

Abstract
13C/12C and D/H stable isotope fractionation during aerobic degradation was determined for Pseudomonas putida strain mt-2, Pseudomonas putida strain F1, Ralstonia pickettii strain PKO1, and Pseudomonas putida strain NCIB 9816 grown with toluene, xylenes, and naphthalene. Different types of initial reactions used by the respective bacterial strains could be linked with certain extents of stable isotope fractionation during substrate degradation.
Intrinsic microbial degradation is an important process in elimination of contaminants in polluted aquifers, which can be used for the sustainable cleanup of contaminated sites. However, cost-effective remediation strategies such as natural attenuation require a profound knowledge of the microbial degradation processes in the subsurface. Although biodegradation of aromatic hydrocarbons by aerobic and anaerobic bacteria has been investigated in detail in laboratory systems (11, 32), assessment at field sites remains difficult. Stable carbon isotope analysis is one approach to quantify microbial activities in situ. For laboratory cultures, isotope fractionation has been shown to occur during degradation of aromatic hydrocarbons, such as toluene (1, 19, 20), or chlorinated hydrocarbons, such as trichloroethene (3, 7, 26). In addition, in contaminated field sites, carbon isotope fractionation could be observed and was interpreted to be indicative of microbial degradation in situ (15, 24). For toluene as a model compound, it has been demonstrated that isotope fractionation is caused mainly by the first enzyme reaction in the degradation pathway, whereas transport to and into the cell appears not to be relevant for fractionation. The extent of isotope fractionation is considered to be independent of differences in the growth kinetics of the bacteria (20). Isotope fractionation during anaerobic degradation of toluene was on the same order of magnitude for denitrifying, iron(III)-reducing, sulfate-reducing, and fermenting bacteria (1, 19), probably because, in these cases, degradation was initiated by benzylsuccinate synthase. This finding suggests that, in anoxic environments, isotope fractionation could be applied to assess biological degradation quantitatively, as has been worked out recently for several aquifers (23, 25).
The objective of this study was to examine whether carbon and hydrogen isotope fractionation could be used to quantify intrinsic biodegradation as well in oxic environments. Previous studies with the aerobic bacterium Pseudomonas putida strain mt-2 showed an extent of isotope fractionation similar to that of anaerobic toluene-degrading strains (19), whereas isotope fractionation during toluene degradation by undefined aerobic microbial communities was not detected (26). Therefore, we started a systematic investigation of the effects of different oxygenase enzymes and stable isotope fractionation.
P. putida strain mt-2 (20), Ralstonia pickettii strain PKO1 (J. J. Kukor, Rutgers University, New Brunswick, N.J.), and P. putida strain F1 (A. M. Cook, Konstanz, Germany) were taken as representatives of bacteria by using different toluene degradation pathways in polluted aquifers. Strains were grown in 200-ml batch cultures with 15 μl of toluene, m-xylene, or p-xylene (analytical grade quality; Fluka, Buchs, Switzerland) as described previously (20). Cultures for isotope fractionation experiments were inoculated with 5% (vol/vol) of precultures (optical density at 578 nm [OD578] = 0.25), and growth was monitored by measuring the OD578. Hydrocarbon concentrations were determined by high-performance liquid chromatography (20). During degradation of toluene by P. putida strain mt-2 (methyl monooxygenase), the 13C/12C isotope ratios in the nondegraded toluene were determined by gas chromatography-combustion-isotope ratio monitoring mass spectrometry (GC-C-IRMS) (20) and shifted from δ 13C = −29.18‰ ± 0.35‰ to more positive values of δ 13C = −23.22‰ ± 0.32‰ after 80% of the substrate was degraded (equation 4) (Fig. 1). Analysis of the carbon isotope ratios ln(Rt/R0) and the respective concentrations ln(ct/c0) according to the Rayleigh equation for closed systems (equation 1) (13, 22) resulted in an isotope fractionation factor: αC = 0.9967 ± 0.0003 (Fig. 2A). Rt and ct are the 13C/12C isotope ratio and the residual substrate concentration at time t, and RStd is the isotope ratio of an international standard (Vienna PDB).
FIG. 1.

