Saccharomyces cerevisiae. Genetics 2014, 196:853–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Qi Q, Lin Y, Guo Y, Liu Y, Wang Q: QTL analysis reveals genomic variants linked to high-temperature fermentation performance in the industrial yeast. Biotechnol Biofuels 2019, 12:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bloom JS, Ehrenreich IM, Loo WT, Lite T- LV, Kruglyak L: Finding the sources of missing heritability in a yeast cross. Nature 2013, 494:234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.*.Renganaath K, Cheung R, Day L, Kosuri S, Kruglyak L, Albert FW: Systematic identification of cis-regulatory variants that cause gene expression differences in a yeast cross. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors used a massive parallel reporter assay to examine eQTLs in the promoter regions between BY and RM strains. They found that most allele-specific expression differences are subtle, and only 6% of causal variants altered expression by over two-fold. This is consistent with the idea of many loci of small effects that contribute to the complex trait.
- 24.Sirr A, Scott AC, Cromie GA, Ludlow CL, Ahyong V, Morgan TS, Gilbert T, Dudley AM: Natural Variation in SER1 and ENA6 Underlie Condition-Specific Growth Defects in Saccharomyces cerevisiae. G3 2018, 8:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duveau F, Vande Zande P, Metzger BP, Diaz CJ, Walker EA, Tryban S, Siddiq MA, Yang B, Wittkopp PJ: Mutational sources of trans-regulatory variation affecting gene expression in Saccharomyces cerevisiae. Elife 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih C-H, Fay J: Cis-regulatory variants affect gene expression dynamics in yeast. Elife 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.*.Brion C, Lutz SM, Albert FW: Simultaneous quantification of mRNA and protein in single cells reveals post-transcriptional effects of genetic variation. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study developed a CRISPR-based system that quantifies mRNA and protein of a given gene using fluorescent reporters simultaneously in the yeast cell. They mapped 86 trans-acting loci affecting the expression of ten genes, and found only a small proportion (20%) have concordant effects of mRNA and proteins.
- 28.Albert FW, Treusch S, Shockley AH, Bloom JS, Kruglyak L: Genetics of single-cell protein abundance variation in large yeast populations. Nature 2014, 506:494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson CA, Castro DM, Saldi G-A, Bonneau R, Gresham D: Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voordeckers K, De Maeyer D, van der Zande E, Vinces MD, Meert W, Cloots L, Ryan O, Marchal K, Verstrepen KJ: Identification of a complex genetic network underlying Saccharomyces cerevisiae colony morphology. Mol Microbiol 2012, 86:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nogami S, Ohya Y, Yvert G: Genetic complexity and quantitative trait loci mapping of yeast morphological traits. PLoS Genet 2007, 3:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins MA, Avery RR, Albert FW: Substrate-Specific Effects of Natural Genetic Variation on Proteasome Activity. bioRxiv 2021, doi: 10.1101/2021.11.23.469794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eder M, Sanchez I, Brice C, Camarasa C, Legras J-L, Dequin S: QTL mapping of volatile compound production in Saccharomyces cerevisiae during alcoholic fermentation. BMC Genomics 2018, 19:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torabi N, Kruglyak L: Genetic basis of hidden phenotypic variation revealed by increased translational readthrough in yeast. PLoS Genet 2012, 8:e1002546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fournier T, Abou Saada O, Hou J, Peter J, Caudal E, Schacherer J: Extensive impact of low-frequency variants on the phenotypic landscape at population-scale. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharon E, Chen S- AA, Khosla NM, Smith JD, Pritchard JK, Fraser HB: Functional Genetic Variants Revealed by Massively Parallel Precise Genome Editing. Cell 2018, 175:544–557.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy KR, Smith JD, Vonesch SC, Lin G, Tu CS, Lederer AR, Chu A, Suresh S, Nguyen M, Horecka J, et al. : Multiplexed precision genome editing with trackable genomic barcodes in yeast. Nat Biotechnol 2018, 36:512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo X, Chavez A, Tung A, Chan Y, Kaas C, Yin Y, Cecchi R, Garnier SL, Kelsic ED, Schubert M, et al. : High-throughput creation and functional profiling of DNA sequence variant libraries using CRISPR–Cas9 in yeast. Nat Biotechnol 2018, 36:540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Després PC, Dubé AK, Seki M, Yachie N, Landry CR: Perturbing proteomes at single residue resolution using base editing. Nat Commun 2020, 11:1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lutz S, Brion C, Kliebhan M, Albert FW: DNA variants affecting the expression of numerous genes in trans have diverse mechanisms of action and evolutionary histories. PLoS Genet 2019, 15:e1008375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.*.Lian J, Schultz C, Cao M, HamediRad M, Zhao H: Multi-functional genome-wide CRISPR system for high throughput genotype–phenotype mapping. Nat Commun 2019, 10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pooling CRISPR plasmid libraries that individually upregulate, downregulate, and knockout all yeast genes simultaneously in one assay revealed not only the genes involved in a trait, but also what genetic changes genome-wide can enhance or reduce fitness under specific conditions. The authors also assembled combinatorial activation, inhibition, or deletion of different genes by cloning two guides on the same plasmid to identify synergistic effects of genetic changes that are not identifiable using vectors that perturb only one gene.
