

Abstract
Objective
Plasma cell-free DNA (cfDNA) methylation has shown potential in the detection and prognostic testing of multiple cancers. Here, we comprehensively investigate the performance of cfDNA methylation for gastric cancer (GC) detection and prognosis.
Methods
GC-specific differentially methylated regions (DMRs) were identified by sequencing 56 GC tissues and 59 normal adjacent tissues (NATs). We then performed targeted bisulfite sequencing of cfDNA from 294 GC and 446 non-gastric cancer (NGC) plasma samples, identifying 179 DMRs that overlapped with those in tissue samples. The efficacy of plasma cfDNA methylation markers for GC detection and prognosis was evaluated.
Results
Based on the 179 DMRs overlapping with those in tissue samples, the random forest (RF) model using 28 DMRs achieved an area under the curve (AUC) of 0.998 in the training cohort, whereas further refinement to the top 6 DMRs resulted in an AUC of 0.985. Consistent results were obtained in the validation cohort (28 DMR AUC: 0.985; 6 DMR AUC: 0.988). Support vector machine (SVM) and logistic regression (LR) models also demonstrated robust performance. Additionally, an 11-DMR signature was developed for prognostic prediction, successfully identifying high-risk GC patients with significantly shorter overall survival.
Conclusions
Our study highlights the potential utility of cfDNA methylation markers for both the detection and prognostication of GC.
Keywords: Gastric cancer, circulating cell-free DNA, detection, prognosis, diagnosis
Introduction
According to the GLOBOCAN 2022 report, gastric cancer (GC) ranks fifth globally in terms of both incidence and cancer-related mortality (1). Remarkably, nearly half of new GC cases and deaths worldwide have occurred in China (2). Between 2002 and 2015, the five-year relative survival rate for patients with GC in China increased from 27.4% to 35.1% (3). However, this rate remains significantly lower than that reported in Japan (80.1%) and South Korea (75.9%), primarily because of differences in the timing of clinical diagnosis (4,5). Currently, GC screening primarily relies on endoscopic examination and serum tumor markers (6). Although endoscopic biopsy is the gold standard for diagnosing GC, it is an expensive and invasive procedure. Moreover, serum markers such as carcinoembryonic antigen (CEA), carbohydrate antigen (CA) 19-9, and alpha-fetoprotein (AFP) have shown poor sensitivity for early-stage GC and lack specificity for GC (7). Therefore, identifying biomarkers associated with GC occurrence and progression has become crucial for the early detection of GC and the assessment of patient prognosis.
In recent years, liquid biopsy has gained prominence for molecular analysis in cancer, serving various functions, such as early detection, prognostic assessment, tumor burden analysis, and predicting response and resistance to targeted therapy, chemotherapy, and immunotherapy, including chimeric antigen receptor T-cell therapy (8). Circulating extracellular nucleic acids, such as cell-free DNA (cfDNA), are key analytes for liquid biopsy and can be isolated from plasma. Numerous studies have demonstrated the potential of cfDNA as a biomarker for cancer diagnosis and screening (9). For instance, Chung et al. reported that blood-based cfDNA testing has a sensitivity of 83.1% for colorectal cancer (CRC) and a specificity of 89.6% for advanced colorectal neoplasia (CRC or advanced precancerous lesions) (10). Liu et al. reported that cfDNA exhibits a specificity of 99.3% [95% confidence interval (95% CI): 98.3%−99.8%] for multicancer detection and a sensitivity of 67.3% (95% CI: 60.7%−73.3%) for stages I−III across 12 cancer types, including GC (11). Yu et al. demonstrated that the area under the curve (AUC) values for stage I−II GC detection using cfDNA in training and validation cohorts range from 0.937 to 0.972, with a specificity of 92.1% and a sensitivity of 88.2% (12). However, most existing studies have relied on noncomprehensive biomarker discovery approaches, failing to translate tumor tissue-derived biomarkers into blood (serum or plasma) and lacking validation in independent clinical sample cohorts (13).
In this study, we prospectively collected 740 blood samples from four centers, including 294 samples from GC patients and 446 samples from non-gastric cancer (NGC) participants. Additionally, we collected 56 GC tissues and 59 normal adjacent tissues (NATs). By analyzing overlapping differentially methylated regions (DMRs) between blood and tissue samples, we aimed to develop and validate these biomarkers to demonstrate their diagnostic and prognostic value in GC. This research holds promise for the development of reliable biomarkers that can be validated in plasma samples, providing valuable insights into the early diagnosis and prognosis of patients with GC.
Materials and methods
Study recruitment and sample collection
This study was conducted in accordance with ethical guidelines and received approval from the Institutional Review Boards at Zhejiang Cancer Hospital, The Sixth Affiliated Hospital of Sun Yat-sen University, and Sichuan Cancer Hospital, as well as from BGI (Approval Nos: IRB-2023-43, 2021ZSLYEC-326, SCCHEC-02-2023-166, and BGI-IRB 23002).