Isotope shifts during degradation of toluene and a toluene-toluene-d8 mixture by P. putida strain mt-2. The decrease in toluene (▪) and concomitant increase in 13C (R = 13C/12C) in the remaining substrate (□) and the decrease in toluene-toluene-d8 concentration (▴) and concomitant increase in deuterium Rt = [toluene-d8]/[toluene] in the residual nondegraded substrate fraction (▵) were monitored over time. In every case, data are shown for one representative experiment out of three performed.
FIG. 2.

Carbon (A) and hydrogen (B) stable isotope fractionation during degradation of toluene by P. putida mt-2 (▪), R. pickettii PKO1 (▴), and P. putida F1 (•) and isotope fractionation during degradation of naphthalene by P. putida NCIB 9816 (⧫). 13C/12C carbon isotope fractionation was plotted according to equation 1. D/H isotope fractionation was determined with equation 3 during growth with equal amounts of per-deuterated toluene-d8 and toluene and per-deuterated naphthalene-d8 and naphthalene.
For direct comparison between the bacterial pure cultures, isotope fractionation will be referred to as “enrichment factor ɛ” (equation 2), which is indicated as ɛn in experiments with nonlabeled toluene. During toluene degradation by P. putida strain mt-2, the enrichment factor was ɛn = −3.3 ± 0.3 for 13C/12C isotope fractionation (Table 1).
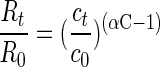 |
(1) |
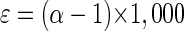 |
(2) |
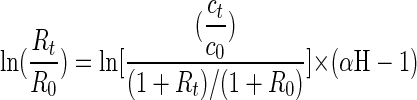 |
(3) |
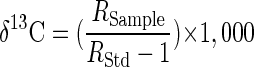 |
(4) |
P. putida. strain mt-2 also degrades m- and p-xylene by using the same enzyme as toluene (methyl monooxygenase) (29). The enrichment factors for carbon isotope fractionation obtained during degradation of m- and p-xylene were slightly smaller than those of toluene (Table 1).
TABLE 1.
13C/12C and D/H isotope fractionation by different aerobic bacterial strains during growth with aromatic hydrocarbonsa
| Strain | Enzyme mechanism | Carbon source | ɛCn | ɛDl |
|---|---|---|---|---|
| P. putida strain mt-2 | Methyl monooxygenase | Toluene | −3.3 ± 0.3 | −905 ± 71 |
| Methyl monooxygenase | m-Xylene | −1.7 ± 0.04 | NDb | |
| Methyl monooxygenase | p-Xylene | −2.3 ± 0.3 | ND | |
| R. pickettii strain PKO1 | Ring monoooxygenase | Toluene | −1.1 ± 0.2 | −16 ± 5.3 |
| P. putida strain F1 | Ring dioxygenase | Toluene | −0.4 ± 0.3 | −28 ± 10 |
| P. putida strain NCIB 9816 | Ring dioxygenase | Naphthalene | −0.1 ± 0.2 | −65 ± 13 |
Stable carbon isotope fractionation is given as the enrichment factor, ɛn, and was calculated with equations 1 and 2. Hydrogen isotope fractionation with mixtures (50:50) of per-deuterated and nonlabeled substrate was determined with equation 3. The enrichment factor for labeled substances, ɛ1, was determined by equation 2. Mean values ± standard deviation are given based on three independent growth experiments with fivefold isotope analysis per data point.
ND, not determined.
Toluene degradation by R. pickettii strain PKO1 (ring monooxygenase) resulted in a carbon isotope fractionation that was three times lower than that with strain mt-2. The third toluene-degrading strain, P. putida strain F1, hydroxylated the substrate with a toluene-dioxygenase to the corresponding catechol. The concomitant 13C/12C isotope fractionation was insignificant. The same was observed with P. putida strain NCIB 9816 in degration experiments with naphthalene (DSM 8368; Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany). Strain NCIB 9816 attacks naphthalene by a dioxygenase reaction similar to that of strain F1. It was grown in batch culture with 150 to 200 μM dissolved naphthalene as a carbon source. Naphthalene was transferred into the culture bottles as a solid crystal and autoclaved in 180 ml of H2O. After cooling, the stock solutions for mineral medium M9 were added. The carbon isotope ratio in the residual naphthalene did not change during degradation over time, and the isotope fractionation was not significant (Fig. 2A and Table 1).