- 42.Momen-Roknabadi A, Oikonomou P, Zegans M, Tavazoie S: An inducible CRISPR interference library for genetic interrogation of Saccharomyces cerevisiae biology. Commun Biol 2020, 3:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGlincy NJ, Meacham ZA, Reynaud KK, Muller R, Baum R, Ingolia NT: A genome-scale CRISPR interference guide library enables comprehensive phenotypic profiling in yeast. BMC Genomics 2021, 22:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alford BD, Tassoni-Tsuchida E, Khan D, Work JJ, Valiant G, Brandman O: ReporterSeq reveals genome-wide dynamic modulators of the heat shock response across diverse stressors. Elife 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sadhu MJ, Bloom JS, Day L, Siegel JJ, Kosuri S, Kruglyak L: Highly parallel genome variant engineering with CRISPR–Cas9. Nat Genet 2018, 50:510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amorosi CJ, Chiasson MA, McDonald MG, Wong LH, Sitko KA, Boyle G, Kowalski JP, Rettie AE, Fowler DM, Dunham MJ: Massively parallel characterization of CYP2C9 variant enzyme activity and abundance. Am J Hum Genet 2021, 108:1735–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ollodart AR, Yeh C- LC, Miller AW, Shirts BH, Gordon AS, Dunham MJ: Multiplexing mutation rate assessment: determining pathogenicity of Msh2 variants in Saccharomyces cerevisiae. Genetics 2021, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeh C- LC, Tsouris A, Schacherer J, Dunham MJ: High-throughput functional analysis of natural variants in yeast. bioRxiv 2021, doi: 10.1101/2021.02.26.433108. [DOI] [Google Scholar]
- 49.Weile J, Roth FP: Multiplexed assays of variant effects contribute to a growing genotype–phenotype atlas. Hum Genet 2018, 137:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rich MS, Payen C, Rubin AF, Ong GT, Sanchez MR, Yachie N, Dunham MJ, Fields S: Comprehensive Analysis of the SUL1 Promoter of Saccharomyces cerevisiae. Genetics 2016, 203:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLure RJ, Radford SE, Brockwell DJ: High-throughput directed evolution: a golden era for protein science. Trends in Chemistry 2022, 4:378–391. [Google Scholar]
- 52.Mehlhoff JD, Ostermeier M: Biological fitness landscapes by deep mutational scanning. Methods Enzymol 2020, 643:203–224. [DOI] [PubMed] [Google Scholar]
- 53.Morton EA, Dorrity MW, Zhou W, Fields S, Queitsch C: Transcriptional re-wiring by mutation of the yeast Hsf1 oligomerization domain. bioRxiv, 2020, doi: 10.1101/2020.05.23.112250. [DOI] [Google Scholar]
- 54.Song L-F, Deng Z-H, Gong Z-Y, Li L-L, Li B-Z: Large-Scale de novo Oligonucleotide Synthesis for Whole-Genome Synthesis and Data Storage: Challenges and Opportunities. Front Bioeng Biotechnol 2021, 9:689797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein JC, Lajoie MJ, Schwartz JJ, Strauch E-M, Nelson J, Baker D, Shendure J: Multiplex pairwise assembly of array-derived DNA oligonucleotides. Nucleic Acids Res 2016, 44:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biot-Pelletier D, Martin VJJ: Seamless site-directed mutagenesis of the Saccharomyces cerevisiae genome using CRISPR-Cas9. J Biol Eng 2016, 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbieri EM, Muir P, Akhuetie-Oni BO, Yellman CM, Isaacs FJ: Precise Editing at DNA Replication Forks Enables Multiplex Genome Engineering in Eukaryotes. Cell 2017, 171:1453–1467.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kapolka NJ, Taghon GJ, Rowe JB, Morgan WM, Enten JF, Lambert NA, Isom DG: DCyFIR: a high-throughput CRISPR platform for multiplexed G protein-coupled receptor profiling and ligand discovery. Proc Natl Acad Sci U S A 2020, 117:13117–13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boonekamp FJ, Knibbe E, Vieira-Lara MA, Wijsman M, Luttik MAH, van Eunen K, den Ridder M, Bron R, Almonacid Suarez AM, van Rijn P, et al. : A yeast with muscle doesn’t run faster: full humanization of the glycolytic pathway in Saccharomyces cerevisiae. bioRxiv 2021, doi: 10.1101/2021.09.28.462164. [DOI] [PubMed] [Google Scholar]
