

Abstract
Ossification of the posterior longitudinal ligament (OPLL) is a degenerative disease characterized by progressive ectopic bone formation process, which can lead to severe neurological impairments and reduced quality of life. While the etiology of OPLL is generally considered multifactorial, there is no consensus regarding these contributing factors including genetic, endocrine, biomechanical, immune and lifestyle factors. Through accumulating evidence from multidisciplinary investigations, the pathophysiological connection between OPLL and endocrine-metabolic dysregulation is becoming increasingly clear. Nevertheless, comprehensive understanding of the relationship between the two is hindered by several problems, such as methodological limitations and inadequate mechanistic studies. This review takes a deep dive into the possible factors contributing to OPLL from all aspects of metabolism, including glucose metabolism, lipid metabolism, bone and mineral metabolism, leptin, vitamin, growth hormone/IGF-1 and sex hormones, highlighting their potential roles in the onset and progression of OPLL. Clarifying the etiology of OPLL and elucidating the underlying pathogenesis are crucial for advancing both early intervention strategies and therapeutic approaches in clinical management. Therefore, the endocrine and metabolic disorders in OPLL patients should become a focus of future research.
Subject terms: Bone, Metabolism
Introduction
Definition, epidemiology, and demography of OPLL
Ossification of the spinal ligament (OSL) represents a spectrum of pathological conditions marked by progressive heterotopic bone formation or calcium deposition in spinal connective tissues. This disease cluster encompasses three main subtypes: ossification of the posterior longitudinal ligament (clinically termed OPLL), ossification of the ligamentum flavum, and other ossified adjacent spinal support structures.1 Notably, OPLL manifests with heightened clinical severity due to the ligament’s unique anatomical positioning directly anterior to the spinal cord within the vertebral canal. The ossification process typically initiates at the posterior vertebral margins, leading to the formation of bone-like structures that progressively enlarge, potentially compressing neural elements. While OPLL may remain asymptomatic or manifest with nonspecific symptoms, even minor trauma can precipitate severe neurological deficits, including sensory and motor impairments, reflex abnormality, and urinary/fecal dysfunction. In extreme cases, paralysis may ensue, significantly impairing quality of life and imposing substantial socioeconomic burdens1,2 (Fig. 1).
Fig. 1.

The spinal cord and nerve roots are compressed by the ossified mass originated from ligament tissue, resulting in various symptoms and signs according to the innervation
OPLL predominantly affects the cervical spine, with the highest incidence observed at the C5 level, followed by C4 and C6. Thoracic and lumbar involvement is less common.3,4 It can sometimes coexist with other musculoskeletal disorders, such as ossification of the ligamentum flavum (OLF), diffuse idiopathic skeletal hyperostosis (DISH), and ankylosing spondylitis.1,3,4 Demographic studies have indicated that OPLL primarily occurs in individuals aged 40–60 years, with a higher prevalence in men (male-to-female ratio of 2:1). Symptomatic cases typically emerge in the fifth to sixth decades of life.5,6 The disease exhibits marked geographic and racial disparities, with higher prevalence rates observed in East Asian populations. For example, the prevalence of cervical OPLL (C-OPLL) has been documented as ranging from 1.9% to 6.3% in Japanese, 0.6% to 5.7% in Korean, with an average prevalence of 4.1% observed in Chinese populations.7 In contrast, prevalence rates in Western populations including the United States and Europe, are much lower, ranging from 0.01% to 1.7%.8 Notably, in a cross-sectional study comparing the prevalence of OPLL between different racial groups, Asian Americans also demonstrate the highest prevalence rates, underscoring the influence of genetic and ethnic factors9 (Fig. 2).
Fig. 2.
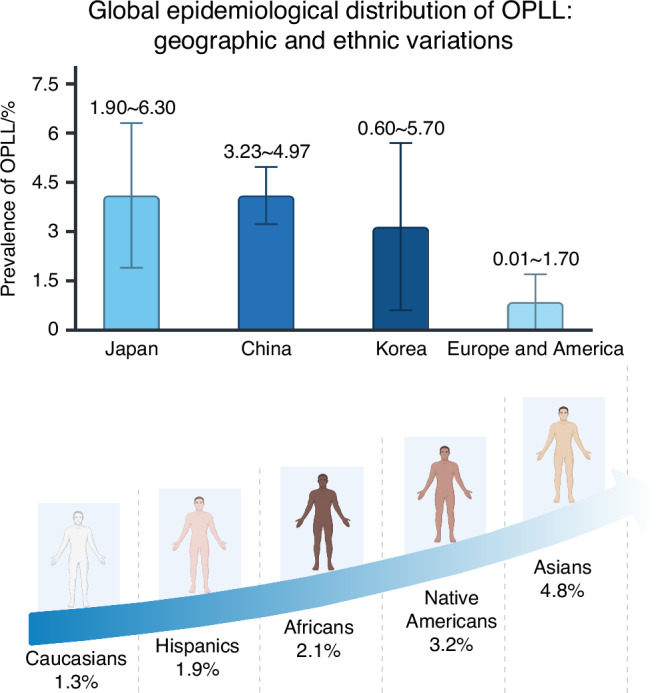
The left is the prevalence of C-OPLL across different regions. According to previous reports, the prevalence of C-OPLL was 1.90% to 6.30% in Japanese, 0.60% to 5.70% in Korean, 3.23% to 4.97% (95% confidence interval) in Chinese, and 0.01% to 1.7% in western populations. The right demonstrates the ethnic differences observed in the USA
Clinical presentation, diagnosis and management of OPLL
OPLL often progresses insidiously, with many patients remaining asymptomatic despite radiographic evidence of ossification.2 In its early stages, patients may experience mild discomfort, pain, or numbness in their hands, but as the disease advances and neural compression intensifies, symptoms worsen. Severe clinical manifestations may involve axial neck pain combined with neurocompressive phenomena such as radicular impairments and spinal cord dysfunction.1 Spine evaluations reveal that over 40% of patients will exhibit signs and/or symptoms of myelopathy.10 Key risk factors for neurological impairment include reduced spinal canal diameter (<6 mm), increased spinal mobility, lateral deviation of OPLL lesions, and spinal canal occupancy exceeding 50%–60% on cross-sectional imaging.11–13
Accurate diagnosis of OPLL relies on advanced imaging modalities, including radiography (X-rays), computed tomography (CT), magnetic resonance imaging (MRI), and three-dimensional (3D) reconstructions. Radiographic classification distinguishes OPLL into focal, segmental, continuous, or mixed types.14 The K-line, a radiographic parameter connecting the midpoints of the anteroposterior canal diameters at C2 and C7, aids in determining surgical approaches.15 CT imaging provides detailed visualization of ossified lesions, including their location, shape, size, and spinal canal occupancy, with severe cases often showing a characteristic “double-layer sign”.16 MRI is invaluable for assessing spinal cord compression, intramedullary signal changes, and predicting surgical outcomes.12
Management strategies for OPLL are tailored to disease severity and neurological status. Conservative treatments, such as cervical traction, bracing, nonsteroidal anti-inflammatory drugs (NSAIDs), activity modification, and physical therapy, are recommended for minimally symptomatic patients or those with high surgical risks.17 However, these measures do not mitigate the risk of future neurologic injury. Surgical intervention becomes necessary when patients exhibit advancing neurological deterioration or demonstrate persistent medullary compression refractory to comprehensive nonsurgical management.18 It aims to decompress neural structures, either by direct resection of ossified lesions or by expanding the spinal canal volume. There are two surgical approaches available: anterior options include discectomy with fusion and corpectomy, while posterior approaches encompass laminectomy with fusion (LF) or laminoplasty (LP). Complex cases may even require combined anterior-posterior decompression.2,19 Although surgical decompression remains the standard treatment for OPLL-induced neurological dysfunction, it carries significant risks of complications and does not halt disease progression, as evidenced by postoperative ossification progression in many cases.20–22
Etiology, pathogenesis, and review objectives
OPLL primarily arises through endochondral ossification, a process involving several stages.23,24 Initially, fibrocartilaginous tissues undergo hypertrophy, driven by the proliferation of SOX9+ chondrocytes. This is followed by matrix mineralization, characterized by hydroxyapatite deposition, leading to the formation of calcified lesions. Hypertrophic chondrocytes within these lesions express RUNX2, a transcription factor essential for osteoblast differentiation. Ultimately, chondrocyte apoptosis, matrix degradation, and neovascularization result in the replacement of calcified cartilage with bone. As for etiology. OPLL is widely regarded as a multifactorial disorder,1 with genetic, endocrine, biomechanical, and lifestyle factors, as well as intracellular and extracellular cytokines all reported to influence its occurrence and progression.25 Despite extensive research, the precise mechanisms remain unclear, and the relative contributions of these factors are debated.
