

Abstract
The dynamical and structural properties of lignin peroxidase and its Trp171Ala mutant have been investigated in aqueous solution using molecular dynamics (MD) simulations. In both cases, the enzyme retained its overall backbone structure and all its noncovalent interactions in the course of the MD simulations. Very interestingly, the analysis of the MD trajectories showed the presence of large fluctuations in correspondence of the residues forming the heme access channel; these movements enlarge the opening and facilitate the access of substrates to the enzyme active site. Moreover, steered molecular dynamics docking simulations have shown that lignin peroxidase natural substrate (veratryl alcohol) can easily approach the heme edge through the access channel.
INTRODUCTION
Lignin peroxidase (LiP), a heme-containing glycoprotein isolated from the ligninolytic cultures of the white rot fungus Phanerochaete chrysosporium, is a very important hydrogen peroxide-dependent enzyme capable of performing the oxidative depolymerization of lignin via an electron transfer (ET) mechanism (Dunford, 1999; ten Have and Teunissen, 2001). LiP is also able to catalyze the oxidation of nonphenolic, electron-rich aromatic lignin model compounds as well as the oxidation of other different classes of organic compounds with a redox potential up to 1.4 V versus normal hydrogen electrode (NHE) (Schoemaker, 1990; Labat and Meunier, 1990; Kersten et al., 1990). Very little is known about the substrate binding site of LiP, and the pathway of electron flux from the substrate to the oxidized heme is still a matter of discussion.
The analysis of LiP crystal structure showed that both the active site and the substrate access channel to it appear almost inaccessible, compared to those of other peroxidases, and a direct interaction between the enzyme active site and a substrate seems quite difficult (Poulos et al., 1993; Piontek et al., 1993; Edwards et al., 1993; English and Tsaprailis, 1995; Banci, 1997). Thus, it has been proposed and generally accepted that small aromatic molecules, such as veratryl alcohol (3,4-dimethoxybenzyl alcohol, VA), an LiP natural substrate, are oxidized at the heme edge via a long-range ET (Poulos et al., 1993; Schoemaker and Piontek, 1996). Recently, it has also been suggested that VA might be oxidized via an ET mediated by the LiP surface residue Trp171 (β-hydroxytryptophan, HTR) (Doyle et al., 1998; Blodig et al., 1998; Choinowski et al., 1999; Sollewijn Gelpke et al., 2002), a hypothesis based on the observation that the mutation of this residue led to the loss of enzymatic activity toward VA oxidation. A very intriguing result was that the mutants were still able to perform the oxidation of two dye substrates, 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) (ABTS), and 4-[(3,5-difluoro-4-hydroxyphenyl)azo]benzenesulfonic acid (DFAD), that had a lower redox potential compared to VA.
However, the pictures described above, both of which do not predict a direct interaction between the active site and the substrate, seem to be contradicted by kinetic deuterium isotope effect and 18O incorporation studies, which instead indicated that small aromatic substrates, such as ring-substituted N,N-dimethylanilines and thioanisoles, can approach the enzymatic active site and that the corresponding substrate radical cations, once formed, can react with the iron(IV)-oxo complex undergoing, respectively, deprotonation or sulfoxidation (oxygen rebound) (Baciocchi et al., 2000, 2001). For example, in the LiP-catalyzed sulfoxidation, a certain degree of stereoselectivity was observed (the enantiomeric excess ranging from 6.8 to 62%) together with a partial (with 4-methoxythioanisole) or absent (with 4-bromothioanisole) 18O incorporation in the sulfoxide when the reaction was carried out in labeled water, indicating that the substrate radical cation receives the oxygen atom predominantly from the iron-oxo complex.
