

Abstract
Kinetics and thermodynamics of the binding of a fluorescent lipid amphiphile, Rhodamine Green™-tetradecylamide (RG-C14:0), to bovine serum albumin were characterized in an equilibrium titration and by stopped-flow fluorimetry. The binding equilibrium of RG-C14:0 to albumin was then used to reduce its concentration in the aqueous phase to a value below its critical micelle concentration. Under these conditions, the only two species of RG-C14:0 in the system were the monomer in aqueous solution in equilibrium with the protein-bound species. After previous determination of the kinetic and thermodynamic parameters for association of RG-C14:0 with albumin, the kinetics of insertion of the amphiphile into and desorption off lipid bilayer membranes in different phases (solid, liquid-ordered, and liquid-disordered phases, presented as large unilamellar vesicles) were studied by stopped-flow fluorimetry at 30°C. Insertion and desorption rate constants for association of the RG-C14:0 monomer with the lipid bilayers were used to obtain lipid/water equilibrium partition coefficients for this fluorescent amphiphile. The direct measurement of these partition coefficients is shown to provide a new method for the indirect determination of the equilibrium partition coefficient of similar molecules between two defined lipid phases if they coexist in the same membrane.
Introduction
Serum albumins, abundant transport proteins found in blood plasma, are known to bind drugs and lipid amphiphiles with a high affinity (Peters, 1997). This binding has been well-studied and crystal structures of albumin and its complexes with fatty acids and drugs have become available in recent years (He and Carter, 1992, Curry et al., 1998, Bhattacharya et al., 2000a, Petitpas et al., 2001). Besides its obvious pharmacological interest, the binding of amphiphiles to serum albumins has been exploited in the labeling of cell surface membranes with fluorescent lipid amphiphiles (FLA) (Lipsky and Pagano, 1985, Pagano and Martin, 1988). The rationale of this utilization of serum albumins in cell biology lies in the fact that FLA form aggregates (microcrystals, micelles) in aqueous solution at concentrations above a critical value (solubility product, critical aggregation concentration or critical micelle concentration). This critical concentration, which we shall refer to generically as a critical aggregation concentration (CAC), can be quite low, so that under the experimental conditions required for an adequate staining of the cell plasma membrane most of the FLA in aqueous solution exists in an aggregated form. Interaction of the aggregate with the plasma membrane of the cell, when it does occur, can result in an undesirable localized (non-homogeneous) staining. When serum albumin is present in the labeling solution, part of the FLA is bound to the albumin. Depending upon the albumin and FLA concentrations as well as on the equilibrium binding constant for the FLA binding to the protein, Ka, a significant reduction of the effective free FLA concentration in aqueous solution can be achieved. Ideally, the free FLA concentration is reduced to a value below its CAC so that the FLA form that labels the cell surface is a monomer and the labeling is homogeneously achieved.
Whereas the above rationale is very commonly used in staining of cell surfaces with FLA for fluorescence microscopy, we are not aware of any systematic study on the binding of FLA to albumin in terms of the binding parameters (equilibrium binding constants, and the binding and desorption rate constants) and the detailed energetics of the process that a study of the temperature-dependence of these parameters can provide. Measurement of the rate constants for transfer of this monomeric FLA species to a lipid bilayer membrane then provide a direct estimation of the equilibrium partition coefficient, KP(L/W), for partitioning of the FLA between the membrane and aqueous phases. This direct measurement of KP(L/W) can be performed for different lipid phases (solid, liquid-ordered, or liquid-disordered) using the same approach, thereby providing an indirect measurement of a hypothetical partition coefficient of the FLA for partitioning between any two of those lipid bilayer phases, KP(L1/L2). The general acceptance of the concept that the biological membrane is a heterogeneous chemical system (has coexisting lipid phases, of which “rafts” may be a manifestation) in which the heterogeneity may have a physiological role (for reviews see Vaz and Almeida, 1993, Simons and Ikonen, 1997, Brown and London, 1998, 2000, Vaz and Melo, 2001) makes such information of particular relevance.
In earlier work we had reported upon the insertion into and desorption off membranes, of an FLA as a monomeric species in aqueous solution (Pokorny et al., 2000, 2001). In that work the FLA used had a high CAC, so that it was possible to do the experiments at FLA concentrations below this value. In this work we have used a Rhodamine Green™ (a Rhodamine-110 derivative marketed by Molecular Probes, Inc., Eugene, Oregon, USA) derivative of tetradecylamine (RG-C14:0), an FLA with a very low critical aggregation concentration, and have utilized its binding to bovine serum albumin (BSA) as a means of reducing its concentration in the aqueous solution below the CAC. The kinetics of the association of RG-C14:0 with BSA has been examined by stopped-flow mixing as a function of temperature. This has allowed us to define the thermodynamics of its binding to BSA. Kinetics of the transfer of RG-C14:0, as a monomer in aqueous solution (obtained by having a large excess of BSA and lipid in the reaction mixture), to lipid bilayer membranes in a variety of phases (solid or gel, liquid-ordered, and liquid-disordered) were also studied using stopped-flow mixing. The insertion and desorption rate constants obtained from these experiments were used to calculate KP(L/W) for the different lipid bilayer phases and to estimate the hypothetical values of KP(L1/L2) for a coexistence of any two of these phases. This approach is proposed as a new method for obtaining information concerning the partitioning of FLA between coexisting lipid phases in heterogeneous lipid bilayers and biological membranes.
Materials and Methods
Bovine serum albumin essentially free of fatty acids (approx. 0.005%), egg yolk sphingomyelin (SpM), tetramethylrhodamine isothiocyanate (TMRITC, isomer R), and tetradecylamine were purchased from Sigma-Aldrich Química S.A., Madrid, Spain. Cholesterol was from Serva/Boehringer Ingelheim, Heidelberg, Germany. Rhodamine Green™ carboxylic acid N-hydroxysuccinimidyl ester hydrochloride “mixed isomers” was purchased from Molecular Probes Europe BV, Leiden, the Netherlands. 1-Palmitoyl-2-oleoylphosphatidylcholine (POPC) was from Avanti Polar Lipids, Inc., Alabaster, Alabama, USA. All reagents were of the highest commercially available purity. Solvents of analytical reagent grade were from Merck Portuguesa, Lisbon, Portugal.
RG-C14:0 was synthesized by addition of a chloroform/methanol (1/1, v/v) solution of tetradecylamine (10-fold molar excess over dye reagent) to a dimethylformamide solution of the dye reagent (Rhodamine Green™ carboxylic acid N-hydroxysuccinimidyl ester hydrochloride “mixed isomers”) which contained a few grains of anhydrous sodium carbonate. The reaction mixture was vortexed and allowed to stand for 24 h at room temperature after which the desired product (RG-C14:0) was isolated and purified by preparative thin layer chromatography on Silica Gel 60 plates (Merck Portuguesa) using chloroform/methanol (1/1, v/v) as eluant.
TMR-BSA was prepared by reacting BSA with fivefold molar excess of TMRITC in 0.01 M sodium bicarbonate, pH 9.5, overnight at room temperature (23°C) and separating the labeled protein (TMR-BSA) from unreacted dye reagent by gel filtration through Sephadex G-25 (Pharmacia). Protein concentrations were determined using the method of Lowry et al. (1951). TMR was determined by measuring the absorption of the SDS-denatured labeled-protein sample at 550 nm using a value of 94900 M−1cm−1 for the molar extinction coefficient. The labeled protein had a dye/protein molar ratio of 1.2.
Suspensions of RG-C14:0 in buffer were prepared by squirting a solution of the FLA in methanol (using a Hamilton syringe) into the desired volume of the buffer, with simultaneous vigorous vortexing, taking care to ensure that the final methanol concentration did not exceed 1%. These suspensions were always freshly prepared before use inasmuch as it was noticed that binding of the FLA to BSA became a very slow process when the FLA suspension was much more than 24 hours old. We attributed this observation to a “maturation” process of the FLA suspension from an initial micellar state to some more ordered aggregate (microcrystals, etc.). The maturation process could be followed by changes in the absorption spectra of the suspension.