Emerging clinical evidence has revealed significant homeostatic imbalances in glucose metabolism (e.g., hyperglycemia with insulin resistance), lipid profiles (e.g., dyslipidemia), and bone remodeling processes (e.g., elevated bone mineral density) among patients with OPLL,26–29 establishing endocrine-metabolic dysregulations as a pathogenic cornerstone (Fig. 3). Nevertheless, current research remains predominantly observational—relying on cross-sectional clinical comparisons and epidemiological correlations, while mechanistic studies elucidating causal relationships between metabolic disorders and OPLL pathogenesis remain critically underexplored. Although recent investigations have identified several candidate biomarkers, such as hormones, calcium-phosphate metabolism markers, and bone turnover indicators,26–29 most of them primarily focus on comparative analyses between OPLL patients and healthy controls, without delving into underlying pathophysiological mechanisms. The inconsistent findings and insufficient mechanistic investigations severely hinder a deeper understanding of the relationship between OPLL and endocrine-metabolic factors.
Fig. 3.

The human body diagram lists systemic endocrine-metabolic factors associated with OPLL
Current therapeutic approaches, whether medical or surgical, fail to halt OPLL progression, underscoring the need for targeted therapies to arrest disease advancement. This review synthesizes and critically evaluates existing research on the interplay between OPLL and endocrine-metabolic dysregulations, offering a mechanistic understanding of disease pathogenesis through metabolic pathways. By identifying clinically actionable biomarkers for early diagnosis/intervention and druggable molecular targets, this work establishes a theoretical foundation for developing targeted therapeutic strategies centered on metabolic modulation, ultimately aiming to optimize OPLL management. Furthermore, as the first comprehensive reexamination of OPLL pathogenesis from an integrated metabolic perspective, this review not only clarifies existing knowledge gaps but also provides valuable insights for advancing future research in molecular diagnostics and personalized treatment approaches.
Role of carbohydrate metabolism disorder
High glucose
Diabetes mellitus, particularly type 2 diabetes mellitus (T2DM), represents one of the most prevalent disorders of carbohydrate metabolism. Epidemiological evidence suggests that T2DM is an independent risk factor for the development of OPLL.30 However, current research gaps persist in delineating the precise pathophysiological pathways linking diabetic dysregulation with ectopic spinal ossification. Hyperglycemia, a hallmark of diabetes, plays a pivotal role in diabetes-associated bone metabolism dysregulation. Experimental models demonstrate that elevated glucose levels potentiate reactive oxygen species (ROS) generation across diverse cellular populations, mediating critical processes including osteoblast differentiation,31,32 vascular calcification,33 and chondrocyte hypertrophy during endochondral ossification.34 Exposure to high glucose concentrations can induce alterations in gene expression, with varying effects on osteogenic differentiation depending on cell types. In vascular smooth muscle lineages, high glucose stimuli upregulate bone morphogenetic regulators (BMP-2/RUNX2), predisposing to pathological mineral deposition.35 In mesenchymal stem cells, glucose overload enhances both osteogenesis and chondrogenesis.36,37 Contrastingly, committed osteoprogenitor cells exhibit significantly attenuated differentiation capacity under high glucose conditions.38,39
Notably, differences in gene expression have been observed between posterior longitudinal ligament cells derived from OPLL patients and those from non-ossified ligaments. Cells from ossified ligaments exhibit osteoblast-like characteristics, including the expression of early osteogenic markers such as Runx2, alkaline phosphatase (ALP), and osteopontin,40 which is essential to obtain an osteoblast phenotype.41 Under hyperglycemic conditions (25 mmol/L glucose), research demonstrates enhanced BMP-2-mediated type I collagen production and upregulated expression of early osteogenic differentiation markers (ALP/RUNX2) in rodent spinal ligament fibroblasts. Mechanistically, the study elucidated three sequential pathological cascades: ROS overproduction initiating oxidative stress, subsequent activation of specific protein kinase C isoforms, and eventual suppression of the p38 MAPK signaling pathway. These sequential responses collectively promote abnormal collagen deposition and pre-osteoblastic genetic programming in these specialized cells.42 While the gluco-ossification correlation remains contentious in certain cohorts,29 chronic hyperglycemia appears to be a plausible etiologic contributor to T2DM-OPLL comorbidity.
Hyperinsulinemia
Extensive population-based research has identified obesity (BMI ≥ 25 kg/m²), impaired glucose tolerance, and non-insulin-dependent diabetes mellitus (NIDDM) as significant risk factors for OPLL.30,43,44 These metabolic disturbances frequently coincide with disrupted insulin signaling pathways and elevated serum insulin levels. This pathophysiological correlation has prompted investigators to explore insulin’s regulatory functions in ectopic bone formation processes. Recently, quantitative analyses have demonstrated that dynamic insulin secretion patterns directly correlate with radiographic progression of spinal ossification, with postprandial insulin production metrics showing particular promise as prognostic biomarkers of OPLL.45 In addition to OPLL patients, hyperinsulinemia is also observed in animal models of OPLL, such as leptin receptor-deficient Zucker rats which demonstrate characteristic hypersecretion of pancreatic hormones.46
These observations in OPLL people and animal models align with cellular-level findings, where supraphysiological insulin concentrations (≥100 nmol/L) are shown to significantly potentiate ectopic mineralization processes in vascular lineage cells.47 Strikingly, mechanistic studies have revealed significant overlap between two key biological pathways: the insulin-responsive PI3K/Akt pathway and BMP-2-activated osteogenic signaling networks. This convergence suggests common molecular mechanisms may simultaneously regulate metabolic processes and bone formation.48,49 Spinal ligament cells possess insulin receptor substrate-1 (IRS-1), enabling them to detect circulating insulin and make a response. Research findings demonstrate that chronically elevated insulin levels direct cellular differentiation processes within spinal ligament cells through a metabolic regulatory system. Specifically, through insulin receptor-triggered PI3K-Akt pathway activation, coupled with the inhibition of ERK-mediated growth regulation, hyperinsulinemia creates conditions favoring both cell proliferation and BMP-2-induced osteogenic differentiation.50 This mechanistic insight provides a potential explanation for how prolonged hyperinsulinemia may drive OPLL progression in individuals with type 2 diabetes.
Advanced glycation end products (AGEs)
Advanced glycation end products (AGEs) represent a class of pathologically modified biomolecules generated through non-enzymatic glycation reactions, involving covalent crosslinking between reducing sugars and macromolecular substrates like proteins, lipids, and nucleic acids. Their systemic accumulation increases with the course of natural aging and diabetes, and exhibits a strong correlation with diabetes severity.51 High AGE concentrations have been linked to aberrant mineralization, ectopic calcification, and impaired osteoblast differentiation.52,53 And emerging metabolomic evidence has demonstrated that circulating pentosidine concentrations, a cardinal AGE biomarker, are quantitatively associated with OPLL presence.54 Histopathological characterization of ossified spinal ligaments confirms the presence of AGEs and their receptors (RAGEs). Crucially, in vitro exposure to AGEs (1 mg/mL) greatly upregulates the expression of osteogenic inducers such as BMP-2 and Cbfa1 in ligament cells from OPLL patients.55 These findings position the AGE-RAGE axis as a critical mediator connecting metabolic dysregulation to pathological ectopic ossification. Clinically, the diagnostic potential of AGEs has prompted adoption of non-invasive detection techniques like skin autofluorescence measurement. Research data reveal individuals with thoracic OPLL exhibit significantly higher AGE scores compared to those without OPLL or with cervical OPLL, underscoring the clinical value of AGE quantification in differential diagnosis of spinal ligament ossification subtypes.56
Overall, the triad of chronic hyperglycemia, compensatory hyperinsulinemia, and advanced glycation end-product deposition, pathognomonic features of type 2 diabetes mellitus, constitute pathophysiological convergence points in both diabetic metabolic dysregulation and OPLL development. Although these metabolic disturbances provide a framework for understanding the link between OPLL and diabetes, comprehensive mechanistic investigations remain imperative to decipher their spatiotemporal interplay and identify therapeutic targets for comorbid disease modulation.