We therefore felt it interesting to investigate in detail the protein behavior to get a better knowledge of both its structure and its dynamical properties in solution. Molecular dynamics (MD) simulations supply a great deal of information with respect to the stability of noncovalent interactions in water and to the mobility of the amino-acidic residues within the protein frame. In fact, MD simulations provide a reliable model of the structural and dynamical behavior in solution that might be different from the static picture of the protein obtained from its crystal structure. For these purposes, we have performed a 5-ns MD simulation of the native enzyme, which is the first long MD simulation carried out on LiP. (In fact, only a 0.5-ns MD simulation of this enzyme has hitherto been reported, but it was aimed at analyzing the effect of structural calcium ions on the stability of the enzyme active site; see Banci et al., 1996.) Our analysis has been focused instead on different structural and dynamical aspects of the protein—in particular, the conformational changes in the access channel have been monitored to investigate possible variations of its size, which is very important in determining the substrate accessibility to the active site. Moreover, a 180-ps steered molecular dynamics docking simulation of VA through the access channel to the heme has been performed to get an indication of how feasible it is for VA to approach the heme edge. In particular, the interaction of VA with the residues forming the access channel and the constraint force have been analyzed. Moreover, a 2.5-ns MD simulation of a LiP mutant, namely Trp171Ala (W171A), has been run to compare the dynamic behavior of native LiP in solution with that of its W171A mutant, the aim being that of determining if the site-directed mutagenesis produces any relevant structural alteration or leads to a different protein mobility in solution.
METHODS
Force field parameters for the protein
Simulations were performed using GROMOS96 force field (van Gunsteren et al., 1998). For nonstandard amino acids and the heme iron, new force field parameters have been introduced. For the heme model a slightly modified GROMO96 heme was used. The partial charge on the iron was changed to 1.4 e.u. This value is comparable to that reported in the literature for other similar systems (Banci et al., 1994). Furthermore, to avoid possible collapse of free water molecules in the active site cavity on the heme iron, a Lennard Jones (LJ) potential for the iron was applied. The LJ parameters taken from the CHARMM19 force field (MacKerell et al., 1998) were used. The distance of the heme iron to Nɛ2of His176 was fixed to 0.215 nm, the crystallographic value. The water molecule coordinated to the iron was covalently bounded and the oxygen iron distance was fixed to the crystallographic value of 0.209 nm. The OH group bonded to the Cβ carbon of Trp171 (HTR171) was parameterized using bonded/nonbonded potential functions and charges similar to those used in the GROMOS96 library for the OH group of other amino acids.
Veratryl alcohol
The VA model (see Fig. 1) was built using the GROMOS96 force field parameters. GROMOS96 atom type and their LJ parameters were considered. Bond distances, angles, and dihedrals, as well as the force constants, were taken for analogy with amino acids having similar functional groups (tyrosine and serine). Explicit polar and apolar hydrogen atoms were considered. Atomic partial charges were obtained from ab initio calculation at HF level, using the 6 − 31 + G* basis set; the Gaussian98 package (Frisch et al., 1998) was used for the calculation. Electrostatic fitting to the electron density by CHELPG (Breneman and Wiberg, 1990) approach was used to obtain the partial charges which were slightly modified to obtain neutral charge groups. In Table 1 the resulting partial charges used in the MD simulations are reported.
FIGURE 1.

Structure of veratryl alcohol.
TABLE 1.