Aqueous phospholipid suspensions were prepared by evaporating a solution of the desired lipid or lipid mixture in chloroform/methanol (1/1, v/v) solution by blowing dry nitrogen over the heated (blowing hot air onto the external surface of the tube) solution and then leaving the residue in a vacuum dessicator for at least 8 h at 23°C. The solvent-free residue, heated in a water bath at 60°C, was then hydrated with deionized water which had been previously heated to the same temperature and the mixture was left to hydrate for ∼10 min at 60°C. The amount of water added was calculated to result in a final lipid concentration of ∼10 mM. The hydrated lipid was vigorously vortexed at room temperature to produce a suspension of multilamellar vesicles which was then extruded, using a minimum of 10 passes, through two stacked polycarbonate filters (Nucleopore) with a pore diameter of 0.1 μm (Hope et al., 1985). During extrusion, the water-jacketed extruder (Lipex Biomembranes, Inc.) was maintained at a temperature that was at least 10°C higher than the transition temperature of the phospholipid with the highest phase transition temperature. Phospholipid concentrations were determined through a modified version of the Bartlett phosphate assay (Bartlett, 1959) and cholesterol concentrations were determined by the Lieberman-Burchard method as described by Taylor et al. (1978). The LUV suspensions obtained after extrusion were diluted in 0.01 M sodium phosphate, pH 7.5, to obtain the desired lipid concentration for fluorimetric stopped-flow experiments.
Absorption spectra were recorded on a Unicam UV530 UV/Vis spectrophotometer and fluorescence spectra were obtained on a Spex DM-3000-F fluorimeter. Kinetic experiments were performed on a thermostated stopped-flow fluorimeter (Hi-Tech model SF-61) by mixing equal volumes of two solutions, as required for each particular case studied. The excitation wavelength was defined by a monochromator and an appropriate band-pass (Thermo Corion) or cutoff (Schott) filter defined the emission wavelength. Data were acquired using the software supplied by Hi-Tech and curve-fitting analyses were done using Microsoft Excel® and Solver®.
Results
Equilibrium titration of RG-C14:0 with BSA
RG-C14:0 in aqueous solution forms an aggregate with a complex absorption spectrum and a very low fluorescence emission. When a large molar excess of BSA is present in the solution, the absorption spectrum changes to one that is qualitatively similar to a solution of RG (without an alkyl chain) in buffer and the fluorescence emission increases by a factor of ∼10-fold (Fig. 1). Inasmuch as RG-C14:0 has to be used as a solution in methanol (pipetting of small volumes of the aqueous dispersion proved to be difficult to do reproducibly), we resolved to titrate a fixed volume of a freshly prepared aqueous dispersion of RG-C14:0 with BSA. This is, in any case, a convenient procedure with fluorescent ligands inasmuch as fluorescence emission intensities are not directly proportional to concentration when there are appreciable inner filter effects. The change in fluorescence emission upon the binding of RG-C14:0 to BSA (increasing BSA concentrations) is shown in Fig. 1. The binding curve (insert, Fig. 1) was analyzed by assuming a single binding site on albumin, which gave us a value of the equilibrium binding constant, Ka, of 1.3 × 106. Binding curves of the type shown in Fig. 1 are difficult to analyze in terms of more than one binding constant if their values are not very different from each other. This result may not, therefore, be interpreted to indicate that there is only a single binding site on BSA for RG-C14:0 and the value of Ka obtained should only serve as a reference for what follows.
FIGURE 1.

Fluorescence emission spectra of a 5 × 10−8 M solution of RG-C14:0 in 0.01 M sodium phosphate, pH 7.7 (lowest trace), and in the presence of BSA at different concentrations (from top to bottom trace, 3.5 × 10−6, 8.6 × 10−7, and 3.3 × 10−7 M). (Insert) Titration of a 5 × 10−8 M solution of RG-C14:0 in 0.01 M sodium phosphate, pH 7.7, with BSA. The solid line is a theoretical titration curve for a single binding site on the protein with Ka = 1.3 × 106.
Stopped-flow study of the binding of RG-C14:0 to BSA
We next examined the binding of RG-C14:0 to BSA in the stopped-flow fluorimeter. To do this, a freshly prepared dispersion of RG-C14:0 was equilibrated overnight with BSA in 0.01 M sodium phosphate, pH 7.7. The final concentrations were 4.2 × 10−8 M for RG-C14:0 and 1.3 × 10−6 M for BSA. Equal volumes of this solution and a solution of TMR-BSA (1.5 × 10−6 M) were mixed in the stopped-flow apparatus and the fluorescence emission was followed over a period of 200 s with a resolution of 0.2 s. Fig. 2 shows the fluorescence emission spectra of RG-C14:0 bound to BSA and to TMR-BSA. Resonance energy transfer between the TMR-BSA-bound RG-C14:0 (acting as an energy donor) and the TMR chromophores covalently attached to the Lys residues on the protein surface (acting as energy acceptors) results in a decrease in the emission intensity of the bound RG-C14:0 and a sensitized increase in the emission intensity of the TMR chromophore. Upon mixing the RG-C14:0–BSA complex with TMR-BSA, a transfer of some of the bound RG-C14:0 from the unlabeled BSA to the TMR-labeled BSA occurs and a decrease in fluorescence of the RG-C14:0 is observed in parallel to an increase in the emission of the TMR. Either of these fluorescence changes can be monitored over time to follow the transfer of the bound ligand from one BSA population to the other. A description of the transfer can be attempted by using kinetic models for binding at a single site on the protein (Model I in the Appendix) or for binding at two independent and nonequivalent binding sites on the protein (Model II in the Appendix). Both models assume that the binding of RG-C14:0 to BSA and TMR-BSA is identical in all respects. Fig. 3 shows the time-dependent evolution of fluorescence resulting from mixing a solution of RG-C14:0 equilibrated with BSA and a solution of TMR-BSA. Two theoretical fits, one assuming that RG-C14:0 binds to a single binding site and the other assuming two independent binding sites on the protein, were attempted. It is evident from the plots of residuals, shown in Fig. 3, that the single-binding-site model does not describe the experimental curve whereas the two-binding-site model describes the experimental curve very well. From this theoretical fit we recovered the rate constants k1 (≡ k4), k−1 (≡ k−4), k2 (≡ k3), and k−2 (≡ k−3); as well as the respective equilibrium association constants Ka1 = k1/k−1 (≡ k4/k−4) and Ka2 = k2/k−2 (≡ k3/k−3).
FIGURE 2.

Fluorescence emission spectra of RG-C14:0 bound to BSA (a); to TMR-BSA (b); and to POPC bilayer vesicles as LUVs in the presence of TMR-BSA as in curves (b) and (c). The transmission characteristics of the cutoff (d) and band-pass (e) filters used in the stopped-flow experiments are also included for reference.
FIGURE 3.

Time-dependent evolution of the fluorescence intensity, measured in the stopped-flow apparatus, of a mixture of an RG-C14:0–BSA complex and TMR-BSA at 25°C and pH 7.7. The experimental trace has been fitted with two theoretical fits, one assuming a single binding site for RG-C14:0 on the protein as described in Model I (see Appendix) and the other assuming two binding sites for RG-C14:0 on the protein as described in Model II (see Appendix). For the second theoretical fit k1 = 7.55 × 105 M−1s−1, k−1 = 0.134 s−1, k2 = 5.99 × 104 M−1s−1, k−2 = 0.017 s−1, Ka1 = 5.64 × 106, and Ka2 = 3.52 × 106. The residuals are shown in the two lower panels: (A) fit to Model I with a single binding site and (B) fit to Model II with two independent binding sites.
Temperature-dependence of the association constants and the rate constants for binding of RG-C14:0 to BSA
Stopped-flow experiments were performed at several temperatures between 15 and 35°C. All experimental curves were fitted to a theoretical expression for the fluorescence intensity using the two-binding-site model (Model II of the Appendix). The results are summarized in Table 1. Arrhenius plots of the rate constants are shown in Fig. 4, A and B. Activation energies for the binding and desorption processes, calculated from the slopes of the respective plots, for each of the two binding sites are given in Table 2. The binding processes show very low activation energy (between 1 and 2 kJ × mol−1) and the dissociation processes are characterized by activation energies on the order of 46–49 kJ × mol−1. From the rate constants obtained at different temperatures the equilibrium association constants at these temperatures may be computed. These data can be plotted as van't Hoff plots (Fig. 5), from the slope of which we obtain the enthalpies for the association processes at both binding sites. Given the free energy change (calculated from the equilibrium association constants) and the enthalpy change (obtained from the van't Hoff plots), we may define all the thermodynamic parameters for the binding processes as in Table 3. Surprisingly, the process of binding is enthalpically driven, contrary to what might be expected for a process that is driven by the “hydrophobic effect.”