Role of lipid metabolism disorder
Lipid metabolism has emerged as a novel and promising area of research in the context of OPLL. Epidemiological analyses document that OPLL patients demonstrate significantly higher prevalence rates of dyslipidemia compared to non-OPLL controls, characterized by elevated serum levels of total cholesterol and triglyceride,57,58 underscoring the potential role of lipid metabolism dysregulation in OPLL pathogenesis. For instance, Fukuda et al. identified dyslipidemia as a novel risk factor for OPLL based on serum lipid profiles, suggesting visceral fat obesity and increased focal mechanical stress due to excess weight may contribute to the condition.58 Zhang et al. utilized Mendelian randomization analysis to reveal that elevated total cholesterol and low-density lipoprotein cholesterol (LDL-C) levels could increase susceptibility to OPLL.59 Additionally, Endo et al. highlighted non-alcoholic fatty liver disease (NAFLD) as a potential risk factor for OPLL progression, with findings showing a higher prevalence of NAFLD in middle-aged OPLL patients and a negative correlation between liver-to-spleen ratio and ossification severity.60
The pathophysiological interplay between lipid dysregulation and ectopic calcification has garnered significant attention. Abnormal dyslipidemia has been linked to the development of vascular calcification. Specifically, through the oxidative damage-mediated apoptosis of vascular smooth muscle cells and downregulation of endogenous calcification inhibitors, dyslipidemia-induced oxidative stress and sustained inflammatory signaling create a permissive microenvironment for cellular phenotype transformation, ultimately driving osteoblastic transdifferentiation and subsequent matrix mineralization.61 Interestingly, these cellular events exhibit striking similarities with the conditions observed in spinal ligament calcification. Central to these mineralization processes is the Wnt/β-catenin signaling pathway, which has been identified as a shared regulatory axis in both vascular calcification and bone formation.62,63 Among numerous signaling molecules in Wnt network, LRP5, a transmembrane co-receptor integral to Wnt signal transduction cascades, functions as a molecular nexus connecting lipid metabolism with skeletal homeostasis.64 In osseous tissues, LRP5 associates with Wnt ligands and Frizzled receptors to form a ternary signaling complex. This molecular assembly initiates a downstream cascade culminating in β-catenin accumulation and nuclear translocation, thereby resulting in the transcription of osteogenic master regulators such as Cbfa-1.65,66 Studies have shown mild dyslipidemia can trigger canonical Wnt/β-catenin pathway via LRP5.67 Furthermore, pro-oxidative conditions induced by hyper-LDLemia are mechanistically linked to vascular calcification processes via dual LRP5/LRP6-dependent Wnt pathway activation.66,68 These discoveries collectively suggest a potential mechanism by which dysregulated lipid profiles, particularly elevated LDL concentrations, may predispose to spinal ligament ossification through Wnt signaling pathway-mediated disruption of bone homeostasis. While these findings provide compelling mechanistic insights, direct experimental evidence linking lipid-mediated Wnt signaling dysregulation to OPLL pathogenesis remains limited. Further investigations are warranted to delineate the exact role of Wnt signaling in lipid metabolism-associated ectopic ossification of the posterior longitudinal ligament.
Metabolomic profiling studies have provided additional insights into the role of fatty acid metabolism in OPLL. Tsuji et al. illuminated distinct fatty acid (FA) profiles with significant elevation of fatty acid-carnitine conjugates in OPLL patients, reinforcing the involvement of lipid metabolism dysregulation in the pathophysiology of this disease.69 Previous investigations have reported that fatty acid derivatives can modulate bone remodeling processes through dual receptor-mediated pathways, including the activation of free fatty acid receptor 4 (FFAR4) signaling cascades and potentiation of parathyroid hormone type 1 receptor (PTH1R) activity. These interactions create a pro-osteogenic microenvironment by enhancing osteoblastgenesis while suppressing osteoclastogenesis.69 Therefore, these elevated lipid intermediates may function as molecular mediators, thus promoting ligamentous ossification processes in OPLL patients. Mead acid and n-3 polyunsaturated fatty acids (PUFAs) are also hypothesized to play a role in OPLL pathogenesis, but comparative lipidomic analyses failed to demonstrate significant concentration differences between OPLL cohorts and control groups.70 Conversely, quantitative profiling uncovers marked elevation of palmitic acid levels in OPLL patients, with a positive correlation with OPLL risk. However, the reason is unclear and their true relationship remains doubtful.70 The precise biological significance of specific fatty acids in OPLL pathogenesis still requires further research.
Role of mineral and bone disorder
Minerals
The relationship between mineral metabolism disorders and spinal ligament ossification was first reported by Adams and Davis.71 Calcium and phosphorus represent essential mineral components crucial for bone health. In normal physiological states, the balance of these minerals is predominantly maintained by calcium-modulating hormones, which are vital for proper bone homeostasis. However, clinical observations have identified disrupted mineral metabolism in OPLL patients, especially those with coexisting endocrine disorders affecting mineral modulation, such as hypoparathyroidism and vitamin D-resistant rickets,71–74 suggesting a potential role of aberrant calcium and phosphate metabolism in this pathogenesis.
Although the co-occurrence of Vitamin D-resistant hypophosphatemic rickets and OPLL has been well documented,75 the accurate incidence of OPLL in patients with this rickets variant remains uncertain due to the limited samples. Vitamin D is well known for its indispensable role in maintaining the mineral and bone homeostasis. While serum measurements show comparable levels of vitamin D metabolites (25-OHD and 1,25-(OH)2D) and circulating calcium between OPLL patients and healthy individuals,27,28,76 functional evaluations reveal distinct abnormalities.77 Specifically, certain OPLL patients demonstrate markedly diminished calciuric responses following oral calcium administration. Furthermore, subsequent investigations disclose a strong association between this hypocalciuric response and the advancement of OPLL.78 The urinary calcium response to oral calcium intake indeed serves as an indicator of intestinal calcium absorption efficiency, which is primarily regulated by active vitamin D metabolites. These observations have led researchers to hypothesize that localized vitamin D activity impairment or tissue-level active vitamin D deficiency, rather than systemic alterations, may underlie OPLL pathogenesis.8 Comparative analysis of bone mineral density (BMD) between OPLL patients and individuals with vitamin D-resistant rickets provides additional validation for this hypothesis. In vitamin D-resistant rickets, bone metabolism abnormalities manifest as decreased BMD and increased unmineralized bone matrix. In contrast, OPLL patients typically exhibit elevated spinal and even systemic BMD regardless of their gender.79,80 BMD, an important marker of mineralization status, is known to be influenced by active vitamin D’s ability to regulate both bone-forming and bone-resorbing activities.29 However, the precise cellular and molecular mechanisms through which active vitamin D enhances mineralization while suppressing bone resorption in OPLL remain unclear, highlighting the need for further investigation into this complex metabolic pathway.
In addition to vitamin D-resistant rickets, a notable prevalence of OPLL among patients with hypoparathyroidism has also been identified, though the exact nature of this association remains a matter of debate.73 The biological basis for this potential connection may lie in the physiological functions of parathyroid hormone (PTH), which regulates calcium and phosphate metabolism by increasing serum calcium while decreasing phosphorus levels. In fact, PTH’s role in bone metabolism is particularly complex, exhibiting both catabolic and anabolic effects depending on its temporal pattern and duration of exposure.81 At the cellular level, research has revealed distinct responses to PTH between ossified and normal ligament tissues. Ligament cells from OPLL patients demonstrate osteoblast-like characteristics when exposed to PTH, contrasting with non-ossified ligament cells from healthy individuals, which show no such osteoblast properties.40 This differential response suggests that ossified ligament cells possess a unique capacity to react to PTH in a manner associated with bone formation. Recent comparative studies have added another layer of complexity by examining OPLL patients with and without DISH. These investigations found significantly higher BMD and intact PTH (i-PTH) levels in OPLL patients with DISH compared to those without DISH.82 However, the specific relationship between DISH and PTH levels remains unexplored, and the precise role of PTH in ligament ossification continues to be poorly understood.
Fibroblast growth factor 23 (FGF-23), a phosphotropic hormone primarily secreted by osteocytes and osteoblasts, plays a critical role in phosphate regulation through the FGF23/Klotho signaling pathway. This cytokine exerts its effects by suppressing active vitamin D synthesis, reducing intestinal phosphate absorption, and inhibiting renal phosphate reabsorption, thereby maintaining phosphate homeostasis.83,84 Elevated circulating FGF-23 levels and associated hypophosphatemia have been consistently observed in both OPLL animal models (Enpp1 ttw/ttw mice) and human patients,27,28,85 suggesting a potential involvement of FGF-23-regulated phosphate metabolism in OPLL pathogenesis. Moreover, recent clinical studies even propose high levels of FGF-23 and hypophosphatemia as potential diagnostic markers for OPLL.83 The connection between phosphate metabolism disorders and OPLL is further supported by the frequent co-occurrence of rickets in OPLL patients.75,86 Similar to OPLL, rickets is characterized by reduced serum phosphate levels, indicating that abnormal phosphate metabolism may be a common feature of both conditions. However, this hypothesis remains controversial, as some studies have found no significant differences in serum phosphate levels between OPLL patients and healthy controls, despite elevated FGF-23 concentrations.27 The potential mechanisms linking FGF-23 to OPLL development remain obscure. Experimental evidence suggests that FGF-23 may inhibit chondrogenesis and accelerate the transition of chondrocytes from proliferation to hypertrophy, potentially contributing to endochondral ossification processes observed in OPLL.87 FGF-23-phosphate metabolism axis may as a promising area for future research into OPLL pathogenesis and potential therapeutic targets.
While significant advances have been made in identifying calcium-phosphate disturbances as potential contributors, the mechanisms linking mineral metabolism abnormalities to the development of OPLL remain incompletely understood. The rare co-occurrence of OPLL with endocrine disorders such as vitamin D-resistant rickets and hypoparathyroidism offers valuable insights into calcium-phosphate dysregulation mechanisms. However, the scarcity of well-documented cases and absence of comprehensive case-control studies have hindered systematic investigation of these metabolic associations. Beyond calcium and phosphorus, recent epidemiological data have expanded the investigative scope to fluoride, another essential mineral influencing bone metabolism and mineralization.88 A significant correlation between fluorosis and OPLL incidence has been established, with increased disease severity being associated with elevated urinary fluoride levels,89 suggesting that fluoride’s osteogenic properties may contribute to pathological bone formation in spinal ligaments.