- 60.*.Hamza A, Driessen MRM, Tammpere E, O’Neil NJ, Hieter P: Cross-Species Complementation of Nonessential Yeast Genes Establishes Platforms for Testing Inhibitors of Human Proteins. Genetics 2020, 214:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors systematically identified which human orthologs can complement the phenotype of 112 nonessential yeast genes involved in chromosome stability. Using nonessential gene knockouts in yeast to screen for complementation by human orthologs has previously been challenging because complementation screens require observable phenotypes, and the authors developed a set of novel yeast-based assays for understanding the function of human genes in humanized yeast.
- 61.Sirr A, Lo RS, Cromie GA, Scott AC, Ashmead J, Heyesus M, Dudley AM: A yeast-based complementation assay elucidates the functional impact of 200 missense variants in human PSAT1. J Inherit Metab Dis 2020, 43:758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Newberry RW, Leong JT, Chow ED, Kampmann M, DeGrado WF: Deep mutational scanning reveals the structural basis for α-synuclein activity. Nat Chem Biol 2020, 16:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cervelli T, Lodovichi S, Bellè F, Galli A: Yeast-based assays for the functional characterization of cancer-associated variants of human DNA repair genes. Microb Cell Fact 2020, 7:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.*.Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, et al. : Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182:1295–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors performed a deep mutational scan on the receptor binding domain (RBD) of the spike protein in SARS-CoV-2 and determined each variant’s affinity to the human ACE2 protein using a fluorescent yeast surface display assay. By using a yeast test bed for RBD function, researchers were able to rapidly determine the effects of substitutions in this viral protein domain at a crucial moment in the COVID-19 pandemic.
- 65.Faure AJ, Domingo J, Schmiedel JM, Hidalgo-Carcedo C, Diss G, Lehner B: Mapping the energetic and allosteric landscapes of protein binding domains. Nature 2022, 604:175–183. [DOI] [PubMed] [Google Scholar]
- 66.Dionne U, Bourgault É, Dubé AK, Bradley D, Chartier FJM, Dandage R, Dibyachintan S, Després PC, Gish GD, Pham NTH, et al. : Protein context shapes the specificity of SH3 domain-mediated interactions in vivo. Nat Commun 2021, 12:1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diss G, Lehner B: The genetic landscape of a physical interaction. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Logsdon GA, Vollger MR, Eichler EE: Long-read human genome sequencing and its applications. Nat Rev Genet 2020, 21:597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Boer CG, Vaishnav ED, Sadeh R, Abeyta EL, Friedman N, Regev A: Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat Biotechnol 2020, 38:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cuperus JT, Groves B, Kuchina A, Rosenberg AB, Jojic N, Fields S, Seelig G: Deep learning of the regulatory grammar of yeast 5′ untranslated regions from 500,000 random sequences. Genome Res 2017, 27:2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vaishnav ED, de Boer CG, Molinet J, Yassour M, Fan L, Adiconis X, Thompson DA, Levin JZ, Cubillos FA, Regev A: The evolution, evolvability and engineering of gene regulatory DNA. Nature 2022, 603:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC, Wu Y, Pons C, Wong C, van Lieshout N, et al. : A framework for exhaustively mapping functional missense variants. Mol Syst Biol 2017, 13:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Livesey BJ, Marsh JA: Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol Syst Biol 2020, 16:e9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gray VE, Hause RJ, Luebeck J, Shendure J, Fowler DM: Quantitative Missense Variant Effect Prediction Using Large-Scale Mutagenesis Data. Cell Systems 2018, 6:116–124.