Hydrogen isotope fractionation during degradation of aromatic hydrocarbons was investigated by cultivating the same four bacterial strains with a mixture of nondeuterated toluene and per-deuterated toluene-d8 (8 μl each) or with a mixture of nonlabeled naphthalene and per-deuterated naphthalene-d8 (3 mg each). The ratio of residual labeled to nonlabeled substrate in the culture during growth was analyzed in pentane extracts of liquid samples (2 to 7 ml) by GC (20). Naphthalene and per-deuterated naphthalene-d8 were separated by GC to the baseline at a constant oven temperature of 140°C. Isotope fractionation factors were calculated according to equation 3 with the isotope ratio Rt = [deuterated substrate]/[substrate] (2). For a very high abundance of the heavier isotope, as was used in the experiments for hydrogen isotope fractionation, equation 1 changes to equation 3 (2).
Enrichment factors derived from equation 3 are depicted as ɛl. Degradation of half of the substrate mixture by P. putida strain mt-2 (methyl monooxygenase) was accompanied by a 40-fold enrichment of per-deuterated toluene-d8, correlated with a near-total depletion of nonlabeled toluene (Fig. 1). The D/H isotope fractionation determined in the first growth phase was ɛl = −905 ± 71. R. pickettii strain PKO1 (ring monooxygenase) and P. putida strain F1 (ring dioxygenase) degraded the toluene-toluene-d8 mixture, resulting in D/H isotope fractionations more than 10 and 50 times stronger than the carbon isotope fractionation by the same strains, but still 50 and 30 times lower, respectively, than the D/H fractionation by P. putida strain mt-2. P. putida strain NCIB 9816 (ring dioxygenase) grown with naphthalene-naphthalene-d8 showed a D/H isotope fractionation that was twice as strong as the D/H isotope fractionation by P. putida strain F1 (Fig. 2B and Table 1).
Of the four strains examined, P. putida strain mt-2 showed by far the highest 13C/12C and D/H isotope fractionation during aerobic toluene degradation. The methyl monooxygenase of this bacterium exhibits similarities in its amino acid sequence to the methane monooxygenase of Methylosinus trichosporium OB3b (12, 33). Oxygen bound as an oxene group to the catalytic iron of methane monooxygenase is proposed to lead to a homolytic C-H bond cleavage of the substrate. The reaction is associated with strong isotope effects during methane conversion and minor effects during ethane and propane conversion (4). The authors assume that the reaction mechanism with methane differs slightly from that with other substrates. Similarly, in our study, the xylene monooxygenase of P. putida strain mt-2 caused strong hydrogen and carbon isotope fractionation. Minor differences in the enzymatic reactions with toluene and m- and p-xylene might be the reason for the variations in the respective 13C/12C isotope fractionation factors. Cytochrome P450 is another well-known example of an enzyme that dissociates a C-H bond and inserts oxygen into benzylic or aliphatic substrates (35). In an experimental study, Manchester et al. (18) found the strongest isotope effects with p-xylene and toluene, which showed six times higher rate constants than deuterated p-xylene and toluene.
Toluene degradation by R. pickettii strain PKO1 is catalyzed by a monooxygenase reaction hydroxylating carbon 3 of the aromatic ring (21). The amino acid sequence of this toluene-3-monooxygenase is remarkably similar to that of toluene-4-monooxygenase of Pseudomonas mendocina strain KR1, an enzyme studied in great detail (31). The poor hydrogen isotope fractionation of strain PKO1 is probably due to an enzymatic reaction with the π-electron system of the aromatic substrate. The hydroxylation presumably is a two-step process starting with an electrophilic attack of iron-bound oxygen on the aromatic ring and the formation of a C-O σ-bond. In a faster second step, the hydrogen atom bound to this carbon atom is released as a proton, and the electrons are used to reconstitute the aromaticity of the carbon ring skeleton (5). The weak carbon and hydrogen isotope fractionation observed here might be due to secondary isotope effects caused by atoms of the substrate molecule not directly contributing to the reaction.