Patients eligible for inclusion in the GC study, which occurred between March 1, 2021, and December 31, 2021, met the following specific criteria: 1) age between 18 and 80 years; 2) an Eastern Cooperative Oncology Group (ECOG) performance score of 0 or 1; and 3) GC diagnosis. Additionally, patients had not undergone any prior anticancer treatments (including chemotherapy, radiotherapy, targeted therapy, surgery, or anaesthesia) before blood collection. Prospective participants and their families were required to fully comprehend the study protocol and express willingness to participate by providing written informed consent. The exclusion criteria included: 1) concurrent hereditary diseases or other tumors; 2) acute severe illnesses causing inflammatory reactions or recent steroid treatments within 14 d before blood sampling; 3) receipt of organ, stem cell, or bone marrow transplants; 4) receipt of blood transfusions within the month before enrolment; 5) pregnancy; or 6) engagement in other clinical trials involving medication within the last 60 d, including anaesthesia. Moreover, individuals with severe cardiovascular diseases, uncontrollable infections, or other unmanageable coexisting conditions, as well as those and their families unable to comprehend the study’s conditions and objectives, were also excluded.
Blood was drawn before tumor resection for GC patients and at recruitment for healthy participants and collected in 10 mL K2EDTA tubes (BD, 366643, Franklin Lakes, USA). Plasma was separated from whole blood within 4 h after blood was drawn and stored at −80 °C until DNA extraction. GC tissues and NATs were collected during surgery and immediately frozen at −80 °C.
Clinical data collection and serum biomarkers
Demographic and clinicopathologic variables, including age, sex, tumor location, histological subtype (Lauren classification), tumor differentiation, and pTNM stage (AJCC 8th edition), were recorded. Baseline serum tumor markers, including AFP (ng/mL), CEA (ng/mL), and CA19-9 (U/mL), were measured at the participating hospitals according to local standard operating procedures using routine clinical immunoassays. For GC patients, serum markers were measured prior to surgery and at the same time as plasma was collected. For NGC participants, markers were measured at enrolment.
DNA extraction and quality control
Plasma cfDNA extraction was performed utilizing the Apostle MiniMax High-Efficiency cfDNA Isolation Kit (Apostle, A17622CN, Santa Clara, USA). Genomic DNA (gDNA) from GC tumors and corresponding NATs was extracted using the MagPure Buffy Coat DNA Midi KF Kit (Magen, D3537-02, Guangzhou, China). The quantification of DNA concentration was performed using the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific, Q32854, Waltham, USA). The integrity of the cfDNA was assessed using an Agilent High-Sensitivity DNA Kit (Agilent Technologies, 5067-4626, Santa Clara, USA) on an Agilent 2100 Bioanalyzer (Agilent Technologies).
Library preparation
For bisulfite sequencing, fragmented genomic DNA (achieved through sonication) or cfDNA samples were subjected to sodium bisulfite treatment using the EZ DNA Methylation-Gold™ Kit (Zymo Research, D5006, Irvine, USA). The bisulfite-converted DNA fragments were then ligated to sequencing adaptors using a single-stranded DNA-based library preparation method, as previously described. To enrich genomic regions of interest in cfDNA, targeted capture reactions were conducted using a custom-designed panel with a size of 449k covering 37k CpG sites (Roche, KAPA HyperExplore, Basel, Switzerland). Both libraries, with and without capture, subsequently underwent amplification procedures and were sequenced on the MGISEQ-2000 platform using 2×100 bp paired-end sequencing.
Identification of DMRs
A Bayesian hierarchical model with smoothing was applied to 56 GC tissues and 59 NATs to identify DMRs as described previously (14). The DMRs were defined as the regions satisfying the following criteria: a difference in the absolute methylation ratio between cancer and normal tissues >0.2, a region size ≥50 bp, the presence of ≥3 CpG sites within the region, and a percentage of CpG sites with significant P values ≥80%. DMRs were annotated using the R package annotatr (Version 1.34.0, https://bioconductor.org/packages/annotate) (15). Gene Ontology (GO) analyses were conducted using the R package clusterProfiler (Version 4.16.0; https://bioconductor.org/packages/clusterProfiler) (16).
Correlation analysis of DMR methylation and gene expression
Paired methylation and mRNA expression data of stomach adenocarcinoma tissue samples were obtained from The Cancer Genome Atlas (TCGA) database using the TCGAbiolinks R package (Version 2.36.0; https://bioconductor.org/packages/TCGAbiolinks). The ChAMP R package was used to filter and normalize methylation data, and mRNA expression data were standardized as Transcripts Per Million (TPM). Spearman correlation between the median methylation level in each DMR and the expression level of the nearest gene was then calculated, and a P value <0.05 was considered to indicate statistical significance.
Diagnostic model of plasma DMRs for GC
The regional methylation ratio was calculated per DMR for each cfDNA sample. Tenfold cross-validation (CV) was performed for feature selection using the Out-of-Bag (OOB) error rate in the randomForest package. Three diagnostic models, namely, random forest (RF), support vector machine (SVM), and logistic regression (LR), were constructed using the randomForest, e1071 and stats packages in R. Model robustness was evaluated by 10-fold CV repeated 10 times in the training cohort. Diagnostic performance was evaluated by the area under the receiver operating characteristic (ROC) curve (AUC) and sensitivity analysis. Threshold values were determined using the Youden index. Model performance was validated in the testing set.