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou X, Cai X: Joint eQTL mapping and inference of gene regulatory network improves power of detecting both cis- and trans-eQTLs. Bioinformatics 2021, 38:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reeb J, Wirth T, Rost B: Variant effect predictions capture some aspects of deep mutational scanning experiments. BMC Bioinformatics 2020, 21:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.She R, Jarosz DF: Mapping Causal Variants with Single-Nucleotide Resolution Reveals Biochemical Drivers of Phenotypic Change. Cell 2018, 172:478–490.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Ušaj M, Hanchard J, Lee SD, et al. : A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Costanzo M, Hou J, Messier V, Nelson J, Rahman M, VanderSluis B, Wang W, Pons C, Ross C, Ušaj M, et al. : Environmental robustness of the global yeast genetic interaction network. Science 2021, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Domingo J, Diss G, Lehner B: Pairwise and higher-order genetic interactions during the evolution of a tRNA. Nature 2018, 558:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuzmin E, VanderSluis B, Nguyen Ba AN, Wang W, Koch EN, Usaj M, Khmelinskii A, Usaj MM, van Leeuwen J, Kraus O, et al. : Exploring whole-genome duplicate gene retention with complex genetic interaction analysis. Science 2020, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Caudal E, Friedrich A, Jallet A, Garin M, Hou J, Schacherer J: Population-level survey of loss-of-function mutations revealed that background dependent fitness genes are rare and functionally related in yeast. bioRxiv 2021, doi: 10.1101/2021.08.25.457624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bakerlee CW, Ba ANN, Shulgina Y, Echenique JIR, Desai MM: Idiosyncratic epistasis leads to global fitness–correlated trends. Science 2022, 376:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim HS, Fay JC: A combined-cross analysis reveals genes with drug-specific and background-dependent effects on drug sensitivity in Saccharomyces cerevisiae. Genetics 2009, 183:1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gou L, Bloom JS, Kruglyak L: The Genetic Basis of Mutation Rate Variation in Yeast. Genetics 2019, 211:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Steinmetz LM, Sinha H, Richards DR, Spiegelman JI, Oefner PJ, McCusker JH, Davis RW: Dissecting the architecture of a quantitative trait locus in yeast. Nature 2002, 416:326–330. [DOI] [PubMed] [Google Scholar]
- 87.Deutschbauer AM, Davis RW: Quantitative trait loci mapped to single-nucleotide resolution in yeast. Nat Genet 2005, 37:1333–1340. [DOI] [PubMed] [Google Scholar]
- 88.Dimitrov LN, Brem RB, Kruglyak L, Gottschling DE: Polymorphisms in multiple genes contribute to the spontaneous mitochondrial genome instability of Saccharomyces cerevisiae S288C strains. Genetics 2009, 183:365–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Demogines A, Smith E, Kruglyak L, Alani E: Identification and dissection of a complex DNA repair sensitivity phenotype in Baker’s yeast. PLoS Genet 2008, 4:e1000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Seuma M, Lehner B, Bolognesi B: An atlas of amyloid aggregation: the impact of substitutions, insertions, deletions and truncations on amyloid beta fibril nucleation. bioRxiv 2022, doi: 10.1101/2022.01.18.476804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Macdonald CB, Nedrud D, Grimes PR, Trinidad D, Fraser JS, Coyote-Maestas W: Deep Insertion, Deletion, and Missense Mutation Libraries for Exploring Protein Variation in Evolution, Disease, and Biology. bioRxiv 2022, doi: 10.1101/2022.07.26.501589. [DOI] [PMC free article] [PubMed] [Google Scholar]