P. putida strains F1 and NCIB 9816 both attack toluene through an initial dioxygenase reaction on the aromatic ring (6, 16, 34). Initially, Π-electrons of the aromatic substrate are attracted by activated oxygen, which is bound to a catalytic iron center (28). Primary isotope effects do not accompany this reaction, because there is no distinct bond cleavage involved. The first reaction product of toluene and naphthalene oxidation is a cis-dihydrodiol, which is dehydrogenated in a subsequent step to form a catechol derivative (9). The terminal oxygenase units of toluene and naphthalene dioxygenase of strains F1 and NCIB 9816 are both hexamers (α3β3) with catalytic mononuclear ferrous iron centers (8, 17, 30). The amino acid sequences of their subunits suggest that they derived from a common ancestor (12) and share the same reaction mechanism (10). Accordingly, 13C/12C isotope fractionation was very weak during toluene degradation by P. putida strain F1 and was not significant during naphthalene degradation by P. putida strain NCIB 9816. The more pronounced secondary D/H isotope fractionation effects of P. putida strain NCIB 9816 might be caused by individual features of the naphthalene degradation mechanism.
Recently, Hunkeler et al. (14) found relatively small amounts of 13C/12C and D/H isotope fractionation during benzene degradation by Acinetobacter sp. and Burkholderia sp. Others observed a small amount of 13C/12C isotope fractionation during benzene degradation by an aerobic mixed culture that was enriched from groundwater of a petrochemical site (27). The small isotope fractionation factors and the increase in δ13C for the initial mono- or dioxygenase reactions at the benzene ring that were documented in these papers are in agreement with our observations that oxygenases acting on Π-electron system of the aromatic ring produce only minor isotope effects.
The present study and previous investigations allow some preliminary conclusions to be drawn about when stable isotope fractionation could be used to assess biodegradative activities in contaminated groundwater. Consistent carbon isotope fractionation during toluene degradation by anaerobic microorganisms allows a quantitative assessment of bacterial degradation under anoxic conditions. Hydrogen isotope effects are stronger than carbon isotope effects by 2 or 3 orders of magnitude, but are also more variable between different types of anaerobic bacteria. Small extents of degradation are detectable, but quantification in situ might be difficult. However, D/H and 13C/12C isotope fractionation in oxic zones of contaminated aquifers could be used only as a qualitative indicator of biodegradation. The differences in isotope fractionation between bacterial strains with different oxygenase reactions preclude a quantitative assessment of in situ biodegradation based on isotope fractionation in oxic zones. Nevertheless, combined measurement of carbon isotope fractionation for in situ quantification and hydrogen isotope fractionation as a positive indicator of bacterial activities could be a valuable tool to assess biodegradation in the environment.
Acknowledgments
This work was financially supported by the Bundesministerium für Bildung und Forschung (grant 02WT0022) and by the Deutsche Forschungsgemeinschaft (grant Schi 180/7).
Footnotes
This paper represents publication no. 182 of the Deutsche Forschungsgemeinschaft priority program 546 “Geochemical processes with long-term effects in anthropogenically affected seepage- and groundwater.”
REFERENCES
- 1.Ahad, J. M. E., B. Sherwood Lollar, E. A. Edwards, G. F. Slater, and B. E. Sleep. 2000. Carbon isotope fractionation during anaerobic biodegradation of toluene: implications for intrinsic bioremediation. Environ. Sci. Technol. 34:892-896. [Google Scholar]
- 2.Biegeleisen, J., and M. Wolfsberg. 1958. Theoretical and experimental aspects of isotope effects in chemical kinetics. Adv. Chem. Phys. 1:15-76. [Google Scholar]
- 3.Bloom, Y., R. Aravena, D. Hunkeler, E. Edwards, and S. K. Frape. 2000. Carbon isotope fractionation during microbial degradation of trichloroethene, cis-1,2-dichloroethene, and vinyl chloride: implications for assessment of natural attenuation. Environ. Sci. Technol. 34:2768-2772. [Google Scholar]
- 4.Brazeau, B. J., and J. D. Lipscomb. 2000. Kinetics and activation thermodynamics of methane monooxygenase compound Q formation and reaction with substrates. Biochemistry 39:13503-13515. [DOI] [PubMed] [Google Scholar]
- 5.Carey, F. A., and R. J. Sundberg. 1990. Advanced organic chemistry. Plenum Press, New York, N.Y.