TABLE 1.
Temperature-dependence of the rate constants and equilibrium binding constants for the association of RG-C14:0 to BSA at two binding sites
| 15°C | 20°C | 25°C | 30°C | 35°C | |
|---|---|---|---|---|---|
| k1, M−1 × s−1 | 7.53 × 105 | 7.54 × 105 | 7.55 × 105 | 7.61 × 105 | 7.65 × 105 |
| k−1, s−1 | 7.08 × 10−2 | 9.14 × 10−2 | 1.30 × 10−1 | 1.66 × 10−1 | 2.78 × 10−1 |
| k2, M−1 × s−1 | 5.86 × 104 | 5.90 × 104 | 6.05 × 104 | 6.07 × 104 | 6.12 × 104 |
| k−2, s−1 | 8.58 × 10−3 | 1.50 × 10−2 | 1.66 × 10−2 | 2.29 × 10−2 | 3.29 × 10−2 |
| Ka1 | 1.06 × 107 | 8.25 × 106 | 5.82 × 106 | 4.59 × 106 | 2.75 × 106 |
| Ka2 | 6.83 × 106 | 3.93 × 106 | 3.65 × 106 | 2.66 × 106 | 1.86 × 106 |
FIGURE 4.

Arrhenius plots for the association rate constants (A), and dissociation rate constants (B), in the binding of RG-C14:0 to BSA. The Arrhenius activation energy, Eact, for k1 is 0.6 kJ × mol−1, for k2 is 1.7 kJ × mol−1, for k−1 is 49 kJ × mol−1, and for k−2 is 46 kJ × mol−1.
TABLE 2.
Activation energies for the association and dissociation of RG-C14:0 at its two binding sites on BSA
| Activation energy (Eact), kJ × mol−1
|
||
|---|---|---|
| Association | Dissociation | |
| Binding Site 1 | 0.6 | 49.1 |
| Binding Site 2 | 1.7 | 45.9 |
FIGURE 5.

van't Hoff plots for the binding of RG-C14:0 to the BSA binding sites. The van't Hoff enthalpies obtained from the slopes of these plots are −48.6 kJ mol−1 and −44.2 kJ mol−1, respectively.
TABLE 3.
Thermodynamic parameters for the binding of RG-C14:0 to the two BSA binding sites
| ΔG°(25°C), kJ × mol−1 | ΔH°, kJ × mol−1 | TΔS°(25°C), kJ × mol−1 | |
|---|---|---|---|
| Binding Site 1 | −38.6 | −48.6 | −9.9 |
| Binding Site 2 | −37.5 | −44.2 | −6.8 |
Transfer of the FLA from the protein-bound to the membrane-associated state
We now consider the use of BSA as a vehicle for transport of FLA to membrane surfaces. This is a widely used method in cell surface biology that has its origins, as far as we can make it out, in the work of Lipsky and Pagano (1985). It allows cell-surface labeling with FLA at relatively high concentrations of the latter without danger of the labeling occurring via a fusion of FLA aggregates with the membrane. The concept is simply the elimination of FLA aggregation in aqueous solution by association with the protein which, in consideration of the binding constants involved, is supplied at a concentration that is high enough to reduce the free FLA concentration to a value below its CAC. As far as we are aware, this widely used method has never been characterized kinetically or thermodynamically, so its use is still a matter of an experimental recipe.
For a membrane in contact with an aqueous solution in which we have an FLA monomer in equilibrium with a protein-bound state, we can imagine two reaction paths for the insertion of the FLA into the membrane. 1) Insertion occurs via encounter of an FLA monomer, in aqueous solution, with the membrane surface. In this case the only relevant species is the monomer in aqueous solution. As membrane association depletes the available monomer, fresh monomer from the protein-bound state becomes available in the aqueous phase. In practice, this process will continue up to the limit dictated by the values of KP(L/W) and Ka. 2) Insertion occurs via encounter of the protein–FLA complex with the membrane surface and a simultaneous exchange of the FLA between the protein and the membrane. In this case the most important species is the protein–FLA complex in solution. The two processes have different mechanisms and, consequently, different time-dependence.
Model III in the Appendix describes the kinetics of the FLA-transfer process between the binding protein and the membrane surface via monomer in the aqueous phase. Model IV in the Appendix describes the kinetics of the FLA transfer via a collisional exchange of the FLA between the protein–FLA complex and the membrane surface. In both models we have considered the two protein binding sites to be independent of and equivalent to each other for the sake of simplicity (see below). In these experiments the FLA is originally bound to TMR-BSA so that its fluorescence is significantly quenched. Its transfer to a membrane surface results in an increase in fluorescence emission intensity as seen in Fig. 2. This increase in fluorescence intensity is used in the stopped-flow apparatus to evaluate the kinetic parameters for association of the FLA with the membrane (Model III or IV, Appendix) given that the kinetic parameters for the protein association are independently known. In both models we have imposed the condition that k2 ≤ kB ≤ k1, k−2 ≤ k−B ≤ k−1, and Ka2 ≤ Ka ≤ Ka1, where k1, k−1, k2, k−2, Ka1, and Ka2 are the independently measured values (as described earlier). From the plots of residuals shown in Fig. 6 it becomes evident that Model III (namely, insertion through diffusional interaction of an aqueous phase FLA monomer) describes the experimental result more accurately.
FIGURE 6.

An experimental discrimination between two models for insertion of RG-C14:0 into POPC/cholesterol (6/4; liquid-ordered phase) bilayer membranes. The experimental trace was fitted to two models: 1) a model (Model III, Appendix) in which insertion into the membrane occurs via diffusional contact between the membrane and an aqueous phase FLA monomer and 2) a model (Model IV, Appendix) in which insertion into the membrane occurs via diffusional contact between the membrane and a protein–FLA complex. The lower panels in this figure show the residuals for the first fit (A) and for the second fit (B).
Six lipid bilayer model systems (as LUVs) were investigated, namely, one solid phase (pure sphingomyelin at a temperature below its phase transition temperature), two liquid-disordered phases (pure DMPC and pure POPC at temperatures well above their phase transition temperatures), and three liquid-ordered phases (binary mixtures of sphingomyelin and cholesterol, of POPC and cholesterol, and of DMPC and cholesterol, all at a molar ratio of 6/4). The liquid-ordered phase formed from binary mixtures of sphingomyelin and cholesterol is of particular interest. “Rafts,” proposed lipid inhomogeneities in the plasma membrane of eukaryotic cells (Simons and Ikonen, 1997, Brown and London, 1998), are supposedly rich in sphingolipids and cholesterol. If this is indeed the case, the rest of the membrane outer leaflet (in which almost all the sphingolipids are localized) could be more similar in chemical composition and physical properties to the bilayers formed from the POPC–cholesterol mixture or from pure POPC. The fluorescence increase with time, upon insertion of RG-C14:0 into the membranes, is shown in Fig. 7, A–F, together with the respective theoretical curves based upon the assumptions of Model III (Appendix). It will be noted that most of the fits of the theoretical curves to the experimental data are quite good with the exception of the fits in Fig. 7 A (pure sphingomyelin, solid phase) and Fig. 7 E (DMPC/cholesterol (6/4), liquid-ordered phase). In the case of the pure sphingomyelin solid phase, insertion of RG-C14:0 into the lipid bilayer probably occurs at grain-boundary and point defects. These could provide a multiplicity of nonequivalent “reaction sites” that may explain the poor fits. In fact, the relatively high insertion rate constant for the solid phase sphingomyelin bilayers probably does not reflect insertion into a true solid phase but rather into packing defects in this phase (see also Pokorny et al., 2000). Coexistence of two liquid-ordered phases in the DMPC/cholesterol bilayers at 30°C, analogous to the coexistence of an loα and an loβ phase in DPPC/cholesterol bilayers (McMullen et al., 1995), would mean that the insertion process, in this case, would have to include two lipid phases and would not be correctly described by <Model III (Appendix). This may be the reason for the rather poor fit seen for this lipid mixture in Fig. 7 E. In any case, we do not see any reason to suppose that the assumption of a single binding site on the protein in the theoretical model poses any significant problem in the description of the experimental result. From the fits shown in Fig. 7 we are able to recover the insertion (k+) and desorption (k−) rate constants for the partitioning of RG-C14:0 between the membrane and the aqueous phases. These values are listed in Table 4. As might be expected, the insertion rate constants are highest, and similar, for the two liquid-disordered phases examined. The insertion rate constant is roughly three orders of magnitude lower for insertion into liquid-ordered phases or the SpM solid phase. Among the liquid-ordered phases, insertion is slowest in SpM/cholesterol and the fastest in POPC/cholesterol bilayers (k+ is about a factor of 4 times higher in the latter membranes). Desorption rate constants are similar for the two liquid-disordered phases but considerably higher (by a factor of between 3.5 and 6 times) in the liquid-ordered phases, being roughly the same for desorption from SpM/cholesterol and POPC/cholesterol liquid-ordered phases. Desorption from a solid phase is extremely slow: ∼27 times slower than from a liquid-disordered membrane and between 90 and 160 times slower than desorption from a liquid-ordered phase.