Prognostic model of DMRs for GC
To identify methylation-based prognostic markers, the samples were randomly divided into training and testing sets at a split ratio of 3:1. To construct the prognostic signature, overall survival (OS) was used as the endpoint. Survival-related DMRs were preselected by univariate Cox regression (P<0.001), followed by least absolute shrinkage and selection operator (LASSO) Cox regression with 10-fold CV to determine the optimal λ. A multivariable Cox model was then fitted using the selected DMRs, and an individual risk score was calculated as the linear predictor (risk score = Σβᵢ × DMRᵢ). The optimal cut-off of the risk score was identified in the training set using maximally selected rank statistics (log-rank-based) and was applied unchanged to the testing set to define low- and high-risk groups. Kaplan-Meier curves, hazard ratios (HRs), and the concordance index (C-index) were used to evaluate prognostic performance.
Statistical analysis
Statistical analyses were conducted in R. Spearman’s rank correlation was applied to examine associations between DNA methylation and gene expression. Diagnostic models—RF, SVM, and LR—were assessed by ROC analysis with 10-fold cross-validation. Prognostic modeling used univariable and multivariable Cox proportional hazards regression, with LASSO for feature selection. Survival outcomes were evaluated using Kaplan-Meier estimates. Unless otherwise specified, two-sided P<0.05 were considered statistically significant.
Results
Study design and participants
In this study, a total of 740 blood samples were collected, consisting of 294 samples from GC patients and 446 samples from NGC individuals, who were age- and sex-matched (Figure 1). The training dataset comprised 174 GC samples and 201 NGC samples, all of which were sourced from Zhejiang Cancer Hospital. The independent testing dataset included 64 GC samples and 149 NGC samples from The Sixth Affiliated Hospital of Sun Yat-sen University, 56 GC samples from Sichuan Cancer Hospital, and 96 NGC samples from BGI (Table 1). Additionally, we collected 56 GC tissue samples and 59 NATs from patients who underwent GC surgery. Age and sex distributions were comparable between the GC and NGC groups in both cohorts (training cohort: age 62.79±11.76 vs. 62.72±9.53 years; male 73.6% vs. 79.1%; testing cohort: age 61.04±12.41 vs. 60.60±11.61 years; male 67.5% vs. 61.6%; Table 1). Among the GC cases, the distribution of the pTNM stages in the training cohort was as follows: I (n=57, 32.8%), II (n=47, 27.0%), III (n=47, 27.0%), and IV (n=23, 13.2%). In the testing cohort, the distribution was as follows: I (n=22, 18.3%), II (n=21, 17.5%), III (n=47, 39.2%), and IV (n=30, 25.0%).
Figure 1.

Flow diagram of the study. NAT, normal adjacent tissue; GC, gastric cancer; DMR, differentially methylated region; NGC, non-gastric cancer; SVM, support vector machine; AUC, area under the curve.
Table 1. Clinicopathological characteristics of participants in training and testing cohorts.
| Variables | Training cohort [n (%)] | Testing cohort [n (%)] | |||||
| GC (N=174) | NGC (N=201) | P | GC (N=120) | NGC (N=245) | P | ||
| GC, gastric cancer; NGC, non-gastric cancer; AFP, alpha-fetoprotein; CEA, carcinoembryonic antigen; CA19-9, carbohydrate antigen 19-9. | |||||||
| Age (year) | |||||||
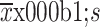
|
62.79±11.76 | 62.72±9.53 | 0.949 | 61.04±12.41 | 60.60±11.61 | 0.438 | |
| ≥65 | 85 (48.9) | 93 (46.3) | 0.618 | 47 (39.2) | 90 (36.7) | 0.652 | |
| <65 | 89 (51.1) | 108 (53.7) | 73 (60.8) | 155 (63.3) | |||
| Sex | 0.207 | 0.274 | |||||
| Male | 128 (73.6) | 159 (79.1) | 81 (67.5) | 151 (61.6) | |||
| Female | 46 (26.4) | 42 (20.9) | 39 (32.5) | 94 (38.4) | |||
| Tumor location | |||||||
| Upper | 31 (17.8) | − | − | 34 (28.3) | − | − | |
| Middle | 39 (22.4) | − | − | 32 (26.7) | − | − | |
| Lower | 98 (56.3) | − | − | 53 (44.2) | − | − | |
| Whole stomach | 1 (0.6) | − | − | − | − | − | |
| Unknown | 5 (2.9) | − | − | 1 (0.8) | − | − | |
| Differentiation | |||||||
| High | 8 (4.6) | − | − | 7 (5.8) | − | − | |
| Median | 26 (14.9) | − | − | 15 (12.5) | − | − | |
| Median-low | 50 (28.7) | − | − | 21 (17.5) | − | − | |
| Low | 64 (36.8) | − | − | 45 (37.5) | − | − | |
| Unknown | 26 (14.9) | − | − | 32 (26.7) | − | − | |
| Lauren type | |||||||
| Diffuse | 23 (13.2) | − | − | 31 (25.8) | − | − | |
| Intestinal | 31 (17.8) | − | − | 24 (20.0) | − | − | |
| Mixed | 13 (7.5) | − | − | 25 (20.8) | − | − | |
| Unknown | 107 (61.5) | − | − | 40 (33.3) | − | − | |
| pTNM stage | |||||||
| I | 57 (32.8) | − | − | 22 (18.3) | − | − | |
| II | 47 (27.0) | − | − | 21 (17.5) | − | − | |
| III | 47 (27.0) | − | − | 47 (39.2) | − | − | |
| IV | 23 (13.2) | − | − | 30 (25.0) | − | − | |
| AFP (ng/mL) | 2.75±5.52 | 2.42±1.81 | 0.428 | 31.24±196.71 | 2.57±3.64 | 0.025 | |
| CEA (ng/mL) | 5.16±23.59 | 1.41±0.96 | 0.028 | 18.86±79.17 | 2.15±1.83 | 0.001 | |
| CA19-9 (U/mL) | 95.39±481.34 | 15.81±2.76 | 0.022 | 72.08±218.57 | 11.69±10.42 | <0.001 | |
Identification of GC-associated epigenomic signatures
To elucidate the epigenomic alterations associated with GC, we performed genome-wide bisulfite sequencing on 56 GC tissues and 59 NATs. We identified 630 DMRs, including 538 hypermethylated and 92 hypomethylated regions (Figure 2A,B). These DMRs were significantly enriched in promoters (66.35%), followed by distal intergenic regions (14.92%), exons (6.67%), introns (6.67%), 5’UTRs (2.22%), 3’UTRs (2.22%), and downstream regions (0.95%) (Figure 2C). On the basis of the results of the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis, genes near these DMRs were enriched in pathways such as the calcium signalling pathway, neuroactive ligand-receptor interaction, cAMP signalling pathway, and proteoglycans in cancer. These findings suggested that these pathways might play crucial roles in the development and progression of GC (Figure 2D).