Atomic partial charges for VA model
| N | Atom type | Charges (q/e) |
|---|---|---|
| 1 | C | −0.20 |
| 2 | H | 0.20 |
| 3 | C | −0.20 |
| 4 | H | 0.20 |
| 5 | C | −0.10 |
| 6 | H | 0.10 |
| 7 | C | 0.24 |
| 8 | O | −0.41 |
| 9 | C | 0.14 |
| 10 | H | 0.01 |
| 11 | H | 0.01 |
| 12 | H | 0.01 |
| 13 | C | 0.10 |
| 14 | C | 0.14 |
| 15 | O | −0.60 |
| 16 | H | 0.40 |
| 17 | H | −0.02 |
| 18 | H | −0.02 |
| 19 | C | 0.24 |
| 20 | O | −0.41 |
| 21 | C | 0.14 |
| 22 | H | 0.01 |
| 23 | H | 0.01 |
| 24 | H | 0.01 |
Starting coordinates
Four major different molecular forms of LiP have been identified with a molecular size of ∼39–43 kDa. The enzyme is formed by two identical monomers in the asymmetric unit, each containing a single iron protoporphyrin IX prosthetic group, two calcium ions, four disulfide bonds, and carbohydrate chains. Each monomer consists of 344 amino-acid residues. The starting structure for simulations was taken from the 0.17-nm resolution refined x-ray LiP crystal structure (Brookhaven Protein Data Bank, PDB code 1QPA; see Choinowski et al., 1999). Only one monomer was used for the simulations. The carbohydrate residues, present in the crystal structure, were cut. This approximation should not alter the results of our simulations, since a molecular engineered glycoside-free variant of the enzyme shows properties identical to the wild-type (Blodig et al., 2001). The four disulfide bridges (2-15, 14-285, 34-120, and 249-317) found in LiP crystal structure were explicitly included. Hydrogen atoms of aromatic side chains and polar groups were added, according to standard geometry rules, to the protein and to crystallization water molecules.
Considering the importance of Trp171 for the alternative ET pathway, two different LiP models were simulated: one with the β-hydroxytryptophan (HTR simulation) and another one with the W171A mutant, which was generated by extracting a frame from the HTR simulation at 2.5 ns and modeling HTR171 into alanine. Finally, VA starting coordinates were obtained from the ab initio calculation.
MD procedure
The simple point charge model (Berendsen et al., 1981) was used to describe water molecules. The protein was solvated in a rectangular periodic box with water, by stacking equilibrated boxes of solvent molecules to form a box of 5.8 × 6.7 × 7.9 nm, large enough to contain the protein and 0.75 nm of solvent on all sides. All solvent molecules with any atom within 0.15 nm of the protein were removed. Since the system has a total charge of −15, sodium counterions were added to neutralize the box charge. The resulting system was formed by the protein, 268 crystallization water molecules, 8419 water molecules, and 15 sodium ions, for a total of ∼29,000 atoms. The spatial arrangement of the solvation molecules were energy-minimized while constraining the protein and the crystallization water molecules at their experimental position. The system was energy-minimized, using the steepest descendent method, for 100 steps. In all the simulations the temperature was maintained close to 300 K by isothermal Gaussian temperature coupling (Allen and Tildesly, 1989). Rotational and translational motions of the protein in the simulation box were removed using the method of Amadei et al. (2000). The LINCS algorithm (Hess et al., 1997) was used to constrain all bond lengths. For water molecules the SETTLE (Miyamoto and Kollman, 1992) algorithm was used. A dielectric permittivity, ɛr = 1, and a time step of 2 fs were used. The nonbonded interactions (electrostatic and LJ) were calculated using the twin-range cutoff method. The long-range cutoff was set to 1.4 nm and the nonbonded interaction pair list was updated every five steps, whereas a short-range cutoff of 1.0 nm was used to calculate nonbonded interactions every step. All atoms were given an initial velocity obtained from a Maxwellian distribution at the desired initial temperature. After the minimization, the system was equilibrated by 100 ps of MD run with position restraints on the protein atoms and the crystallization water molecules to allow relaxation of generated solvent molecules. After the equilibration, the position restraints on the protein were removed and the system was gradually heated from 50 K to 300 K during 50 ps of simulation. All the MD runs and the analysis of the trajectories were performed using a modified version of the GROMACS 3.0 software package (van der Spoel et al., 1994) that implements the isothermal Gaussian temperature coupling and the rototranslational removing of the center of mass of the solvated protein algorithm. In the analysis of the trajectories, the C-terminal nine residues of both native and mutant LiP were not considered since they were found to be very mobile. O⋯H distances shorter than 0.25 nm and O-H⋯O angles larger than 60° were the criteria adopted for acceptance of hydrogen bonds.