Bone turnover markers
OPLL patients typically exhibit increased bone mineral density,80,90 suggesting enhanced bone formation activities linked to this disease. Research has explored connections between OPLL and various biochemical indicators of bone remodeling, such as type I collagen derivatives (PICP, ICTP, PINP), bone-specific proteins (osteocalcin, osteopontin), urinary collagen breakdown products (Pyr, Dpyr), and tartrate-resistant acid phosphatase isoform5b (TRAP5b).27,74,76,91–93 Nevertheless, due to limitations, such as a limited sample size, lack of comparison groups, and insufficient control for variables influencing bone metabolism involving age, gender, BMI, and renal function, these investigations have yielded conflicting outcomes, failing to establish definitive conclusions. Consequently, well-designed studies with standardized protocols and adequate sample sizes are essential for clarification. Despite controversy, it remains evident that abnormal bone turnover marker profiles are associated with OPLL’s pathological mechanisms.
Wnt signaling markers
It is well known that Wnt signaling cascade represents a crucial regulatory system in human physiology. As an osteogenic pathway, the Wnt/β-catenin pathway serves as a key modulator of bone formation through promoting the propagation of osteoprogenitor cells and inhibiting the programmed cell death of osteoblasts.94–96 Among various regulators of this pathway, sclerostin and dickkopf-1 (DKK-1) have emerged as the most extensively studied endogenous inhibitors. Sclerostin, a glycoprotein secreted by osteocytes, exerts its inhibitory effect by interacting with low-density lipoprotein receptor-related proteins (LRPs), thereby blocking Wnt ligand binding.97 DKK-1 also acts as an antagonist of Wnt signaling and similarly regulates the pathway through its binding to LRP5/6 co-receptors, though its mode of action differs from sclerostin.98 In the context of posterior longitudinal ligament ossification, alterations in these Wnt inhibitors have been widely observed. Most studies reported elevated serum sclerostin levels alongside reduced DKK-1 concentrations in OPLL patients, while some investigations found no significant changes in DKK-1 levels.27,74,92,99 Another two studies also investigated the relationship between DKK-1 and DISH, but their findings were conflicting.100,101
Notably, increased sclerostin production in older individuals with OPLL has been linked to both higher bone mass and reduced bone turnover, with decreased DKK-1 levels potentially representing a compensatory response to elevated sclerostin.74 Therefore, the discrepancies in these studies may reflect the complex interplay between Wnt signaling components and systemic factors such as age and bone mass. At the cellular level, research have demonstrated that OPLL ligament cells exhibit reduced DKK-1 expression compared to normal controls. Further explorations reveal this downregulation correlates with enhanced Wnt/β-catenin pathway activity, as indicated by increased β-catenin stability and transcriptional activation. Additionally, DKK-1 can inhibit BMP-2-induced osteogenic differentiation in ligament cells through its suppression of Wnt signaling.99 These findings collectively position DKK-1 as a critical negative regulator of ligament ossification. Targeting the Wnt pathway through DKK-1 modulation may offer a promising therapeutic approach for OPLL management.
The canonical Wnt signaling pathway can also influence bone remodeling through boosting osteoprotegerin (OPG) production while suppressing receptor activator of nuclear factor κB ligand (RANKL) expression in osteoblasts, thereby creating an anti-resorptive environment.102 As a soluble receptor secreted by osteoblasts, osteoprotegerin plays a pivotal role in bone mass regulation by competitively binding to RANKL and preventing its interaction with RANKL receptors, thus inhibiting osteoclast formation and activity.103 Comparative analyses have demonstrated significantly elevated concentrations of both osteopontin and OPG in ligament tissues from OPLL patients compared to non-affected individuals. Conspicuously, tissue OPG levels were found to be sixfold higher than corresponding serum concentrations, suggesting a localized mechanism of bone formation regulation in OPLL.104 However, this tissue-specific upregulation contrasts with systemic measurements, as different studies show no significant difference of serum OPG levels between OPLL and non-OPLL groups.92 Further investigations are needed to elucidate the distinct patterns of molecular expression between local tissue environments and systemic circulation in OPLL pathogenesis.
To date, current understanding of bone metabolism dynamics in OPLL remains incomplete, characterized by conflicting research findings and unresolved questions. A fundamental uncertainty persists regarding the causal relationship between abnormal bone metabolism and OPLL development - whether it serves as a primary driver or secondary consequence of the disease. Addressing these questions demands comprehensive clinical studies combined with deep mechanistic investigations at cellular and molecular levels. Most importantly, advancing our knowledge of bone metabolism in OPLL holds significant clinical potential. A thorough understanding could enable the development of predictive biomarker systems, allowing for early identification of at-risk individuals and monitoring of disease progression. Such biomarkers could revolutionize clinical management by facilitating timely interventions and personalized treatment strategies. As discussed above, the Wnt signaling pathway emerges as a promising target for drug development. Identification of novel regulatory components within this pathway could lead to innovative pharmacological approaches to restore bone homeostasis, potentially offering more effective targeted treatment options than current symptom-focused management strategies.
Other endocrine-metabolic factors
Vitamin
In addition to aforementioned vitamin D, vitamins associated with OPLL pathogenesis also involve vitamin A and vitamin K in spite of conflicting evidence surrounding their roles in this disease. Early clinical observations associated excessive vitamin A intake with heterotopic ossification phenotypes including OPLL and DISH, particularly in patients receiving long-term vitamin A supplementation, suggesting a potential causal relationship.105,106 Preclinical models further corroborated these findings, demonstrating that sustained vitamin A administration induces osteophyte formation and heterotopic ossification in tendons and joint capsules within six-month exposure windows.107 Additionally, biochemical analyses have identified elevated circulating retinol and retinol-binding protein (RBP) concentrations in OPLL patients, with pronounced elevations observed in females.108 However, contemporary epidemiological data present contradictory evidence, revealing reduced dietary vitamin A intake and correspondingly lower serum levels in early-onset OPLL cases. Furthermore, quantitative assessments disclose a negative correlation between serum vitamin A and OPLL severity, challenging previous assumptions about vitamin A’s role in disease progression.109 Retinoic acid signaling is regarded as a potent inhibitor of chondrogenesis,110 and nuclear retinoic acid receptor agonists have been proved to inhibit heterotopic ossification by suppressing endochondral ossification.111 Based on these findings, vitamin A insufficiency is conjectured to facilitate OPLL progression by diminishing endogenous inhibitory effects on endochondral ossification.109 While accumulating evidence has implicated abnormal vitamin A metabolism in OPLL pathophysiology, the role of vitamin A remains obscure. Further exploration and mechanistic studies are warranted to delineate the specific association.
Vitamin K functions as an essential enzymatic cofactor for γ-glutamyl carboxylase, catalyzing the post-translational modification of substrate proteins through the conversion of glutamate residues into γ-carboxyglutamate moieties. This biochemical transformation is critical for the function of vitamin K-dependent proteins (VKDPs) that regulate hemostasis, vascular calcification, and bone metabolism.112 Clinical investigations have identified prolonged clotting times and reduced plasma protein C levels in patients with OPLL, indicating potential vitamin K deficiency in OPLL pathogenesis.24 Besides, experimental studies utilizing ttw mice, a well-characterized cervical OPLL model, demonstrate that dietary vitamin K supplementation significantly attenuates cervical ligament calcification and improves motor function, suggesting vitamin K’s protective effects against ectopic mineralization and its potential as a therapeutic strategy for OPLL.24 However, contrasting evidence emerges from epidemiological analyses, with a recent case-control investigation reporting much higher serum menaquinone-4 (MK-4, a form of vitamin K2) levels in male OPLL patients compared to matched controls.113 While some experimental data confirm vitamin K2’s dual regulatory effects on bone metabolism—enhancing osteoblast function while suppressing osteoclast activity, in vitro assays using OPLL-derived ligament cells demonstrated no significant MK-4-induced ALP elevation or osteoblastic activation.113 These paradoxical findings highlight the need for mechanistic investigations into vitamin K’s tissue-specific effects and its metabolic interplay with VKDPs in ectopic ossification processes.