Figure 2.

DMRs discovered by targeted bisulfite sequencing of GC and NAT tissues. (A) Heatmaps showing DMR methylation levels in tissue data; (B) Circus plot showing the distribution of GC-specific DMRs across the genome. Red points: hyper-DMRs. Blue points: hypo-DMRs. The circles from the outer circle to the inner circle represent the overview of DMRs and the area statistics of hypermethylated regions and hypomethylated regions, respectively; (C) Locations of DMRs in the genome; (D) KEGG term annotation of DMRs; (E) Correlation of methylation rates between plasma and tumor tissue samples from 3 GC patients; (F) Correlation between DMR methylation levels and the expression of associated genes. Each lollipop represents a DMR, with red and blue corresponding to hyper- and hypo-DMRs, respectively. The vertical axis depicts the Spearman correlation coefficient between the DMR methylation level and gene expression (P<0.05). DMR, differentially methylated region; GC, gastric cancer; NAT, normal adjacent tissue; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Correlation analysis and transcriptional regulation of DMRs in plasma and tissue samples
We performed targeted bisulfite sequencing of cfDNA from 294 GC and 446 NGC plasma samples and identified 179 DMRs that overlapped with those in tissue samples (Supplementary Figure S1). Correlation analysis of matched patient samples revealed a significant correlation between the methylation ratios of plasma and GC tissues (Figure 2E).
Among the 179 DMRs in the TCGA dataset, 119 were significantly correlated with the expression levels of nearby genes, including 118 hypermethylated DMRs and 1 hypomethylated DMR. Although 89% of the hypermethylated DMRs (105 out of 118) were negatively correlated with methylation expression, 11% of the hypermethylated DMRs (13 out of 118) were positively correlated with methylation expression (Figure 2F).
GC diagnostic models based on DMR markers
To distinguish between GC and healthy plasma, we developed diagnostic models using DMR methylation ratios as biomarkers. We first tested a RF model, which effectively classified plasma from patients with GC and healthy controls on the basis of 28 DMRs, yielding an AUC of 0.998. Further refinement to the top 6 DMRs resulted in an AUC of 0.985. Consistent results were obtained in the external validation cohort (28 DMRs AUC: 0.985; 6 DMRs AUC: 0.988), highlighting the accuracy of these markers (Figure 3A−C). These outcomes underscore the potential of GC-specific methylation changes in plasma cfDNA as highly effective biomarkers for diagnosing GCs. We further employed two advanced machine learning algorithms: SVM and LR. When the 28 DMRs were used, the SVM model yielded an impressive AUC of 0.990, and the LR model achieved an equally remarkable AUC of 0.999 (Supplementary Figure S2). To evaluate potential overfitting, we performed stratified 10-fold CV repeated 10 times on the training cohort for RF, SVM, and LR. The AUC distributions remained consistently high, with minimal variance across folds and repeats, indicating stable model performance (Supplementary Figure S3).
Figure 3.

Performance of methylation-based GC diagnostic models. (A) Cross-validation error rate for feature selection in RF model by sequentially reducing the number of DMR features on the basis of their importance scores; (B,C) ROC curves of the RF model using 28 DMRs and 6 DMRs as features in training dataset (B) and testing dataset (C); (D) ROC curves of three GC methylation diagnosis models (RF, SVM, and LR) using 28 DMRs in testing dataset; (E−G) GC diagnostic sensitivities by TNM stage (E), tumour location (F) and differentiation (G). GC, gastric cancer; DMR, differentially methylated region; ROC, receiver operating characteristic; RF, random forest; SVM, support vector machine; LR, logistic regression; U, upper; M, middle; L, lower; W, whole stomach.