Principal component analysis of the trajectories
The principal component analysis of the enzyme positional fluctuations along the MD trajectories was carried out to evidence the presence of correlated residue motions. This method, known as essential dynamics (ED) (Amadei et al., 1993), allows the characterization of relevant collective motions (essential motions) occurring during the MD simulation and their distinction from thermal noise motions. The analysis was performed by building the covariance matrix (C) of the positional fluctuations obtained from MD simulations. Upon diagonalization of C, a set of eigenvalues and eigenvectors is generated defining a new set of generalized coordinates. The eigenvectors correspond to directions in a 3N dimensional space (where N is the number of atoms used for the analysis) along which collective fluctuations of atoms occur. The eigenvalues represent the total mean square fluctuation of the system along the corresponding eigenvector.
Steered molecular dynamics docking simulations
Steered molecular dynamics (SMD) method (Izrailev et al., 1998) was used to reduce the timescale required to have a spontaneous diffusion of VA through LiP access channel. This technique has been already used to study ligand binding (Meulenhoff, 1999) and unbinding (Kosztin et al., 1999) to proteins. SMD docking provides a means to accelerate the binding process through the application of external forces that lower the energy barrier and drive the ligand to the active site in a nanosecond timescale. Thus, to obtain a further qualitative evidence of VA accessibility to the heme edge, a 180-ps-long SMD docking simulation was performed. VA has been chosen for this docking simulation because it is a natural substrate of LiP. It is reasonable to think that the results obtained with VA also hold for thioanisole and N,N-dimethylaniline, inasmuch as all these molecules have a similar dimension. As starting LiP conformation, the one at 2500 ps of the HTR simulation was used. VA molecule was positioned in the simulation box, in front of the access channel, by manual docking, replacing few overlapping water molecules. After a preliminary minimization and equilibration procedure, in which VA molecule was kept at a fixed distance from the heme-iron atom, the SMD simulation was started. In the pulling simulation, the iron-VA benzylic carbon atom (C14) was chosen as constraint coordinate. The C14 atom was harmonically restrained to a point moving with a constant velocity along the constraint coordinate. During the simulation, the external force exerted on VA was F = k(vt−x), were x is the displacement of the restraint atom with respect to its original position and t is the time elapsed from the beginning of the simulation. As values for k and v, 100 kJ mol−1 nm−2 and 0.02 nm ps−1 were used.
RESULTS AND DISCUSSION
Wild-type enzyme
General structural properties
The time evolution of the backbone-backbone root mean square deviation (RMSD) of LiP relative to the crystal structure is shown in Fig. 2 a for HTR simulation (black line). The protein atoms did not significantly deviate from the crystal structure and the backbone RMSD is stabilized to an average value of 0.2 nm. Although the RMSD appears to rise slightly even after 1.5 ns, the fluctuation amplitudes obtained are relatively small. In Fig. 2 b the radius of gyration (Rg) of the protein for the HTR simulation (black line) is reported. The average value, 1.95 nm, in the last 3 ns shows a small deviation from the crystallographic one (1.925 nm, Fig. 2 b, dashed line), which indicates a negligible crystallographic packing effect as well as a small influence of the second monomer on the overall enzyme stability. The secondary structure observed in the x-ray crystal structure is maintained during the simulation (Table 2). All the structurally defined features are retained.
FIGURE 2.

Backbone-backbone RMSD, with respect to the crystallographic structure of LiP (a) and radius of gyration (b) along the simulations. HTR (black) and W171A (gray) simulations are reported. (Dashed line) The crystallographic value of the gyration radius.
TABLE 2.
Summary of the enzyme structural properties obtained from the analysis of the last 2.5 ns of the HTR and W171A simulations and comparison with the properties of crystal LiP
| % Helix | % Strand | % Loop | % Coil | Surface area (nm2) | No. of hydrogen bonds | |
|---|---|---|---|---|---|---|
| Crystal | 41 | 7 | 26 | 26 | 50*/92† | 281 |
| HTR | 38 | 6 | 27 | 29 | 57*/89† | 286 |
| W171A | 39 | 5 | 27 | 29 | 58*/90† | 295 |
Hydrophobic surface area.