- 6.Carredano, E., A. Karlsson, B. Kauppi, D. Choudhury, D. T. Gibson, H. Eklund, and S. Ramaswamy. 2000. Substrate binding site of naphthalene 1,2-dioxygenase: functional implications of indole binding. J. Mol. Biol. 296:701-712. [DOI] [PubMed] [Google Scholar]
- 7.Ertl, S., F. Seibel, L. Eichinger, F. H. Frimmel, and A. Kettrup. 1996. Determination of the 13C/12C isotope ratio of organic compounds for the biological degradation of tetrachloroethene (PCE) and trichloroethene (TCE). Acta Hydrochim. Hydrobiol. 24:16-21. [Google Scholar]
- 8.Gassner, G. T., M. L. Ludwig, D. L. Gatti, C. C. Correll, and D. P. Ballou. 1995. Structure and mechanism of the iron-sulfur flavoprotein phthalate dioxygenase reductase. FASEB J. 9:1411-1418. [DOI] [PubMed] [Google Scholar]
- 9.Gibson, D. T., G. E. Cardini, F. C. Maseles, and R. E. Kallio. 1970. Incorporation of oxygen-18 into benzene by Pseudomonas putida. Biochemistry 9:1631-1635. [DOI] [PubMed] [Google Scholar]
- 10.Gibson, D. T., and R. E. Parales. 2000. Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 11:236-243. [DOI] [PubMed] [Google Scholar]
- 11.Gibson, D. T., and V. Subramanian. 1984. Microbial degradation of aromatic hydrocarbons, p. 181-252. In D. T. Gibson (ed.), Microbial degradation of organic compounds, vol. 13. Dekker, New York, N.Y.
- 12.Harayama, S., M. Kok, and E. L. Neidle. 1992. Functional and evolutionary relationships among diverse oxygenases. Annu. Rev. Microbiol. 46:565-601. [DOI] [PubMed] [Google Scholar]
- 13.Hoefs, J. 1997. Stable isotope geochemistry, 4th ed. Springer-Verlag, Berlin, Germany.
- 14.Hunkeler, D., N. Andersen, R. Aravena, S. M. Bernasconi, and B. J. Butler. 2001. Hydrogen and carbon isotope fractionation during aerobic biodegradation of benzene. Environ. Sci. Technol. 35:3462-3467. [DOI] [PubMed] [Google Scholar]
- 15.Hunkeler, D., R. Aravena, and B. J. Butler. 1999. Monitoring microbial dechlorination of tetrachloroethene (PCE) in groundwater using compound-specific stable carbon isotope ratios: microcosm and field studies. Environ. Sci. Technol. 33:2733-2738. [Google Scholar]
- 16.Jeffrey, A. M., H. J. C. Yeh, D. M. Jerina, T. R. Patel, J. F. Davey, and D. T. Gibson. 1975. Initial reactions in the oxidation of naphthalene by Pseudomonas putida. Biochemistry 14:575-584. [DOI] [PubMed] [Google Scholar]
- 17.Kauppi, B., K. Lee, E. Carredano, R. E. Parales, D. T. Gibson, H. Eklund, and S. Ramaswamy. 1998. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6:571-586. [DOI] [PubMed] [Google Scholar]
- 18.Manchester, J. I., J. P. Dinnocenzo, L. Higgins, and J. P. Jones. 1997. A new mechanistic probe for cytochrome P450: an application of isotope effect profiles. J. Am. Chem. Soc. 119:5069-5070. [Google Scholar]
- 19.Meckenstock, R. U., B. Morasch, R. Warthmann, B. Schink, E. Annweiler, W. Michaelis, and H. H. Richnow. 1999. 13C/12C isotope fractionation of aromatic hydrocarbons during microbial degradation. Environ. Microbiol. 1:409-414. [DOI] [PubMed] [Google Scholar]
- 20.Morasch, B., H. H. Richnow, B. Schink, and R. U. Meckenstock. 2001. Stable hydrogen and carbon isotope fractionation during microbial toluene degradation: mechanistic and environmental aspects. Appl. Environ. Microbiol. 67:4842-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsen, R. H., J. J. Kukor, and B. Kaphammer. 1994. A novel toluene-3-monooxygenase pathway cloned from Pseudomonas pickettii PKO1. J. Bacteriol. 176:3749-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rayleigh, J. W. S. 1896. Theoretical considerations respecting the separation of gases by diffusion and similar processes. Philos. Mag. 42:493-498. [Google Scholar]
- 23.Richnow, H. H., M. Gehre, M. Kästner, B. Morasch, and R. U. Meckenstock. 2001. Characterisation of microbial in situ degradation of aromatic hydrocarbons, p. 99-108. In V. S. Magar, T. M. Vogel, M. Aelion, and A. Leeson (ed.), Innovative methods in support of bioremediation. Battelle Press, Columbus, Ohio.