Validation using an independent testing set confirmed the model’s effectiveness, revealing comparable AUCs (0.985 with the RF model, 0.978 with the SVM model, and 0.959 with the LR model; Figure 3D). The RF model exhibited exceptional sensitivity (93.3%) and specificity (96.3%). Similarly, the performance of the other models was comparable, underscoring the reproducibility and reliability of the selected features (Supplementary Figure S4). Intriguingly, this sensitivity remained consistent across different stages, locations, differentiation and Lauren types of GC (Figure 3E−G, Supplementary Figure S5).
Performance of serum protein markers and methylation-based diagnostic models
We evaluated AFP, CEA, and CA19-9 individually and in combination as a serum 3-marker panel using multivariable LR. Individually, the AUCs of the markers were less than 0.70 across cohorts. The combined serum 3-marker panel achieved AUCs of 0.626 in the training cohort and 0.712 in the testing cohort. In contrast, the cfDNA DMR-based models performed substantially better. The 28-DMR model achieved AUCs of 0.998 and 0.985 in the training and testing cohorts, respectively, and the 6-DMR model achieved 0.984 and 0.987, respectively. This superiority was maintained in early disease. For stage I vs. NGC, the serum 3-marker panel achieved AUCs of 0.573 in the training cohort and 0.615 in the testing cohort, whereas the 28-DMR and 6-DMR models achieved AUCs of 0.999 and 0.959 and 0.994 and 0.968, respectively. For stage I−II disease vs. NGC, the corresponding AUCs were 0.600 in the training cohort and 0.617 in the testing cohort for the serum 3-marker panel, compared with 0.998 and 0.976 for the 28-DMR model and 0.987 and 0.980 for the 6-DMR model, respectively (Figure 4).
Figure 4.

Comparison of serum protein markers and DMRs in diagnosis of GC. (A,B) Diagnostic performance of serum markers and DMRs for GC in training cohort (A) and testing cohort (B); (C,D) Diagnostic performance of serum markers and DMRs for stage I GC in training cohort (C) and testing cohort (D); (E,F) Diagnostic performance of serum markers and DMRs for stage I−II GC in training cohort (E) and testing cohort (F). GC, gastric cancer; NGC, non-gastric cancer; DMR, differentially methylated region; AUC, area under the curve; AFP, alpha-fetoprotein; CEA, carcinoembryonic antigen; CA19-9, carbohydrate antigen 19-9.
Prognosis model for GC
A total of 230 stomach adenocarcinoma patients with complete OS data were randomly assigned in a 3:1 ratio to a training cohort (n=172) or a testing cohort (n=58) (Figure 5A). Survival-related DMRs were first screened by univariate Cox regression (P<0.001), yielding 159 candidates. Using LASSO Cox regression, we derived an 11-DMR prognostic signature (Supplementary Figure S6). A multivariable Cox model was then fitted to compute an individual risk score (C-index=0.78; Figure 5B). Among the eleven DMRs, seven were associated with increased risk (HR>1), and four were protective (HR<1). With the use of maximally selected rank statistics in the training cohort, the optimal risk score cut-off was −1.11 (Supplementary Figure S7), and this was applied to the testing cohort to define low- and high-risk groups. In the training cohort, 49 patients were classified as high risk, and 123 were classified as low risk. In the testing cohort, 21 patients were high risk, and 37 were low risk. Patients in the high-risk group had significantly shorter OS than those in the low-risk group in both cohorts (log-rank P<0.01; Figure 5C,D).
Figure 5.

GC prognosis model. (A) Flow chart depicting the survival analysis workflow based on cfDNA methylation in GC patients; (B) Forest plot of the multivariate Cox regression analysis; (C,D) Kaplan-Meier curves illustrating OS for patients in the high-risk and low-risk score groups in the training (C) and testing (D) datasets. DMR, differentially methylated region; GC, gastric cancer; OS, overall survival.
Discussion
Despite significant recent advancements in treatment strategies, the mortality rate associated with GC remains high, primarily because of late-stage diagnosis and limited treatment options (17). Although endoscopic screening, as a secondary preventive measure, has reduced GC-related mortality by 40% (18), its invasiveness, high cost, and limited benefits for low-risk individuals have constrained its global implementation (19,20). Given the high incidence and mortality rates of GC, there is an urgent need to develop a convenient, cost-effective, and noninvasive method to increase early detection efficiency. cfDNA, discovered in 1948, has gained widespread attention in recent years for its clinical application in cancer diagnosis, largely because of the high cost, invasiveness, and complexity associated with tissue biopsies and radiological examinations (21,22). In this study, we employed a systematic and comprehensive biomarker discovery and validation approach to develop a cfDNA methylation profile in plasma as a minimally invasive biomarker for GC detection and prognosis assessment.