FIGURE 7.

Time-dependent evolution of the fluorescence of RG-C14:0 as a consequence of its transfer from a TMR-BSA bound state to a lipid membrane via the monomeric form in the aqueous phase. Experimental data are presented, at 30°C, for (A), pure sphingomyelin LUVs in the solid phase; (B), pure DMPC LUVs in the liquid-disordered phase; (C), pure POPC LUVs in the liquid-disordered phase; (D), a binary mixture of sphingomyelin and cholesterol (6/4 molar ratio) in the liquid-ordered phase; (E), a binary mixture of DMPC and cholesterol (6/4 molar ratio) in the liquid-ordered phase; and (F), a binary mixture of POPC and cholesterol (6/4 molar ratio) in the liquid-ordered phase. The concentration of lipid was 30 μM for the pure lipids and 100 μM for the binary mixtures. The RG-C14:0 concentration was 5 × 10−8 M and the TMR-BSA concentration was 1.3 × 10−6 M in all cases.
TABLE 4.
Rate constants and equilibrium partition coefficients for the partitioning of RG-C14:0 between lipid membrane phases and the aqueous phase at 30°C
| k+, M−1 × s−1 | k−, s−1 | KP(L/W)† | |
|---|---|---|---|
| Sphingomyelin (solid) | 3.45 × 105 | 0.0075 | 5.76 × 107 |
| DMPC (liquid-disordered) | 5.41 × 108 | 0.2167 | 3.01 × 109 |
| POPC (liquid-disordered) | 6.78 × 108 | 0.2023 | 4.21 × 109 |
| Sphingomyelin/cholesterol (6/4) (liquid-ordered) | 2.77 × 105 | 0.7014 | 4.96 × 105 |
| DMPC/cholesterol (6/4) (liquid-ordered) | 4.80 × 105 | 1.204 | 5.02 × 105 |
| POPC/cholesterol (6/4) (liquid-ordered) | 1.15 × 106 | 0.7486 | 1.93 × 106 |
The molar volume, 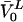 , was assumed to be 0.795 L × mol−1 (Wiener and White, 1992) for all the membranes used in this work.
, was assumed to be 0.795 L × mol−1 (Wiener and White, 1992) for all the membranes used in this work.
Inasmuch as KP(L/W) is obtained independently for each lipid phase, the hypothetical partition coefficient for partitioning of this FLA between any two of these lipid phases if they coexist (including in the same membrane), can be written as KP(L1/L2) = KP(L1/W)/KP(L2/W). As far as we are aware, this is a novel utilization of this method. The results are summarized in Table 5. From these results it is evident that RG-C14:0 prefers the more disordered phases: KP(L1/L2) is on the order of 10−4 for partitioning between a liquid-ordered phase and a liquid-disordered phase made from the same phospholipid, and has a fourfold preference for the POPC/cholesterol liquid-disordered phase compared to the SpM/cholesterol liquid-ordered phase. In utilizing this method for determination of KP(L1/L2), caution should be exercised with regard to interpretations concerning the chemical composition and physical properties of a phase (or domains) in biological membranes. It is highly improbable that a pure SpM/cholesterol or POPC/cholesterol phase exists in natural membranes. Also, solid phase domains, if they do exist in the biological system, probably exist as mesoscopic domains of solid-phase lipid in which grain boundary and point defects are unlikely. As stated earlier, the partitioning into a pure solid phase bilayer (as is the case for sphingomyelin bilayers in Table 4) is probably a phenomenon related exclusively to the existence of these defects.
TABLE 5.
Hypothetical equilibrium partition coefficients, KP(L1/L2), for partitioning of RG-C14:0 between two hypothetically coexisting lipid phases at 30°C
| Hypothetically coexisting lipid phases | KP(L1/L2) |
|---|---|
| [DMPC] / [POPC] [liquid-disordered] / [liquid-disordered] | 0.71 |
| [DMPC/cholesterol (6/4)] / [DMPC] [liquid-ordered] / [liquid-disordered] | 1.7 × 10−4 |
| [POPC/cholesterol (6/4)] / [POPC] [liquid-ordered] / [liquid-disordered] | 4.6 × 10−4 |
| [Sphingomyelin/cholesterol (6/4)] / [POPC] [liquid-ordered] / [liquid-disordered] | 1.2 × 10−4 |
| [Sphingomyelin/cholesterol (6/4)] / [POPC/cholesterol (6/4)] [liquid-ordered] / [liquid-ordered] | 0.26 |
Discussion
The present work provides a detailed kinetic and thermodynamic characterization of the binding of FLA to bovine serum albumin and of the subsequent transfer of the protein-bound amphiphile to a lipid bilayer membrane. The motivation for this work arises from our interest in devising quantitatively understood methods for transfer of FLA to membrane surfaces and for the determination of the partitioning behavior of FLA once these are incorporated into membranes with coexisting phases. BSA seems to be used routinely in most laboratories of cell surface biology in protocols for plasma membrane labeling with FLA following the initial suggestion of Pagano and co-workers (Lipsky and Pagano, 1985, Pagano and Martin, 1988). A rigorous quantitative understanding of this commonly used methodology in cell biology can only serve to make its use more predictable. We are not aware of any detailed kinetic and thermodynamic characterization of the process and have, therefore, attempted to fill this gap. In addition, we have extended the use of this method to recover rate constants for FLA insertion into and desorption off membranes. From these, it is possible to directly obtain the values of lipid phase/aqueous phase partition coefficients, KP(L/W), for partitioning of the FLA between these two phases, and the activation energies involved in the insertion/desorption processes. Kinetic studies on the transfer of fatty acids and bilirubin between serum albumins and membranes have been reported by several laboratories over the years (Daniels et al., 1985; Noy et al., 1986; Leonard et al., 1989; Pownall et al., 1991; Zucker et al., 1995; Massey et al., 1997; Zakim, 2000; Pownall, 2001; Zucker, 2001). Other laboratories (Storch and Bass, 1990, Kim and Storch, 1992a, 1992b, Wootan and Storch, 1994, Richieri et al., 1994, 1995, 1996) have studied the transfer of fatty acids and their fluorescent derivatives from fatty acid binding proteins to membranes. The kinetic models presented in some of this work are very similar, if not identical, to the kinetic models presented by us here. The major difference between the present work and the work from other laboratories lies in the analysis of the kinetic data. Our present analysis of the experimental kinetic data permits us to directly obtain the forward and reverse reaction rate constants for each of the individual steps in the kinetic models without any previous assumptions. Previous work from our laboratory on this theme has attempted to characterize the kinetics and thermodynamics of FLA insertion into lipid bilayers (Pokorny et al., 2000, 2001) and the partitioning of amphiphiles between coexisting lipid phases (Mesquita et al., 2000). We now use the kinetics of transfer of FLA between the FLA-binding protein (bovine serum albumin) and different membranes to obtain a hypothetical partition coefficient, KP(L1/L2), for partitioning of the FLA between those membrane phases if they were in coexistence in the same membrane. As far as we are aware, this is the first time that this approach is being used to gather this useful information (for a recent review, see Vaz and Melo, 2001).