GH/IGF-1
Clinical observations have documented co-occurrence of spinal ligament ossification disorders with acromegaly, which is characterized by excessive circulating growth hormone (GH) and insulin-like growth factor 1 (IGF-1).114–116 The GH/IGF-1 paracrine signaling axis is well established as a critical regulator of bone formation and remodeling processes. An in vitro investigation has revealed pronounced IGF-1 expression patterns in ossified posterior longitudinal ligament tissues compared to non-pathological tissues from normal individuals.117 When exposed to IGF-1, the upregulation of DNA replication and type I collagen precursor synthesis were observed in both OPLL and non-OPLL cell lines. Crucially, significantly enhanced ALP induction was identified in OPLL-derived cells following IGF-1 stimulation,117 indicating that IGF-1 signaling preferentially activates osteogenic programming in these ossification-prone ligament cells, potentially contributing to localized ectopic mineralization characteristic of OPLL pathology. However, contemporary analyses show no statistical differences in circulating GH/IGF-1 concentrations between acromegaly patients exhibiting spinal ligament ossification and those without, nor a significant correlation between ossification index and GH or IGF-1 levels. Intriguingly, elevated serum growth hormone receptor (GHBP) levels are detected in OPLL patients.116 While the anabolic effects of GH signaling in skeletal development are well elucidated, its contributions to spinal ligament ossification processes remain poorly defined, necessitating further mechanistic investigations into this endocrine-osseous axis.
Leptin
Obesity has been widely recognized as a significant contributor to the development of OPLL.118–120 The metabolic state of obesity is frequently accompanied by elevated circulating leptin, a hormone encoded by the obesity gene and predominantly secreted by fat cells. This hormone plays a pivotal role in translating adipose tissue signals into bone metabolic responses, with multifaceted mechanisms involving both central and peripheral pathways towards bone remodeling. Centrally, leptin exerts inhibitory effects on osteogenesis and bone resorption through hypothalamic-sympathetic neural pathways, while peripherally, it stimulates bone formation by enhancing the multiplication and specialization of bone marrow mesenchymal cells into osteoblasts.121 Numerous studies have linked leptin to OPLL pathogenesis, with abnormally high leptin levels detected in both genetically obese Zucker rat models and female OPLL patients.122,123 Research by Ikeda and colleagues establishes a direct relationship between circulating leptin concentrations and the spinal regions affected by OPLL. Furthermore, these increased leptin levels show a positive association with insulin concentrations, indicating that the combined effects of elevated leptin and insulin may drive pathological ossification processes in spinal ligaments.122 Another study by Takahata’s group reveals that patients with extensive OPLL involvement present with greater body mass indices (BMI), altered adipokine ratios, and elevated osteocalcin levels compared to those with single-region OPLL. And adiponectin-to-leptin ratio negatively correlated with OPLL severity is proposed as a potential marker for adipokine dysregulation that may underlie widespread spinal ligament ossification in obese populations.121
Leptin’s involvement in spinal ligament ossification processes has been explored a lot in these years. Previous experimental evidence demonstrates leptin’s capacity to promote cell proliferation and osteogenic differentiation in various cell types, including embryonic cells, bone marrow stromal cells, and osteoblasts.124–127 A separate investigation by Fan’s team discloses that leptin specifically upregulates bone-specific gene markers (ALP and OCN) in ligamentum flavum cells from ossification patients, with no similar effects observed in normal ligament cells. These osteogenic responses exhibit concentration- and time-dependent patterns, though only at supraphysiological doses.124 Further mechanistic studies by Feng et al. show that leptin treatment in ossified ligament cells from cervical OPLL samples not only enhances osteogenic marker (ALP and OCN) expression and mineralization capacity but also activates multiple signaling cascades including ERK1/2, p38 MAPK, and JNK pathways.128 Chen’s group provides additional insights by examining the combined effects of leptin signaling and mechanical stress on posterior longitudinal ligament cells, demonstrating their synergistic promotion of osteogenic differentiation through MAPK, JAK2-STAT3, and PI3K/Akt pathways. In their studies, distinct expression patterns of relevant molecules are verified, with elevated leptin receptor and reduced ligand levels in ossified ligament tissues.129 These findings strongly support leptin’s significant contribution to the pathological mechanisms underlying spinal ligament ossification. Modulating the leptin signaling may represent a promising approach to retard the development of OPLL.
Sex hormones
The relation between estrogen and bone metabolism has been extensively studied, particularly in the context of osteoporosis. However, its role in spinal ligament ossification has received comparatively less attention. Previous clinical observations reveal a distinct pattern of sex hormone dysregulation in male patients with OPLL, marked by elevated total estrogen levels and decreased 5a-(OH)2 testosterone concentrations. The results also show a positive correlation between these hormonal alterations with the degree of ossification progression.130 Nevertheless, this phenomenon appears specific to males, as female patients, despite experiencing more dramatic hormonal fluctuations during menstrual cycles and menopause, do not exhibit similar changes.130 Experiments have demonstrated that ligament cells from OPLL patients possess estrogen receptors with enhanced binding affinity for estrogen compared to those from non-OPLL individuals.131 Genetic analyses further indicate that variations in estrogen receptor genes may influence both OPLL predisposition and disease severity.132 Supporting evidence from animal models suggests that sex hormone imbalances can affect the development of chondrocytes and fibroblasts during the integration of the posterior longitudinal ligament with vertebral structures.133 Notably, recent research on human periodontal ligament cells provides additional mechanistic insights, showing estrogen’s capacity to promote osteogenic differentiation.134,135 Despite these findings, the precise involvement of sex hormones in pathological ossification processes remains unconfirmed, highlighting the need for further investigation.
Challenges and perspectives
Ossification of the posterior longitudinal ligament represents a complex multifactorial disorder with polygenic inheritance, which may account for the current limitations in elucidating its fundamental pathogenic mechanisms. Despite extensive investigation, research has yet to achieve sufficient depth or convergence to fully characterize the disease’s pathophysiology. Existing epidemiological studies examining the association between endocrine-metabolic factors and OPLL are significantly constrained by several limitations, including inadequate sample sizes, uncontrolled confounding variables, selection bias, geographic and ethnic restrictions, and inconsistent assessment protocols. These methodological shortcomings have resulted in divergent conclusions across studies, ultimately compromising their scientific validity and cross-study comparability. To address these issues, there is an urgent need for standardized, large-scale epidemiological investigations across diverse populations and geographic regions to validate the endocrine-metabolic factors implicated in the onset and progression of OPLL.
Despite significant advancements in OPLL research, the precise molecular mechanisms through which various endocrine-metabolic factors contribute to OPLL development remain incompletely understood, highlighting the necessity for more fundamental mechanistic studies. Emerging evidence indicates that leptin interacts with components of the insulin signaling cascade through recruitment of insulin receptor substrates,136–138 suggesting potential crosstalk between leptin and insulin signaling pathways. Clinical observations further support this interaction, as demonstrated by the positive correlation between leptin/BMI ratio and serum insulin levels in female OPLL patients.122 These findings suggest that elevated levels of both leptin and insulin may synergistically enhance downstream signaling pathway, thereby contributing to OPLL pathogenesis. Given the frequent co-occurrence of various metabolic disorders such as obesity and diabetes mellitus in OPLL patients, the interaction between leptin and insulin pathways presents a constructive perspective for future research to focus on the synergistic effects of multiple metabolic regulators. Beyond endocrine-metabolic factors, comprehensive understanding of OPLL pathogenesis will require systematic investigation of the interplay between genetic predisposition, metabolic dysregulation, and biomechanical stress factors.
From a clinical perspective, elucidating the precise roles of endocrine-metabolic factors in OPLL development could significantly advance both early intervention strategies and therapeutic approaches. Detailed characterization of metabolic marker dynamics throughout OPLL progression may facilitate the identification of reliable biomarkers for disease monitoring and prognosis prediction. Mechanistic understanding of these factors’ specific roles in OPLL pathogenesis could inform the development of targeted therapies directed against key molecular mediators. Besides, the timely correction of endocrine-metabolic disturbances through restoration of metabolic homeostasis may offer a promising therapeutic strategy for OPLL management.
Conclusion
This systematic review synthesizes contemporary research from domestic and international sources to critically examine the role of endocrine-metabolic factors in the pathogenesis and progression of OPLL. Substantial progress has been achieved in elucidating the roles of metabolic disorders, such as dysregulated glucose metabolism (e.g., hyperglycemia, insulin resistance, AGEs), lipid metabolism (e.g., dyslipidemia), and bone-mineral homeostasis (e.g., calcium-phosphate disturbance, aberrant bone remodeling) in OPLL development. While the precise molecular mechanisms underlying their contribution to OPLL pathogenesis remain incompletely characterized, current investigations have significantly advanced our understanding of OPLL from a metabolic perspective, providing critical insights into its multifactorial etiology. Continued exploration of these endocrine-metabolic relationships promises to deepen fundamental insights into OPLL pathogenesis while simultaneously informing the development of targeted therapeutic strategies, including metabolic modulation and precision interventions for clinical management.