- 24.Richnow, H. H., and R. U. Meckenstock. 1999. Isotopen-geochemisches Konzept zur in situ Erfassung des biologischen Abbaus in kontaminiertem Grundwasser. TerraTech 5:38-41. [Google Scholar]
- 25.Richnow, H. H., A. Vieth, M. Kästner, M. Gehre, and R. U. Meckenstock. 2002. Isotope fractionation of toluene: a perspective to characterise microbial in situ degradation. Sci. World J. 2:7227-7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherwood Lollar, B., G. F. Slater, J. Ahad, B. Sleep, J. Spivack, M. Brennan, and P. MacKenzie. 1999. Contrasting carbon isotope fractionation during biodegradation of trichloroethylene and toluene: implications for intrinsic bioremediation. Org. Geochem. 30:813-820. [Google Scholar]
- 27.Stehmeier, L. G., M. M. Francis, T. R. Jack, E. Diegor, L. Winsor, and T. A. Abrajano. 1999. Field and in vitro evidence for in-situ bioremediation using compound-specific 13C/12C ratio monitoring. Org. Geochem. 30:821-833. [Google Scholar]
- 28.Subramanian, V., T.-N. Liu, W. K. Yeh, and D. T. Gibson. 1979. Toluene dioxygenase: purification of an iron-sulfur protein by affinity chromatography. Biochem. Biophys. Res. Commun. 91:1131-1139. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki, M., T. Hayakawa, J. P. Shaw, M. Rekik, and S. Harayama. 1991. Primary structure of xylene monooxygenase: similarities to and differences from the alkane hydroxylation system. J. Bacteriol. 173:1690-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torok, D. S., S. M. Resnick, J. M. Brand, D. L. Cruden, and D. T. Gibson. 1995. Desaturation and oxygenation of 1,2-dihydronaphthalene by toluene and naphthalene dioxygenase. J. Bacteriol. 177:5799-5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whited, G. M., and D. T. Gibson. 1991. Toluene-4-monooxygenase, a three-component enzyme system that catalyzes the oxidation of toluene to p-cresol in Pseudomonas mendocina KR1. J. Bacteriol. 173:3010-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Widdel, F., and R. Rabus. 2001. Anaerobic biodegradation of saturated and aromatic hydrocarbons. Curr. Opin. Biotechnol. 12:259-276. [DOI] [PubMed] [Google Scholar]
- 33.Worsey, M. J., and P. A. Williams. 1975. Metabolism of toluene and xylenes by Pseudomonas putida (arvilla) mt-2: evidence for a new function of the TOL plasmid. J. Bacteriol. 124:7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh, W. K., D. T. Gibson, and T.-N. Liu. 1977. Toluene dioxygenase: a multicomponent enzyme system. Biochem. Biophys. Res. Commun. 78:401-410. [DOI] [PubMed] [Google Scholar]
- 35.Yoshizawa, K., Y. Kagawa, and Y. Shioto. 2000. Kinetic isotope effects in a C-H bond dissociation by the iron-oxo species of cytochrome P450. J. Phys. Chem. 104:12365-12370. [Google Scholar]