We conducted whole-genome bisulfite sequencing on GC tumor tissues and corresponding NATs and identified DMRs closely associated with GC. Notably, most DMRs were located in promoter regions, suggesting that these areas may play a critical role in GC initiation and progression through the regulation of gene expression (23). These DMRs were particularly enriched in several cancer-related signalling pathways, such as calcium signalling, cAMP signalling, and neuroactive ligand-receptor interactions, further supporting their potential role in the pathophysiology of GC (24). From a mechanistic perspective, this pattern is consistent with the classical model in which promoter hypermethylation drives transcriptional silencing, a hallmark frequently observed in GC (25). TCGA classification similarly highlights this mechanism, as the Epstein-Barr virus (EBV)-positive subtype is characterized by a CpG island methylator phenotype, and the microsatellite instability (MSI) subtype is often driven by MLH1 promoter methylation, underscoring the central role of promoter methylation in GC biology (26). Consistent with our KEGG results, enrichment in calcium, cAMP/G-protein-coupled receptor (GPCR)-cAMP response element-binding protein (CREB), and neuroactive ligand-receptor signalling suggests that certain promoter-centric DMRs may influence GC proliferation, invasion, and metastasis through calcium-sensing receptor (CaSR)/transient receptor potential (TRP)-mediated Ca2+ influx and the GPCR-cAMP/protein kinase A (PKA)-CREB axis (27,28). The enrichment of “proteoglycans in cancer” further implies that epigenetic regulation may modulate the tumor microenvironment and growth factor availability, both of which are known to shape GC aggressiveness and therapeutic response (29). Targeted methylation sequencing of plasma cfDNA revealed 179 DMRs consistent with those identified in tissue samples, indicating that these DMRs possess stable epigenetic marker characteristics across different biological sample types. Previous studies have confirmed that the DMRs in plasma cfDNA are highly correlated with the differential methylation of CpGs between tumor and normal tissues (30), which aligns with our findings. These results suggest that targeted bisulfite sequencing of plasma cfDNA can be used to effectively detect tumor-derived DNA methylation events in circulating tumor DNA (ctDNA).
Aberrant DNA methylation patterns are a hallmark of many cancers, and these changes often occur early in cancer development. Systematic analyses of cfDNA methylation profiles for early cancer detection are currently under exploration (31). Chemi et al. demonstrated that DMRs in plasma cfDNA could predict small cell lung cancer (SCLC) with impressive accuracy, yielding a mean area under the receiver operating characteristic curve (AUROC) of 0.986 for limited-stage SCLC (n=29) and 1.0 for extensive-stage SCLC (n=49) (32). Similarly, Luo et al. developed a cfDNA methylation-based model to predict CRC, achieving an AUC of 0.96, with a sensitivity and specificity of 87.9% and 89.6%, respectively (33). The PATHFINDER study reported a positive predictive value of 38% (35 out of 92) for cancer detection in asymptomatic individuals over 50 years old, underscoring the feasibility of using cfDNA for cancer screening (34). In the field of GC, prior studies have reported diagnostic models based on methylation markers derived from tissue or from cfDNA, which generally show high accuracy (35,36). Building on this literature, our study strengthens the evidence chain from tissue to plasma. We first performed whole-genome bisulfite sequencing in paired GC and NATs to identify GC-specific DMRs, then confirmed 179 overlapping sites in plasma, and finally evaluated the models in independent multicentre cohorts. Using these sites, we constructed diagnostic models based on regional methylation ratios. The RF model using 28 DMRs achieved an AUC of 0.998 in the training cohort and 0.985 in the validation cohort. After reduction to six DMRs, the AUCs were 0.985 and 0.988 in the training and validation cohorts, respectively. SVM and LR built on the same feature sets yielded comparable results. The models maintained stable sensitivity and specificity across stages, tumor locations, and differentiation subgroups, indicating particular utility for early detection and as a noninvasive complement for individuals in whom traditional approaches have limited sensitivity. Compared with prior studies, our approach provides an integrated discovery and validation pipeline from tissue discovery to plasma confirmation to external multicenter verification and achieves comparable accuracy with a smaller 6-DMR panel, which is advantageous for clinical translation. To further address potential overfitting and assess robustness, we performed stratified 10-fold CV repeated 10 times in the training cohort. The AUCs remained consistently high with minimal variability across repeats, supporting the stability of the models.
In addition to GC detection, we explored the potential clinical application of cfDNA methylation in prognostic stratification. Previous studies have shown that cfDNA methylation markers may play a role in predicting the prognosis of patients with various malignancies, such as ovarian cancer, CRC, and advanced biliary tract cancer (33,37,38). In our study, we developed an 11-DMR marker classifier to assess the prognosis of GC patients. These findings indicate that cfDNA methylation markers can be used to predict prognosis in GC patients and can serve as independent risk factors for disease progression. Prognostic stratification analysis could help identify patients who may benefit from aggressive treatment and more frequent monitoring.
This study has several limitations. First, our research primarily included patient samples from Asian cohorts. Given the known genomic differences across populations, the generalizability of our model needs to be further investigated in larger and more diverse cohorts. Second, the study was mainly based on cross-sectional data and lacked long-term follow-up of patients, limiting our ability to assess the effectiveness of the model in predicting GC prognosis or recurrence. Future studies should incorporate longitudinal follow-up data to more comprehensively evaluate the model’s predictive ability at different time points and stages of the disease. Additionally, although the independent test set achieved high AUC values for GC prediction, there is a potential risk of overfitting in the model. Therefore, further validation in larger-scale and more diverse populations is necessary to ensure the stability and reliability of these models in real-world applications. Third, patients with benign gastric diseases, precancerous lesions, or multiple comorbidities were not included in our training set. Future studies are needed to validate the model in these populations to better assess its stability and specificity in more complex clinical contexts. Finally, the study focused on the detection and diagnosis of GC, but it did not sufficiently explore whether the DMRs used exhibit similar diagnostic capabilities in other types of cancer. The lack of comparative analysis may limit the model’s specificity, and future research should include samples from other common cancer types to evaluate the broader applicability of the DMRs and their specificity for GC.