Author contributions
Junfeng Wang and Ziheng Wei are responsible for the writing of the manuscript. Junfeng Wang, Ziheng Wei, Qingjie Kong and Yanqing Sun are responsible for the search and screening of references. Zhichao Zhang and Haiyuan Yang provide suggestions for improving the content and viewpoints of the article. Xiongsheng Chen is responsible for financial support.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Junfeng Wang, Ziheng Wei
References
- 1.Boody, B. S., Lendner, M. & Vaccaro, A. R. Ossification of the posterior longitudinal ligament in the cervical spine: a review. Int. Orthop.43, 797–805 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Le, H. V., Wick, J. B., Van, B. W. & Klineberg, E. O. Ossification of the posterior longitudinal ligament: pathophysiology, diagnosis, and management. J. Am. Acad. Orthop. Surg.30, 820–830 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Fujimori, T. et al. Prevalence, concomitance, and distribution of ossification of the spinal ligaments: results of whole spine CT scans in 1500 Japanese patients. Spine41, 1668–1676 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Tsuji, T. et al. Epidemiological survey of ossification of the posterior longitudinal ligament by using clinical investigation registration forms. J. Orthop. Sci.21, 291–294 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Choi, B. W., Song, K. J. & Chang, H. Ossification of the posterior longitudinal ligament: a review of literature. Asian Spine J.5, 267–276 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan, L. et al. The pathogenesis of ossification of the posterior longitudinal ligament. Aging Dis.8, 570–582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang, H. et al. Epidemiology of ossification of the spinal ligaments and associated factors in the Chinese population: a cross-sectional study of 2000 consecutive individuals. BMC Musculoskelet. Disord.20, 253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velan, G. J., Currier, B. L., Clarke, B. L. & Yaszemski, M. J. Ossification of the posterior longitudinal ligament in vitamin D-resistant rickets: case report and review of the literature. Spine26, 590–593 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Fujimori, T. et al. Ossification of the posterior longitudinal ligament of the cervical spine in 3161 patients: a CT-based study. Spine40, E394–E403 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Chang, H., Song, K. J., Kim, H. Y. & Choi, B. W. Factors related to the development of myelopathy in patients with cervical ossification of the posterior longitudinal ligament. J. Bone Jt. Surg. Br.94, 946–949 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Matsunaga, S., Komiya, S. & Toyama, Y. Risk factors for development of myelopathy in patients with cervical spondylotic cord compression. Eur. Spine J.24, 142–149 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Matsunaga, S. et al. Radiographic predictors for the development of myelopathy in patients with ossification of the posterior longitudinal ligament: a multicenter cohort study. Spine (Philos. Pa 1976)33, 2648–2650 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Matsunaga, S. et al. Pathogenesis of myelopathy in patients with ossification of the posterior longitudinal ligament. J. Neurosurg.96, 168–172 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Tetreault, L. et al. A systematic review of classification systems for cervical ossification of the posterior longitudinal ligament. Glob. Spine J.9, 85–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koda, M. et al. Comparison of clinical outcomes between laminoplasty, posterior decompression with instrumented fusion, and anterior decompression with fusion for K-line (-) cervical ossification of the posterior longitudinal ligament. Eur. Spine J.25, 2294–2301 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Yang, H. et al. Implications of different patterns of “double-layer sign” in cervical ossification of the posterior longitudinal ligament. Eur. Spine J.24, 1631–1639 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Abiola, R., Rubery, P. & Mesfin, A. Ossification of the posterior longitudinal ligament: etiology, diagnosis, and outcomes of nonoperative and operative management. Glob. Spine J.6, 195–204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng, B. W., Tan, J. A., Sabri, S., Baharuddin, A. & Muhamad Ariffin, M. H. Surgical management of cervical ossification of posterior longitudinal ligament: the treatment algorithm and outcome. Cureus15, e36517 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An, H. S., Al-Shihabi, L. & Kurd, M. Surgical treatment for ossification of the posterior longitudinal ligament in the cervical spine. J. Am. Acad. Orthop. Surg.22, 420–429 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Smith, Z. A., Buchanan, C. C., Raphael, D. & Khoo, L. T. Ossification of the posterior longitudinal ligament: pathogenesis, management, and current surgical approaches. A Rev. Neurosurg. Focus30, E10 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Lee, C. H. et al. Are there differences in the progression of ossification of the posterior longitudinal ligament following laminoplasty versus fusion? A meta-Analysis. Spine42, 887–894 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Onari, K. et al. Long-term follow-up results of anterior interbody fusion applied for cervical myelopathy due to ossification of the posterior longitudinal ligament. Spine26, 488–493 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Sato, R. et al. Ossification of the posterior longitudinal ligament of the cervical spine: histopathological findings around the calcification and ossification front. J. Neurosurg. Spine7, 174–183 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Kimura, A. et al. Possible involvement of vitamin K insufficiency in the progression of cervical ossification of the posterior longitudinal ligament. Sci. Rep.15, 2608 (2025). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saetia, K., Cho, D., Lee, S., Kim, D. H. & Kim, S. D. Ossification of the posterior longitudinal ligament: a review. Neurosurg. Focus30, E1 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi, Y. Biomarkers of ossification of the spinal ligament. Glob. Spine J.9, 650–657 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai, G. D. et al. Multiplex analysis of serum hormone and cytokine in patients with cervical cOPLL: towards understanding the potential pathogenic mechanisms. Growth Factors35, 171–178 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Katsumi, K. et al. Predictive biomarkers of ossification progression and bone metabolism dynamics in patients with cervical ossification of the posterior longitudinal ligament. Eur. Spine J.32, 1282–1290 (2023). [DOI] [PubMed] [Google Scholar]
- 29.Li, H., Jiang, L. S. & Dai, L. Y. Hormones and growth factors in the pathogenesis of spinal ligament ossification. Eur. Spine J.16, 1075–1084 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobashi, G. et al. High body mass index after age 20 and diabetes mellitus are independent risk factors for ossification of the posterior longitudinal ligament of the spine in Japanese subjects: a case-control study in multiple hospitals. Spine29, 1006–1010 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Bai, X. C. et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun.314, 197–207 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Bai, X. C. et al. Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J. Biol. Chem.280, 17497–17506 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Mody, N., Parhami, F., Sarafian, T. A. & Demer, L. L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med.31, 509–519 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Morita, K. et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med.204, 1613–1623 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen, N. X., Duan, D., O’Neill, K. D. & Moe, S. M. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol. Dial. Transpl.21, 3435–3442 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Li, Y. M. et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem. Biophys. Res. Commun.363, 209–215 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Han, Y. S. et al. High dose of glucose promotes chondrogenesis via PKCalpha and MAPK signaling pathways in chick mesenchymal cells. Cell Tissue Res.318, 571–578 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Balint, E., Szabo, P., Marshall, C. F. & Sprague, S. M. Glucose-induced inhibition of in vitro bone mineralization. Bone28, 21–28 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Inaba, M. et al. Influence of high glucose on 1,25-dihydroxyvitamin D3-induced effect on human osteoblast-like MG-63 cells. J. Bone Miner. Res.10, 1050–1056 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Ishida, Y. & Kawai, S. Characterization of cultured cells derived from ossification of the posterior longitudinal ligament of the spine. Bone14, 85–91 (1993). [DOI] [PubMed] [Google Scholar]
- 41.Nam, D. C., Lee, H. J., Lee, C. J. & Hwang, S. C. Molecular pathophysiology of ossification of the posterior longitudinal ligament (OPLL). Biomol. Ther.27, 342–348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, H., Jiang, L. S. & Dai, L. Y. High glucose potentiates collagen synthesis and bone morphogenetic protein-2-induced early osteoblast gene expression in rat spinal ligament cells. Endocrinology151, 63–74 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Shingyouchi, Y., Nagahama, A. & Niida, M. Ligamentous ossification of the cervical spine in the late middle-aged Japanese men. Its relation to body mass index and glucose metabolism. Spine21, 2474–2478 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Singh, M., Kuharski, M., Balmaceno-Criss, M., Diebo, B. G. & Daniels, A. Hyperlipidemia, obesity, and diabetes, and risk of ossification of the posterior longitudinal ligament. World Neurosurg.188, e642–e647 (2024). [DOI] [PubMed] [Google Scholar]