In principle, KP(L/W) may be determined by equilibrium measurements of transfer of the FLA between the protein-bound and membrane-associated states as long as Ka for binding of the FLA to the protein is known, or through a knowledge of the insertion and desorption rate constants, as has been done in this work. Both approaches have their advantages and limitations. The advantage of the equilibrium approach is that it is simple to perform and analyze. Its main limitation lies in the range of KP(L/W) values that can be reliably measured. For KP(L/W) determinations to be of significance, the FLA/lipid ratio in the bilayers should not exceed the limit where the FLA ceases to be an “impurity” and begins to act as a chemical constituent of the system. In general experience, this ratio should not exceed ∼1% and should, by preference, be considerably less. Exceedingly high KP(L/W) values, that may be expected from FLA that are derived from lipids, and are therefore perhaps the most interesting ones, would lead to an almost complete partitioning of the FLA into the lipid phase so that its concentration in the aqueous phase (including that which is bound to the protein) would be difficult, if not impossible, to reliably measure. This difficulty is shown in Fig. 8 where we have assumed Ka = 1 × 106 and a protein concentration of 1 × 10−4 M (∼6.7 mg/mL for BSA). Lower protein con-centrations or lower values of Ka lower the maximum limits of KP(L/W) that can be reliably determined. For the case shown in Fig. 8, KP(L/W) ≈ 107 could be reliably measured using the equilibrium approach. In a kinetic analysis, on the other hand, if we assume that the insertion rate constant is diffusion-limited (k+ ≈ 1010 M−1s−1), that the desorption rate constants, k− , lie between the extremes listed in Table 4, and that the lipid concentration is of the order of 1 × 10−3 M, the characteristic kinetic curves shown in Fig. 7 would have half-times on the order of ∼20 s. Given the time-resolution of our stopped-flow apparatus (1 ms) this curve would be quite reliably measurable and its half-time could be further reduced, if necessary, if lipid concentration were lowered. In effect, there is no upper limit for the experimental determination of KP(L/W) using the kinetic approach described here. In this approach, however, values of KP(L/W) ≪ Ka would imply the use of impractically high lipid concentrations. However, it is also probable that FLA with very low KP(L/W) values aggregate in aqueous solutions at reasonably high CAC so that KP(L/W) may be directly determined without the necessity of using an FLA-binding protein, as we have done elsewhere (Pokorny et al., 2000). An added advantage of the kinetic approach used by us is that the insertion and desorption rate constants may be studied at varying temperatures so as to provide activation energies for these processes if desired. This information can, in principle, only be obtained from a kinetic study.
FIGURE 8.

Equilibrium concentrations of FLA in the lipid phase, for a protein concentration of 1 × 10−4 M (6.7 mg/mL, in the case of BSA) and Ka = 1 × 106, as a function of the lipid concentration. (A) Fraction of total FLA in the lipid phase and (B) loading of the lipid phase with FLA. For both A and B, the values of KP(L/W) assumed were: 105 (curve 1), 106 (curve 2), 107 (curve 3), 108 (curve 4), 109 (curve 5), and 1010 (curve 6).
Serum albumins are known to bind amphiphiles of various types, in particular, fatty acids, but also various drugs and anesthetics (Peters, 1997). The binding sites for fatty acids, anesthetics, and drugs have been characterized by high-resolution x-ray diffraction structures in the case of human serum albumin (He and Carter, 1992, Curry et al., 1998, Bhattacharya et al., 2000a, 2000b, Petitpas et al., 2001). There seems to be some degree of promiscuity with regard to ligand binding at least in some of these sites (Bhattacharya et al., 2000b). Considering the molecular structure of RG-C14:0 it could be expected that this FLA would bind either to a typical fatty acid binding site or to a typical drug-binding site on BSA. We tested the second hypothesis by trying to displace the bound RG-C14:0 with several drugs (W.L.C. Vaz, unpublished results) that are known to bind with considerable affinity primarily to one (and in some cases, both) of two well-described binding sites on the protein (Sudlow et al., 1975, Peters, 1997). The displacement results indicated an association constant of these drugs to the site or sites at which RG-C14:0 was bound on the order of 102 to 103, which is considerably less than the binding constants (on the order of 105) known for the binding of these drugs to the well-characterized drug-binding sites. We therefore conclude that RG-C14:0 does not bind to either of the known drug-binding sites on albumin. Alternative binding sites for this FLA on albumin are the high affinity fatty acid binding sites (Curry et al., 1998). The equilibrium association constants obtained by us in this work for the binding of RG-C14:0 to BSA are compatible with some of the association constants for myristic (Curry et al., 1998, Spector, 1975) and parinaric acids (Sklar et al., 1977). We conclude that RG-C14:0 probably binds to fatty acid binding sites on BSA and are presently studying the competitive displacement of RG-C14:0 from its binding sites on BSA by myristate. This will be the subject of a future communication. The molecular structural prerequisites (alkyl chain and terminal negative charge) for binding to the fatty-acid binding sites are available in RG-C14:0. The kinetic analysis that we have used clearly indicates that there are at least two binding sites (Fig. 3). Any further refinement of this type of analysis in terms of increasing the number of possible binding sites would be quite complex (see Model II, Appendix) without necessarily improving our understanding of the system for purposes of the present work and was, therefore, not attempted.
From the results presented in Table 3, it is apparent that the driving force for the binding of RG-C14:0 to BSA is of an enthalpic nature. There is, in fact, an apparent decrease in the entropy of the system upon binding of the FLA to albumin. The association of amphiphiles, like RG-C14:0, or fatty acids with proteins is generally understood to be due to the “hydrophobic effect” and the driving force for this process is generally understood to be entropic (Tanford, 1991). A related study on the binding of fatty acids to cytosolic fatty acid binding proteins (Richieri et al., 1994, 1995, 1996) also indicated that the major contribution to the free energy of binding was enthalpic. We present two possible explanations for the lack of an entropic contribution to the free energy of binding in our results. 1) A decrease in entropy of the system resulting from the reduction of conformational states of the FLA in water compared to its protein-bound state could offset the entropy gain resulting from a breakdown of the aqueous cage around the apolar molecule upon binding to the protein. 2) Our experiment may be blind to a diffusion-limited rapid step that involves the actual transfer of the FLA from the aqueous phase to the protein surface. This possibility is discussed further below.
A noteworthy result is the low values of activation energy in the binding of the FLA to the albumin binding sites (Fig. 4 A). A simple explanation for this observation could be as follows: binding of the FLA to the albumin binding sites is a two-step process of the type:
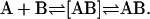 |
In this scheme [AB] is an intermediate product whose formation/dissociation is diffusion-limited and, therefore, too fast to be observed by us. It could be a nonspecific asso-ciation of the aqueous-phase FLA monomer with the protein surface, the complex then undergoing a slow (rate-limiting) reaction in which the FLA associates with its final binding site. This second reaction rate would be the only one we are able to see and the one for which the rate constants are reported in Table 4.
The rather high negative binding enthalpy observed experimentally also requires to be understood. One possible explanation is a dipole–dipole interaction between the RG-C14:0 dipole (μ = 15.7 D, Estronca et al., 2002) and a protein dipole resulting from helical segments in the proximity of the binding site. Perhaps the same type of dipole–dipole interaction may also be invoked in the case of fatty acid binding to fatty acid binding proteins.