Conclusions
Our study demonstrated the rationale and accuracy of using cfDNA methylation markers for GC detection and prognosis prediction. However, further validation in larger and more diverse populations is needed to confirm these findings and ensure their broader applicability.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
Acknowledgements
This work was supported by National Key R&D Program of China (No. 2021YFA0910100), National Natural Science Foundation of China (No. 82374544, 82204828, 82422078), Healthy Zhejiang One Million People Cohort (No. K-20230085), Program of Zhejiang Provincial TCM Sci-tech Plan (No. GZY-ZJ-KJ-230003, No. GZY-ZJ-KJ-23048), and Natural Science Foundation of Zhejiang Province (No. LHDMY22H160008).
Acknowledgments
Footnote
Conflicts of Interest: Jiaxi Peng, Fengming Zhang, Chun Song, and Yuying Wang are employees of BGI Genomics. All other authors have no conflicts of interest to declare.
Funding Statement
This work was supported by National Key R&D Program of China (No. 2021YFA0910100), National Natural Science Foundation of China (No. 82374544, 82204828, 82422078), Healthy Zhejiang One Million People Cohort (No. K-20230085), Program of Zhejiang Provincial TCM Sci-tech Plan (No. GZY-ZJ-KJ-230003, No. GZY-ZJ-KJ-23048), and Natural Science Foundation of Zhejiang Province (No. LHDMY22H160008).
Contributor Information
Yuying Wang, Email: wangyuying@bgi.com.
Lei Lian, Email: lianlei2@mail.sysu.edu.cn.
Xiaodong Chen, Email: gis_sch@163.com.
Xiangdong Cheng, Email: chengxd@zjcc.org.cn.
Author contributions
Study concepts: L Yuan, XD Cheng; Study design: XD Cheng, YY Wang, L Lian, XD Chen; Data acquisition: XH Liu, PC Yu, Y Wang, ZH Bao, YH Xia, KL Yin; Quality control of data and algorithms: JX Peng, FM Zhang, C Song; Data analysis and interpretation: YN Wang, L Yuan, LY Jin, WY He; Statistical analysis: JX Peng, YY Wang; Manuscript preparation: YN Wang, LY Jin, WY He, JX Peng; Manuscript editing: XD Cheng, L Yuan; Manuscript review: YY Wang, L Lian, XD Chen. All authors read and approved the final manuscript.
References
- 1.Bray F, Laversanne M, Sung H, et al Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63. doi: 10.3322/caac.21834. [DOI] [PubMed] [Google Scholar]
- 2.Qi C, Chong X, Zhou T, et al Clinicopathological significance and immunotherapeutic outcome of claudin 18. 2 expression in advanced gastric cancer: A retrospective study. Chin J Cancer Res. 2024;36:78–89. doi: 10.21147/j.issn.1000-9604.2024.01.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng H, Chen W, Zheng R, et al Changing cancer survival in China during 2003-15: a pooled analysis of 17 population-based cancer registries. Lancet Glob Health. 2018;6:e555–67. doi: 10.1016/S2214-109X(18)30127-X. [DOI] [PubMed] [Google Scholar]
- 4.Ito Y, Miyashiro I, Ishikawa T, et al Determinant Factors on Differences in Survival for Gastric Cancer Between the United States and Japan Using Nationwide Databases. J Epidemiol. 2021;31:241–8. doi: 10.2188/jea.JE20190351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang MJ, Won YJ, Lee JJ, et al Cancer statistics in Korea: Incidence, mortality, survival, and prevalence in 2019. Cancer Res Treat. 2022;54:330–44. doi: 10.4143/crt.2022.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma S, Zhou M, Xu Y, et al Clinical application and detection techniques of liquid biopsy in gastric cancer. Mol Cancer. 2023;22:7. doi: 10.1186/s12943-023-01715-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimada H, Noie T, Ohashi M, et al Clinical significance of serum tumor markers for gastric cancer: a systematic review of literature by the Task Force of the Japanese Gastric Cancer Association. Gastric Cancer. 2014;17:26–33. doi: 10.1007/s10120-013-0259-5. [DOI] [PubMed] [Google Scholar]
- 8.Nikanjam M, Kato S, Kurzrock R Liquid biopsy: current technology and clinical applications. J Hematol Oncol. 2022;15:131. doi: 10.1186/s13045-022-01351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medina JE, Dracopoli NC, Bach PB, et al Cell-free DNA approaches for cancer early detection and interception. J Immunother Cancer. 2023;11:e006013. doi: 10.1136/jitc-2022-006013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung DC, Gray DM 2nd, Singh H, et al A cell-free DNA blood-based test for colorectal cancer screening. N Engl J Med. 2024;390:973–83. doi: 10.1056/NEJMoa2304714. [DOI] [PubMed] [Google Scholar]