- 45.Akune, T. et al. Insulin secretory response is positively associated with the extent of ossification of the posterior longitudinal ligament of the spine. J. Bone Jt. Surg. Am.83, 1537–1544 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Okano, T. et al. Orthotopic ossification of the spinal ligaments of Zucker fatty rats: a possible animal model for ossification of the human posterior longitudinal ligament. J. Orthop. Res.15, 820–829 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Olesen, P., Nguyen, K., Wogensen, L., Ledet, T. & Rasmussen, L. M. Calcification of human vascular smooth muscle cells: associations with osteoprotegerin expression and acceleration by high-dose insulin. Am. J. Physiol. Heart Circ. Physiol.292, H1058–H1064 (2007). [DOI] [PubMed] [Google Scholar]
- 48.White, M. F. & Kahn, C. R. The insulin signaling system. J. Biol. Chem.269, 1–4 (1994). [PubMed] [Google Scholar]
- 49.Xiao, G. et al. Bone morphogenetic proteins, extracellular matrix, and mitogen-activated protein kinase signaling pathways are required for osteoblast-specific gene expression and differentiation in MC3T3-E1 cells. J. Bone Miner. Res.17, 101–110 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Li, H., Liu, D., Zhao, C. Q., Jiang, L. S. & Dai, L. Y. Insulin potentiates the proliferation and bone morphogenetic protein-2-induced osteogenic differentiation of rat spinal ligament cells via extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. Spine33, 2394–2402 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Aubert, C. E. et al. Association of peripheral neuropathy with circulating advanced glycation end products, soluble receptor for advanced glycation end products and other risk factors in patients with type 2 diabetes. Diabetes Metab. Res. Rev.30, 679–685 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Yamagishi, S., Fujimori, H., Yonekura, H., Tanaka, N. & Yamamoto, H. Advanced glycation endproducts accelerate calcification in microvascular pericytes. Biochem. Biophys. Res. Commun.258, 353–357 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Kume, S. et al. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J. Bone Miner. Res.20, 1647–1658 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Yoshimura, N. et al. Prevalence and progression of radiographic ossification of the posterior longitudinal ligament and associated factors in the Japanese population: a 3-year follow-up of the ROAD study. Osteoporos. Int.25, 1089–1098 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Yokosuka, K. et al. Immunohistochemical demonstration of advanced glycation end products and the effects of advanced glycation end products in ossified ligament tissues in vitro. Spine32, E337–E339 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Doi, T. et al. Noninvasive skin autofluorescence of advanced glycation end products for detecting ossification of the posterior longitudinal ligament in the thoracic spine. Spine48, E40–E45 (2023). [DOI] [PubMed] [Google Scholar]
- 57.Endo, T. et al. Strong relationship between dyslipidemia and the ectopic ossification of the spinal ligaments. Sci. Rep.12, 22617 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukada, S. et al. Dyslipidemia as a novel risk for the development of symptomatic ossification of the posterior longitudinal ligament. Spine J.23, 1287–1295 (2023). [DOI] [PubMed] [Google Scholar]
- 59.Zhang, R., Yang, Q., Wang, Y. & Zhao, Y. Investigation of the association between hyperlipidemia and ossification of the posterior longitudinal ligament through two-sample mendelian randomization analysis. Spine50, 163–171 (2025). [DOI] [PubMed] [Google Scholar]
- 60.Endo, T. et al. Close association between non-alcoholic fatty liver disease and ossification of the posterior longitudinal ligament of the spine. Sci. Rep.11, 17412 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parisi, V. et al. The lipid theory in the pathogenesis of calcific aortic stenosis. Nutr. Metab. Cardiovasc. Dis.25, 519–525 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Bundy, K., Boone, J. & Simpson, C. L. Wnt signaling in vascular calcification. Front. Cardiovasc. Med.8, 708470 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu, L., Chen, W., Qian, A. & Li, Y. P. Wnt/beta-catenin signaling components and mechanisms in bone formation, homeostasis, and disease. Bone Res.12, 39 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He, X., Semenov, M., Tamai, K. & Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development131, 1663–1677 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Karner, C. M. & Long, F. Wnt signaling and cellular metabolism in osteoblasts. Cell Mol. Life Sci.74, 1649–1657 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alekos, N. S., Moorer, M. C. & Riddle, R. C. Dual effects of lipid metabolism on osteoblast function. Front. Endocrinol.11, 578194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borrell-Pages, M., Romero, J. C. & Badimon, L. LRP5 deficiency down-regulates Wnt signalling and promotes aortic lipid infiltration in hypercholesterolaemic mice. J. Cell Mol. Med.19, 770–777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Albanese, I., Khan, K., Barratt, B., Al-Kindi, H. & Schwertani, A. Atherosclerotic calcification: Wnt is the hint. J. Am. Heart Assoc.7, e007356 (2018). [DOI] [PMC free article] [PubMed]
- 69.Tsuji, T. et al. Metabolite profiling of plasma in patients with ossification of the posterior longitudinal ligament. J. Orthop. Sci.23, 878–883 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Hamazaki, K. et al. Mead acid (20:3n-9) and n-3 polyunsaturated fatty acids are not associated with risk of posterior longitudinal ligament ossification: results of a case-control study. Prostaglandins Leukot. Ess. Fat. Acids96, 31–36 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Adams, J. E. & Davies, M. Paravertebral and peripheral ligamentous ossification: an unusual association of hypoparathyroidism. Postgrad. Med. J.53, 167–172 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soehle, M. & Casey, A. T. Cervical spinal cord compression attributable to a calcified intervertebral disc in a patient with X-linked hypophosphatemic rickets: case report and review of the literature. Neurosurgery51, 239–242 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Okazaki, T. et al. Ossification of the paravertebral ligaments: a frequent complication of hypoparathyroidism. Metabolism33, 710–713 (1984). [DOI] [PubMed] [Google Scholar]
- 74.Kashii, M. et al. Circulating sclerostin and dickkopf-1 levels in ossification of the posterior longitudinal ligament of the spine. J. Bone Miner. Metab.34, 315–324 (2016). [DOI] [PubMed] [Google Scholar]
- 75.Taguchi, T. in OPLL: Ossification of the Posterior Longitudinal Ligament (eds Kazuo Yonenobu, Kozo Nakamura, & Yoshiaki Toyama) 29–31 (Springer, 2006).
- 76.Sasaki, K. et al. Bone turnover markers in patients with ossification of the posterior longitudinal ligament in the thoracic spine. Spine49, E100–E106 (2024). [DOI] [PubMed] [Google Scholar]
- 77.Takuwa, Y. et al. Calcium metabolism in paravertebral ligamentous ossification. Acta Endocrinol.109, 428–432 (1985). [DOI] [PubMed] [Google Scholar]
- 78.Seichi, A., Hoshino, Y., Ohnishi, I. & Kurokawa, T. The role of calcium metabolism abnormalities in the development of ossification of the posterior longitudinal ligament of the cervical spine. Spine (Philos. Pa 1976)17, S30–S32 (1992). [DOI] [PubMed] [Google Scholar]
- 79.Yamauchi, T., Taketomi, E., Matsunaga, S. & Sakou, T. Bone mineral density in patients with ossification of the posterior longitudinal ligament in the cervical spine. J. Bone Miner. Metab.17, 296–300 (1999). [DOI] [PubMed] [Google Scholar]
- 80.Fujita, R. et al. High whole-body bone mineral density in ossification of the posterior longitudinal ligament. Spine J.23, 1461–1470 (2023). [DOI] [PubMed] [Google Scholar]
- 81.Silva, B. C. & Bilezikian, J. P. Parathyroid hormone: anabolic and catabolic actions on the skeleton. Curr. Opin. Pharm.22, 41–50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Horie, S. et al. Factors associated with bone metabolism in patients with cervical ossification of the posterior longitudinal ligament accompanied with diffuse idiopathic skeletal hyperostosis. Sicot J.4, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawaguchi, Y. et al. Increase of the serum FGF-23 in ossification of the posterior longitudinal ligament. Glob. Spine J.9, 492–498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fukumoto, S. & Yamashita, T. FGF23 is a hormone-regulating phosphate metabolism–unique biological characteristics of FGF23. Bone40, 1190–1195 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Mackenzie, N. C. et al. Altered bone development and an increase in FGF-23 expression in Enpp1-/- mice. PLoS One7, e32177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ikegawa, S. Genomic study of ossification of the posterior longitudinal ligament of the spine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci.90, 405–412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marie, P. J., Miraoui, H. & Severe, N. FGF/FGFR signaling in bone formation: progress and perspectives. Growth Factors30, 117–123 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Wang, W., Kong, L., Zhao, H. & Jia, Z. Ossification of the transverse atlantal ligament associated with fluorosis: a report of two cases and review of the literature. Spine29, E75–E78 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Reddy, K. V. S., Mudumba, V. S., Tokala, I. M. & Reddy, D. R. Ossification of posterior longitudinal ligament and fluorosis. Neurology66, 1394–1399 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Sohn, S. & Chung, C. K. Increased bone mineral density and decreased prevalence of osteoporosis in cervical ossification of the posterior longitudinal ligament: a case-control study. Calcif. Tissue Int.92, 28–34 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Matsui, H., Yudoh, K. & Tsuji, H. Significance of serum levels of type I procollagen peptide and intact osteocalcin and bone mineral density in patients with ossification of the posterior longitudinal ligaments. Calcif. Tissue Int.59, 397–400 (1996). [DOI] [PubMed] [Google Scholar]