The next step in this study was analysis of the kinetics of transfer of the protein-bound FLA to different homogeneous lipid phases (solid, liquid-ordered, and liquid-disordered). In the first place, we established that transfer of FLA to the membranes occurred via the FLA monomer in the aqueous phase and not through collisional transfer of the FLA–protein complex and the membrane surface (Fig. 6). This has also been shown to be the case for transfer of bilirubin between albumin and small unilamellar vesicles (Zucker et al., 1995). This mechanism of transfer of the FLA to the membrane from its protein-bound state allowed us to measure the rate constants for FLA insertion into and desorption off the membrane surface unambiguously. The results obtained are similar to our previous results on FLA insertion into membranes in which the FLA was used at concentrations below its CAC (Pokorny et al., 2000, 2001). If the membrane is treated as a solvent phase, these rate constants can be used to calculate an equilibrium partition coefficient, KP(L/W), for partitioning of the FLA between the membrane and the aqueous phases. The ratio of KP(L/W) values between two lipid-water systems provides an indirect estimation of the partition coefficient of the FLA between those two lipid phases, KP(L1/L2), if they coexisted in the same membrane. From the results summarized in Table 5 we note that RG-C14:0 would appear to prefer the more dis-ordered phases. This result agrees with the solubility of this amphiphile in different lipid bilayer membrane phases (Estronca et al., 2002). Considering the interest in cell membrane heterogeneity manifested in the current literature (Simons and Ikonen, 1997; Brown and London, 1998), we obtained KP(L1/L2) values for RG-C14:0 partitioning between sphingomyelin-cholesterol (6/4 molar ratio) and POPC-cholesterol (6/4 molar ratio) liquid-ordered phases and between a sphingomyelin-cholesterol (6/4 molar ratio) liquid-ordered phase and a pure POPC liquid-disordered phase (see Table 5). If “rafts” are indeed sphingolipid-cholesterol-rich membrane domains, the sphingomyelin-cholesterol (6/4) bilayer phase may be expected to simulate the physical properties of a “raft” domain in a cell plasma membrane outer leaflet. The rest of that leaflet may be expected to have properties that are similar to or intermediate between those of a POPC-cholesterol (6/4) bilayer and a pure POPC bilayer.
In general terms, the present work reports a new method for the determination of membrane phase/aqueous phase partition coefficients for substances that form molecular aggregates in water. The prerequisite is that these substances should also bind to proteins with a reasonably high affinity so that their aqueous phase concentrations can be reduced to values below their critical aggregation concentrations. The method has been specifically applied to amphiphiles but is, in principle, equally applicable to the study of partitioning of drugs and other molecules such as porphyrins between the aqueous phase and membranes. The principle of the method reported here does not differ significantly from previous attempts, notably that of Feigenson (1997), to study similar phenomena. The present method has the advantage that, as long as the dispersing agent (in our case, albumin) does not interact with the membranes, the only phenomenon of con-cern is the partitioning process between the aqueous phase and the membrane. We are presently extending this approach to the study of partitioning of other amphiphiles between the membrane and aqueous phases and between (hypothetically) coexisting membrane phases.
Acknowledgments
This work was supported in part by projects funded by the Portuguese Ministry for Science and Technology (Fundação para a Ciência e a Tecnologia) through the Praxis and Sapiens programs. Magda Abreu and Luís Estronca acknowledge support in the form of stipends for initiation into scientific research (BIC) from the Fundação para a Ciência e a Tecnologia.
APPENDIX
Model I
We consider the association of an FLA to BSA as occurring at a single binding site (see text). In this case the experimental system in the stopped-flow experiment may be defined by the following two equilibria:
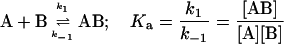 |
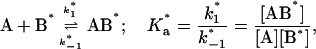 |
where A, B, and B* are the FLA, BSA, and TMR-BSA, respectively. It is understood that [A], [B], [B*], [AB], and [AB*] are the equilibrium concentrations of the respective entities. If [B] and [B*] are in a large molar excess compared to [A] the system becomes pseudo-first order so that k1[B] and k1*[B*] are constants. It is assumed that k1 = k*1 and k−1 = k*−1; i.e., labeling of BSA with TMRITC does not alter its affinities for the FLA.
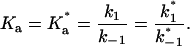 |
The following system of differential equations describe the kinetics of the model:
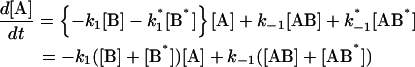 |
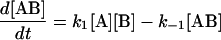 |
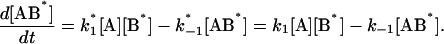 |
The above system of differential equations was resolved using matrix algebra with the help of MapleV®-Release 4.0 (Gutfreund, 1995). The general solution for the temporal evolution of each species can be written in the form:
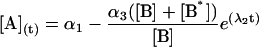 |
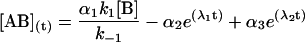 |
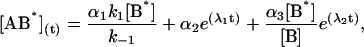 |
where αi (i = 1..3) are the amplitudes and are explicit functions of the initial conditions of the experiment (concentrations at t = 0) and the rate constants. λi are the eigenvalues of the matrix and have the following values:
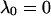 |
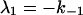 |
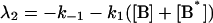 |
The temporal evolution of the measured fluorescence intensity F(t), using the cutoff filter (see Fig. 2) is a function of the initial concentrations and of the rate constants:
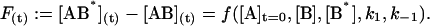 |
Model II
Here we consider the simultaneous association of an FLA to BSA and TMR-BSA as occurring at two independent binding sites in each case (see text). In this case, the experimental system may be defined by the following four equilibria:
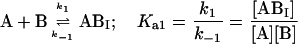 |
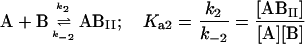 |
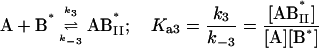 |
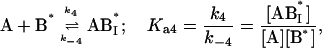 |
where the terms A, B, and B* have the same meanings as in Model I and it is understood that [A], [B], and [B*] are the equilibrium concentrations of the respective entities. [ABI] and [AB*I] are the equilibrium concentrations of FLA bound to the first protein binding site, and [ABII] and [AB*II] are the concentrations of FLA bound to the second protein binding site in BSA and TMR-BSA, respectively. If [B] and [B*] are in a large molar excess compared to [A] the system becomes pseudo-first order so that k1[B], k2[B], k3[B*], and k4[B*] are constants. A result of having the protein in large molar excess compared to the FLA also makes the probability of a single protein molecule having both binding sites occupied by the FLA negligibly small. As in Model I, it is further assumed that k1 = k4, k−1 = k−4, k2 = k3, and k−2 = k−3; i.e., that the labeling of BSA with TMRITC does not change its affinities for the FLA. We may, therefore, write:
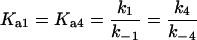 |
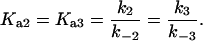 |
The following system of differential equations describes the kinetics of the model:
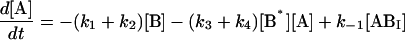 |
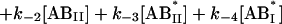 |
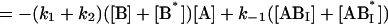 |
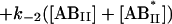 |
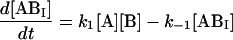 |
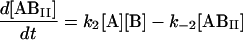 |
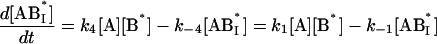 |
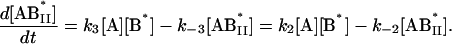 |
The general solution for the temporal evolution of each species can be written in the form:
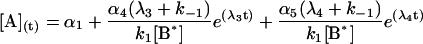 |
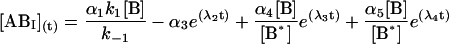 |
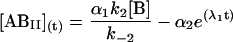 |
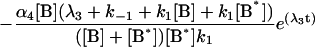 |
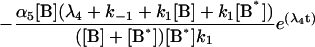 |
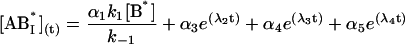 |
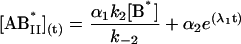 |
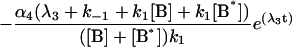 |
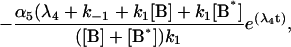 |
where αi (i = 1..5) and λi have the same significance as in Model I, and λi have the following values:
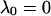 |
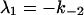 |
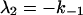 |
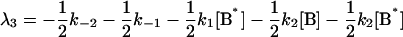 |
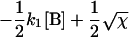 |
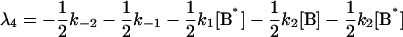 |
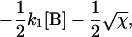 |
where
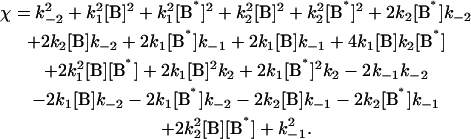 |
The temporal evolution of the 0measured fluorescence intensity F(t), is a function of the initial concentrations and of the rate constants and has the following form:
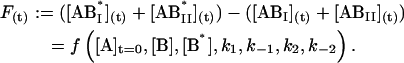 |
Model III
In this model we consider the simultaneous equilibria of a monomeric FLA species in the aqueous phase between its binding site(s) on a protein and its insertion into a lipid bilayer membrane that is present in the mixture. The equilibrium between the FLA in the aqueous phase and the membrane phase can be considered a simple process of partitioning between the two phases. The protein is TMR-BSA (B*). In the case of the association of RG-C14:0 with TMR-BSA, the protein has two binding sites for the FLA (see text), so that under conditions where the protein concentration is very much higher than the FLA concentration, double binding of the FLA to the protein can be ignored and the system may be defined by three equilibria that describe the kinetics of the FLA-transfer process between the binding protein and the membrane surface via monomer in the aqueous phase:
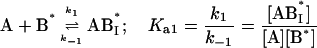 |
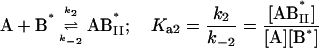 |
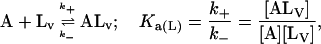 |
where [A] and [ALV] are the equilibrium concentrations of the aqueous FLA and FLA in the lipid bilayer, respectively, [LV] is the concentration of lipid vesicles, which, assuming 105 lipid molecules per vesicle, can be written as [LV] = [L] × 10−5. k+ and k− are the rate constants for the FLA insertion into and desorption off the membrane, and Ka(L) represents the equilibrium association constant for the association of the FLA with the lipid bilayer vesicles. It is related to the equilibrium partition coefficient, KP(L/W), for partitioning of the FLA between the lipid and the aqueous phases (Pokorny et al., 2002) by the expression:
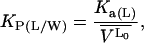 |
where 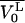 is the molar volume of the lipid. For the sake of simplicity we have assumed the two binding sites on the protein to be independent and equivalent so that the system may now be defined by just two equilibria:
is the molar volume of the lipid. For the sake of simplicity we have assumed the two binding sites on the protein to be independent and equivalent so that the system may now be defined by just two equilibria:
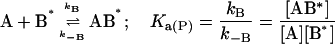 |
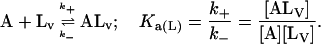 |
The condition imposed in the analysis was that k2 ≤ kB ≤ k1, k−2 ≤ k−B ≤ k−1, and Ka2 ≤ Ka(P) ≤ Ka1, where k1, k−1, k2, k−2, Ka1, and Ka2 are the independently experimentally measured values (see text). In the second equilibrium above, ALV and LV function in an analogous manner with respect to the association of A with the lipid vesicles so that, effectively, [LV] does not change in the course of the reaction. Inasmuch as [B*] is in a large molar excess compared to [A] and [LV] remains constant, both equilibria become pseudo-first order so that kB[B*] and k+[LV] are constants.
The following system of differential equations describes the kinetics of the model:
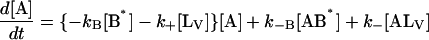 |
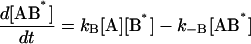 |
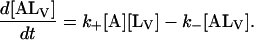 |
The general solution for the temporal evolution of each species can be written in the form:
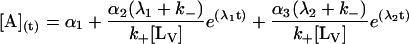 |
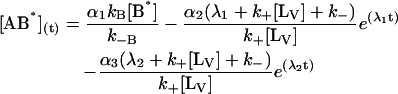 |
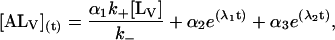 |
where αi (i = 1..3) are the amplitudes and are explicit functions of the initial conditions of the experiment (concentrations at t = 0 s) and the rate constants.
λi are the eigenvalues of the matrix and have the following values:
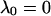 |
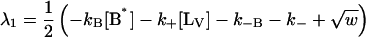 |
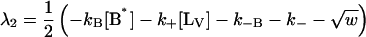 |
with
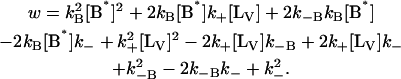 |
The temporal evolution of the measured fluorescence intensity F(t), is a function of the initial concentrations and of the rate constants:
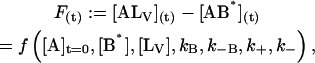 |
k+ and k− are the only fitting parameters.
Model IV
This model describes the kinetics of the FLA transfer via a collision between protein–FLA complex and the membrane surface. Once again, for the sake of simplicity we have assumed the two binding sites on the protein to be independent and equivalent so that the system may now be defined by just two equilibria:
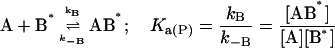 |
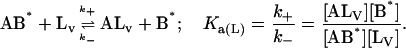 |
The same conditions as in Model III are imposed in the analysis and the following system of differential equations describes the kinetics of the model:
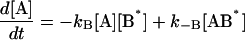 |
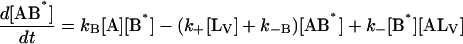 |
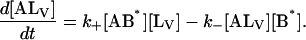 |
The general solution for the temporal evolution of each species can be written in the form:
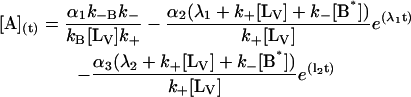 |
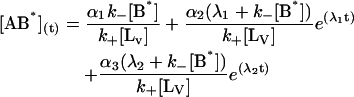 |
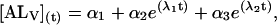 |
where αi (i = 1..3) are the amplitudes and are explicit functions of the initial conditions of the experiment (concentrations at t = 0 s) and the rate constants. λi are the eigenvalues of the matrix and have the following values:
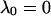 |
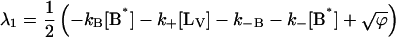 |
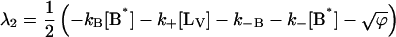 |
with
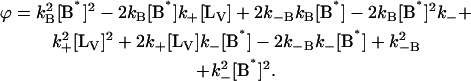 |
The temporal evolution of the measured fluorescence intensity F(t), is a function of the initial concentrations and of the rate constants:
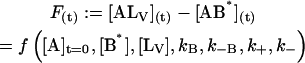 |
k+ and k− are, again, the only fitting parameters.
Abbreviations used: BSA, bovine serum albumin; CAC, critical aggregation concentration, used here synonymously with solubility product or critical micelle concentration; FLA, fluorescent lipid amphiphile(s); Ka, equilibrium binding constant for FLA to protein; KP(L/W), equilibrium partition coefficient for partitioning of FLA between a membrane and aqueous phase; KP(L1/L2), equilibrium partition coefficient for partitioning of FLA between two lipid phases; LUV, large unilamellar vesicles with an average diameter of 0.1 μm; POPC, 1-palmitoyl-2-oleoylphosphatidyl choline; RG-C14:0, Rhodamine Green™-carboxylic acid tetradecylamide; SpM, Egg yolk sphingomyelin; TMRITC, Tetramethylrhodamine isothiocyanate (isomer R); TMR-BSA, BSA labeled covalently with TMRITC at an average molar labeling ratio of 1.
References
- Bartlett, G. R. 1959. Phosphorous assay in column chromatography. J. Biol. Chem. 234:466–468. [PubMed] [Google Scholar]
- Bhattacharya, A. A., T. Grüne, and S. Curry. 2000a. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 303:721–732. [DOI] [PubMed] [Google Scholar]
- Bhattacharya, A. A., S. Curry, and N. P. Franks. 2000b. Binding of the general anesthetics Propofol and Halothane to human serum albumin. J. Biol. Chem. 275:38731–38738. [DOI] [PubMed] [Google Scholar]
- Brown, D. A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- Brown, D. A., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- Curry, S., H. Mandelkow, P. Brick, and N. Francks. 1998. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 5:827–835. [DOI] [PubMed] [Google Scholar]
- Daniels, C., N. Noy, and D. Zakim. 1985. Rates of hydration of fatty acids bound to unilamellar vesicles of phosphatidylcholine or to albumin. Biochemistry. 24:3286–3292. [DOI] [PubMed] [Google Scholar]
- Estronca, L. M. B. B., M. J. Moreno, M. S. C. Abreu, E. Melo, and W. L. C. Vaz. 2002. Solubility of amphiphiles in membranes: influence of phase properties and amphiphile head group. Biochem. Biophys. Res. Commun. 296:596–603. [DOI] [PubMed] [Google Scholar]
- Feigenson, G. W. 1997. Partitioning of a fluorescent phospholipid between fluid bilayers: dependence on host lipid acyl chains. Biophys. J. 73:3112–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutfreund, H. 1995. Kinetics for the Life Sciences: Receptors, Transmitters and Catalysts.Cambridge University Press, Cambridge, UK.