- 11.Liu MC, Oxnard GR, Klein EA, et al Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31:745–59. doi: 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu P, Chen P, Wu M, et al Multi-dimensional cell-free DNA-based liquid biopsy for sensitive early detection of gastric cancer. Genome Med. 2024;16:79. doi: 10.1186/s13073-024-01352-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy S, Kanda M, Nomura S, et al Diagnostic efficacy of circular RNAs as noninvasive, liquid biopsy biomarkers for early detection of gastric cancer. Mol Cancer. 2022;21:42. doi: 10.1186/s12943-022-01527-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng H, Conneely KN, Wu H A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014;42:e69. doi: 10.1093/nar/gku154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavalcante RG, Sartor MA annotatr: genomic regions in context. Bioinformatics. 2017;33:2381–3. doi: 10.1093/bioinformatics/btx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu G, Wang LG, Han Y, et al clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth EC, Nilsson M, Grabsch HI, et al Gastric cancer. Lancet. 2020;396:635–48. doi: 10.1016/S0140-6736(20)31288-5. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Li M, Chen S, et al Endoscopic screening in Asian countries is associated with reduced gastric cancer mortality: A meta-analysis and systematic review. Gastroenterology. 2018;155:347–54.e9. doi: 10.1053/j.gastro.2018.04.026. [DOI] [PubMed] [Google Scholar]
- 19.Gupta N, Bansal A, Wani SB, et al Endoscopy for upper GI cancer screening in the general population: a cost-utility analysis. Gastrointest Endosc. 2011;74:610–24.e2. doi: 10.1016/j.gie.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Saumoy M, Schneider Y, Shen N, et al Cost effectiveness of gastric cancer screening according to race and ethnicity. Gastroenterology. 2018;155:648–60. doi: 10.1053/j.gastro.2018.05.026. [DOI] [PubMed] [Google Scholar]
- 21.Mandel P, Metais P Nuclear acids in human blood plasma. C R Seances Soc Biol Fil. 1948;142:241–3. [PubMed] [Google Scholar]
- 22.Song P, Wu LR, Yan YH, et al Limitations and opportunities of technologies for the analysis of cell-free DNA in cancer diagnostics. Nat Biomed Eng. 2022;6:232–45. doi: 10.1038/s41551-021-00837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel KB, Padhya TA, Huang J, et al Plasma cell-free DNA methylome profiling in pre- and post-surgery oral cavity squamous cell carcinoma. Mol Carcinog. 2023;62:493–502. doi: 10.1002/mc.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang S, Zhang T, Liu H, et al Tracking the evolution of untreated high-intermediate/high-risk diffuse large B-cell lymphoma by circulating tumour DNA. Br J Haematol. 2022;196:617–28. doi: 10.1111/bjh.17894. [DOI] [PubMed] [Google Scholar]
- 25.Padmanabhan N, Ushijima T, Tan P How to stomach an epigenetic insult: the gastric cancer epigenome. Nat Rev Gastroenterol Hepatol. 2017;14:467–78. doi: 10.1038/nrgastro.2017.53. [DOI] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie R, Xu J, Xiao Y, et al Calcium promotes human gastric cancer via a novel coupling of calcium-sensing receptor and TRPV4 channel. Cancer Res. 2017;77:6499–512. doi: 10.1158/0008-5472.CAN-17-0360. [DOI] [PubMed] [Google Scholar]
- 28.Yan H, Zhang JL, Leung KT, et al An update of G-protein-coupled receptor signaling and its deregulation in gastric carcinogenesis. Cancers (Basel) 2023;15:736. doi: 10.3390/cancers15030736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iozzo RV, Sanderson RD Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 2011;15:1013–31. doi: 10.1111/j.1582-4934.2010.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen SY, Singhania R, Fehringer G, et al Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–83. doi: 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 31.Luo H, Wei W, Ye Z, et al Liquid biopsy of methylation biomarkers in cell-free DNA. Trends Mol Med. 2021;27:482–500. doi: 10.1016/j.molmed.2020.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Chemi F, Pearce SP, Clipson A, et al cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat Cancer. 2022;3:1260–70. doi: 10.1038/s43018-022-00415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo H, Zhao Q, Wei W, et al Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med. 2020;12:eaax7533. doi: 10.1126/scitranslmed.aax7533. [DOI] [PubMed] [Google Scholar]
- 34.Schrag D, Beer TM, McDonnell CH 3rd, et al Blood-based tests for multicancer early detection (PATHFINDER): a prospective cohort study. Lancet. 2023;402:1251–60. doi: 10.1016/S0140-6736(23)01700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson BW, Suh YS, Choi B, et al Detection of gastric cancer with novel methylated DNA markers: discovery, tissue validation, and pilot testing in plasma. Clin Cancer Res. 2018;24:5724–34. doi: 10.1158/1078-0432.CCR-17-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi J, Hong B, Wang S, et al Plasma cell-free DNA methylome-based liquid biopsy for accurate gastric cancer detection. Cancer Sci. 2024;115:3426–38. doi: 10.1111/cas.16284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang L, Zhang Y, Li C, et al Plasma cfDNA methylation markers for the detection and prognosis of ovarian cancer. EBioMedicine. 2022;83:104222. doi: 10.1016/j.ebiom.2022.104222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berchuck JE, Facchinetti F, DiToro DF, et al The clinical landscape of cell-free DNA alterations in 1671 patients with advanced biliary tract cancer. Ann Oncol. 2022;33:1269–83. doi: 10.1016/j.annonc.2022.09.150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online.