- 92.Niu, C. C. et al. Correlation of blood bone turnover biomarkers and Wnt signaling antagonists with AS, DISH, OPLL, and OYL. BMC Musculoskelet. Disord.18, 61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ishihara, C. et al. The efficacy of biochemical markers in patients with ossification of posterior longitudinal ligament of the spine. Spinal Cord.38, 211–213 (2000). [DOI] [PubMed] [Google Scholar]
- 94.Westendorf, J. J., Kahler, R. A. & Schroeder, T. M. Wnt signaling in osteoblasts and bone diseases. Gene341, 19–39 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Piters, E., Boudin, E. & Van Hul, W. Wnt signaling: a win for bone. Arch. Biochem. Biophys.473, 112–116 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Baron, R. & Kneissel, M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med.19, 179–192 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Li, X. et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem.280, 19883–19887 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Mao, B. et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature417, 664–667 (2002). [DOI] [PubMed] [Google Scholar]
- 99.Dong, J., Xu, X., Zhang, Q., Yuan, Z. & Tan, B. Dkk1 acts as a negative regulator in the osteogenic differentiation of the posterior longitudinal ligament cells. Cell Biol. Int.44, 2450–2458 (2020). [DOI] [PubMed] [Google Scholar]
- 100.Senolt, L. et al. Low circulating Dickkopf-1 and its link with severity of spinal involvement in diffuse idiopathic skeletal hyperostosis. Ann. Rheum. Dis.71, 71–74 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Aeberli, D., Schett, G., Eser, P., Seitz, M. & Villiger, P. M. Serum Dkk-1 levels of DISH patients are not different from healthy controls. Jt. Bone Spine78, 422–423 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Maeda, K., Takahashi, N. & Kobayashi, Y. Roles of Wnt signals in bone resorption during physiological and pathological states. J. Mol. Med.91, 15–23 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Simonet, W. S. et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell89, 309–319 (1997). [DOI] [PubMed] [Google Scholar]
- 104.Fay, L. Y. et al. Comparative study of the cytokine profiles of serum and tissues from patients with the ossification of the posterior longitudinal ligament. Biomedicines11, 2021 (2023). [DOI] [PMC free article] [PubMed]
- 105.DiGiovanna, J. J., Helfgott, R. K., Gerber, L. H. & Peck, G. L. Extraspinal tendon and ligament calcification associated with long-term therapy with etretinate. N. Engl. J. Med.315, 1177–1182 (1986). [DOI] [PubMed] [Google Scholar]
- 106.Pennes, D. R., Martel, W. & Ellis, C. N. Retinoid-induced ossification of the posterior longitudinal ligament. Skelet. Radio.14, 191–193 (1985). [DOI] [PubMed] [Google Scholar]
- 107.Ishida, Y. & Kawai, S. Effects of bone-seeking hormones on DNA synthesis, cyclic AMP level, and alkaline phosphatase activity in cultured cells from human posterior longitudinal ligament of the spine. J. Bone Miner. Res.8, 1291–1300 (1993). [DOI] [PubMed] [Google Scholar]
- 108.Kodama, T., Matsunaga, S., Taketomi, E. & Sakou, T. Retinoid and bone metabolic marker in ossification of the posterior longitudinal ligament. Vivo12, 339–344 (1998). [PubMed] [Google Scholar]
- 109.Endo, T. et al. Association between Vitamin A intake and disease severity in early-onset heterotopic ossification of the posterior longitudinal ligament of the spine. Glob. Spine J.12, 1770–1780 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weston, A. D., Chandraratna, R. A., Torchia, J. & Underhill, T. M. Requirement for RAR-mediated gene repression in skeletal progenitor differentiation. J. Cell Biol.158, 39–51 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shimono, K. et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat. Med.17, 454–460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Willems, B. A., Vermeer, C., Reutelingsperger, C. P. & Schurgers, L. J. The realm of vitamin K dependent proteins: shifting from coagulation toward calcification. Mol. Nutr. Food Res.58, 1620–1635 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Yamada, K. et al. High serum levels of menatetrenone in male patients with ossification of the posterior longitudinal ligament. Spine28, 1789–1793 (2003). [DOI] [PubMed] [Google Scholar]
- 114.Schmidt, R. F., Goldstein, I. M. & Liu, J. K. Ossified ligamentum flavum causing spinal cord compression in a patient with acromegaly. J. Clin. Neurosci.20, 1599–1603 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Kamakura, D. et al. Acromegaly presenting with myelopathy due to ossification of posterior longitudinal ligament: a case report. BMC Musculoskelet. Disord.22, 353 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ikegawa, S. et al. Increase of serum growth hormone-binding protein in patients with ossification of the posterior longitudinal ligament of the spine. Spine18, 1757–1760 (1993). [DOI] [PubMed] [Google Scholar]
- 117.Goto, K. et al. Involvement of insulin-like growth factor I in development of ossification of the posterior longitudinal ligament of the spine. Calcif. Tissue Int.62, 158–165 (1998). [DOI] [PubMed] [Google Scholar]
- 118.Chaput, C. D., Siddiqui, M. & Rahm, M. D. Obesity and calcification of the ligaments of the spine: a comprehensive CT analysis of the entire spine in a random trauma population. Spine J.19, 1346–1353 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Zhao, Y., Xiang, Q., Lin, J., Jiang, S. & Li, W. High body mass index is associated with an increased risk of the onset and severity of ossification of spinal ligaments. Front. Surg.9, 941672 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Katsumi, K. et al. Natural history of the ossification of cervical posterior longitudinal ligament: a three dimensional analysis. Int. Orthop.42, 835–842 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Takahata, M. et al. Adipokine dysregulation as an underlying pathology for diffuse ectopic ossification of spinal posterior longitudinal ligament in patients with obesity. Spine J.25, 80–90 (2025). [DOI] [PubMed] [Google Scholar]
- 122.Ikeda, Y. et al. Association between serum leptin and bone metabolic markers, and the development of heterotopic ossification of the spinal ligament in female patients with ossification of the posterior longitudinal ligament. Eur. Spine J.20, 1450–1458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Iida, M. et al. Substitution at codon 269 (glutamine –> proline) of the leptin receptor (OB-R) cDNA is the only mutation found in the Zucker fatty (fa/fa) rat. Biochem. Biophys. Res. Commun.224, 597–604 (1996). [DOI] [PubMed] [Google Scholar]
- 124.Fan, D., Chen, Z., Chen, Y. & Shang, Y. Mechanistic roles of leptin in osteogenic stimulation in thoracic ligament flavum cells. J. Biol. Chem.282, 29958–29966 (2007). [DOI] [PubMed] [Google Scholar]
- 125.Zheng, B. et al. Increased osteogenesis in osteoporotic bone marrow stromal cells by overexpression of leptin. Cell Tissue Res.361, 845–856 (2015). [DOI] [PubMed] [Google Scholar]
- 126.Lamghari, M., Tavares, L., Camboa, N. & Barbosa, M. A. Leptin effect on RANKL and OPG expression in MC3T3-E1 osteoblasts. J. Cell Biochem.98, 1123–1129 (2006). [DOI] [PubMed] [Google Scholar]
- 127.Jiang, H. et al. Leptin accelerates the pathogenesis of heterotopic ossification in rat tendon tissues via mTORC1 signaling. J. Cell Physiol.233, 1017–1028 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Feng, B. et al. Roles and mechanisms of leptin in osteogenic stimulation in cervical ossification of the posterior longitudinal ligament. J. Orthop. Surg. Res.13, 165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen, S. et al. Combined use of leptin and mechanical stress has osteogenic effects on ossification of the posterior longitudinal ligament. Eur. Spine J.27, 1757–1766 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Okada Y, M. M., Fujita, L., Furufu, T., Yuji, M. & Tabe, S. Association of ossification of the spinal ligaments and sex hormones. Orthop. MOOK, 12–25 (1987).
- 131.Wada, A. Affinity of estrogen binding in the cultured spinal ligament cells: an in vitro study using cells from spinal ligament ossification patients]. Nihon Seikeigeka Gakkai Zasshi69, 440–449 (1995). [PubMed] [Google Scholar]
- 132.Ogata, N. et al. Association of bone metabolism regulatory factor gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the spine and its severity. Spine27, 1765–1771 (2002). [DOI] [PubMed] [Google Scholar]
- 133.Morisu, M. Influence of foods on the posterior longitudinal ligament of the cervical spine and serum sex hormones. Nihon Seikeigeka Gakkai Zasshi68, 1056–1067 (1994). [PubMed] [Google Scholar]
- 134.Liang, L., Yu, J. F., Wang, Y., Wang, G. & Ding, Y. Effect of estrogen receptor beta on the osteoblastic differentiation function of human periodontal ligament cells. Arch. Oral. Biol.53, 553–557 (2008). [DOI] [PubMed] [Google Scholar]
- 135.Shu, L. et al. Estrogen modulates cytokine expression in human periodontal ligament cells. J. Dent. Res.87, 142–147 (2008). [DOI] [PubMed] [Google Scholar]
- 136.Fruhbeck, G. & Salvador, J. Relation between leptin and the regulation of glucose metabolism. Diabetologia43, 3–12 (2000). [DOI] [PubMed] [Google Scholar]
- 137.Niswender, K. D. & Schwartz, M. W. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol.24, 1–10 (2003). [DOI] [PubMed] [Google Scholar]
- 138.Hegyi, K., Fulop, K., Kovacs, K., Toth, S. & Falus, A. Leptin-induced signal transduction pathways. Cell Biol. Int.28, 159–169 (2004). [DOI] [PubMed] [Google Scholar]