- He, X. M., and D. C. Carter. 1992. Atomic structure and chemistry of human serum albumin. Nature. 358:209–215. [DOI] [PubMed] [Google Scholar]
- Hope, M. J., M. B. Bally, G. Webb, and P. R. Cullis. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure—characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta. 812:55–65. [DOI] [PubMed] [Google Scholar]
- Kim, H. K., and J. Storch. 1992a. Free fatty acid transfer from rat liver fatty acid-binding protein to phospholipid vesicles. Effect of ligand and solution properties. J. Biol. Chem. 267:77–89. [PubMed] [Google Scholar]
- Kim, H. K., and J. Storch. 1992b. Mechanism of free fatty acid transfer from rat heart fatty acid-binding protein to phospholipid membranes. Evidence for a collisional process. J. Biol. Chem. 267:20051–20056. [PubMed] [Google Scholar]
- Leonard, M., N. Noy, and D. Zakim. 1989. The interactions of bilirubin with model and biological membranes. J. Biol. Chem. 264:5648–5652. [PubMed] [Google Scholar]
- Lipsky, N. G., and R. E. Pagano. 1985. A vital stain for the Golgi apparatus. Science. 228:745–747. [DOI] [PubMed] [Google Scholar]
- Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275. [PubMed] [Google Scholar]
- Massey, J. B., D. H. Bick, and H. J. Pownall. 1997. Spontaneous transfer of monoacyl amphiphiles between lipid and protein surfaces. Biophys. J. 72:1732–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen, T. P. W., R. N. A. H. Lewis, and R. N. McElhaney. 1995. New aspects of the interaction of cholesterol with dipalmitoylphosphatidylcholine bilayers as revealed by high-sensitivity differential scanning calorimetry. Biochim. Biophys. Acta. 1234:90–98. [DOI] [PubMed] [Google Scholar]
- Mesquita, R. M. R. S., E. Melo, T. E. Thompson, and W. L. C. Vaz. 2000. Partitioning of amphiphiles between coexisting ordered and disordered phases in two-phase lipid bilayer membranes. Biophys. J. 78:3019–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy, N., T. M. Donnelly, and D. Zakim. 1986. Physical-chemical model for the entry of water-insoluble compounds into cells. Studies of fatty acid uptake by liver. Biochemistry. 25:2013–2021. [DOI] [PubMed] [Google Scholar]
- Pagano, R. E., and O. C. Martin. 1988. A series of fluorescent N-acylsphingosines: synthesis, physical properties, and studies in cultured cells. Biochemistry. 27:4439–4445. [DOI] [PubMed] [Google Scholar]
- Peters, T., Jr. 1997. All About Albumin: Biochemistry, Genetics, and Medical Applications. Academic Press,San Diego, California.
- Petitpas, I., A. A. Bhattacharya, S. Twine, M. East, and S. Curry. 2001. Crystal structure analysis of Warfarin binding to human serum albumin: anatomy of drug site I. J. Biol. Chem. 276:22804–22809. [DOI] [PubMed] [Google Scholar]
- Pokorny, A., P. F. F. Almeida, E. C. C. Melo, and W. L. C. Vaz. 2000. Kinetics of amphiphile association with two-phase lipid bilayer vesicles. Biophys. J. 78:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorny, A., P. F. F. Almeida, and W. L. C. Vaz. 2001. Association of a fluorescent amphiphile with lipid bilayer vesicles in regions of solid-liquid-disordered phase coexistence. Biophys. J. 80:1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pownall, H. J. 2001. Cellular transport of nonesterified fatty acids. J. Mol. Neurosci. 16:109–115. [DOI] [PubMed] [Google Scholar]
- Pownall, H. J., D. L. Bick, and J. B. Massey. 1991. Spontaneous phospholipid transfer: development of a quantitative model. Biochemistry. 30:5696–5700. [DOI] [PubMed] [Google Scholar]
- Richieri, G. V., R. T. Ogata, and A. M. Kleinfeld. 1994. Equilibrium constants for the binding of fatty acids with fatty acid-binding proteins from adipocyte, intestine, heart, and liver measured with the fluorescent probe ADIFAB. J. Biol. Chem. 269:23918–23930. [PubMed] [Google Scholar]
- Richieri, G. V., R. T. Ogata, and A. M. Kleinfeld. 1995. Thermodynamics of fatty acid binding to fatty acid-binding proteins and fatty acid partition between water and membranes measured using the fluorescent probe ADIFAB. J. Biol. Chem. 270:15076–15084. [DOI] [PubMed] [Google Scholar]
- Richieri, G. V., R. T. Ogata, and A. M. Kleinfeld. 1996. Kinetics of fatty acid interactions with fatty acid binding proteins from adipocyte, heart, and intestine. J. Biol. Chem. 271:11291–11300. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Sklar, L. A., B. S. Hudson, and R. D. Simoni. 1977. Conjugated polyene fatty acids as fluorescent probes: binding to bovine serum albumin. Biochemistry. 16:5100–5108. [DOI] [PubMed] [Google Scholar]
- Spector, A. 1975. Fatty acid binding to plasma albumin. J. Lipid Res. 16:165–179. [PubMed] [Google Scholar]
- Storch, J., and N. M. Bass. 1990. Transfer of fluorescent fatty acids from liver and heart fatty acid binding proteins to model membranes. J. Biol. Chem. 265:7827–7831. [PubMed] [Google Scholar]
- Sudlow, G., D. J. Birkett, and D. N. Wade. 1975. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 11:824–832. [PubMed] [Google Scholar]
- Tanford, C. 1991. The Hydrophobic Effect: Formation of Micelles and Biological Membranes, 2nd ed. Krieger Publishing Company, Malabar, Florida.
- Taylor, R. P., A. V. Broccoli, and C. M. Grisham. 1978. Enzymatic and colorimetric determination of total serum cholesterol. J. Chem. Educ. 55:63–64. [DOI] [PubMed] [Google Scholar]
- Vaz, W. L. C., and E. Melo. 2001. Fluorescence studies on phase heterogeneity in lipid bilayer membranes. J. Fluorescence. 11:255–271. [Google Scholar]
- Vaz, W. L. C., and P. F. F. Almeida. 1993. Phase topology and percolation in multi-phase lipid bilayers. Is the biological membrane a domain mosaic? Curr. Opin. Struct. Biol. 3:482–488. [Google Scholar]
- Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. II. Distribution and packing of terminal methyl groups. Biophys. J. 61:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootan, M. G., and J. Storch. 1994. Regulation of fluorescent fatty acid transfer from adipocyte and heart fatty acid-binding proteins by acceptor membrane lipid composition and structure. J. Biol. Chem. 269:10517–10523. [PubMed] [Google Scholar]
- Zakim, D. 2000. Topical review: thermodynamics of fatty acid transfer. J. Membr. Biol. 176:101–109. [DOI] [PubMed] [Google Scholar]
- Zucker, S. D. 2001. Kinetic model of protein-mediated ligand transport: Influence of soluble binding proteins on the intermembrane diffusion of a fluorescent fatty acid. Biochemistry. 40:977–986. [DOI] [PubMed] [Google Scholar]
- Zucker, S. D., W. Goessling, and J. L. Gollan. 1995. Kinetics of bilirubin transfer between serum albumin and membrane vesicles. J. Biol. Chem. 270:1074–1081. [DOI] [PubMed] [Google Scholar]