Hydrophilic surface area.
To provide a more detailed description of the mobility of the protein residues, the backbone RMSD per residue is shown in Fig. 3 a (full line). The gray bands indicate the helix regions of the protein. It is clear from this representation that the most ample deviations do not involve residues forming structurally important regions. In particular, the residues showing the largest displacements are those in the C- and N-termini of the protein, regions characterized by a low structural organization. Moreover, other residues that experience a large displacement are those forming loops that are mainly located on the LiP surface. In particular, the two monomeric LiP units are not covalently linked, but they are in contact through one loop (from residue 292 to 299 and from 326 to 332) that resulted to be quite flexible during the simulation. From all these graphics it is evident that the enzyme mostly retained the overall backbone structure in the course of the simulation. This finding is consistent with the presence of an extensive secondary structure in LiP with several α-helices and β-sheets that make the protein rigid.
FIGURE 3.

Backbone RMSD (a) and RMSF (b) with respect to the crystallographic structure of LiP residues. HTR (black) and W171A (dashed) simulations are reported. (a, gray bands) The α-helix regions of the protein. (b, vertical lines) The residues forming the access channel to the heme.
Active site: hydrogen bonds
The heme in LiP is not covalently linked to the protein, but held in place by a network of hydrogen bonds. In particular, the carboxylate group of the outer propionate on pyrrole ring A forms a hydrogen bond with the carboxylate of Asp183. The inner propionate group on pyrrole ring D interacts with Ala180. The side-chain nitrogen atoms of Arg43 and of Asn182 form hydrogen bonds with, respectively, the propionates of rings A and D. Moreover, hydrogen bonds in LiP are important not only from a structural point of view, but also for the fundamental role in determining the enzyme catalytic properties. Accordingly, a strong hydrogen bond between Ser177 and Asp201 weakens the bond between the heme iron and its proximal ligand, His176, so that the heme iron becomes more electron-deficient and the enzyme redox potential gets higher, compared to that of other similar peroxidases (Piontek et al., 1993).
The importance of hydrogen bonds is even evident considering that, as with other peroxidases, the rate of formation of iron(IV)-oxo porphyrin radical cation (LiP Compound I), the catalytic active intermediate of the enzyme, is relatively insensitive to pH (Andrawis et al., 1988), while its reduction is pH-sensitive and exhibits an optimum near pH 3 (Marquez et al., 1988). Such a low pH value suggests that at higher pH the protonation of a carboxylate, forming a hydrogen bond, would break this interaction leading to a destabilization and a change in the heme pocket crucial to catalysis. The hydrogen bond responsible for this behavior is supposed to be the one between the carboxylate group of Asp183 and the carboxylate group of the outer propionate on pyrrole ring A or the other one between the Nδ1 atom of His82 and the carboxylate group Glu146 (Edwards et al., 1993). The analysis of this extensive network of hydrogen bonds showed that all these interactions are retained during the MD simulations.
Active site: structure
The heme iron fifth ligand is the proximal histidine, His176, while the sixth coordination site is occupied by a water molecule, hydrogen-bonded to the Nɛ2 of His47. The structure of the active site in LiP is very similar to that of other peroxidases such as horseradish peroxidase and cytochrome C peroxidase. On the distal helix there are three key catalytic residues, Arg43, Phe46, and His47, that form the peroxide-binding pocket where hydrogen peroxide interacts with the heme iron leading to the formation of LiP Compound I. Arg43 and His47 are conserved residues in peroxidases, whereas Phe46, which is almost coplanar to the heme pyrrole ring C, is not (English and Tsaprailis, 1995). These two invariant residues are thought to play an important role in the formation and stabilization of LiP Compound I. Accordingly, it has been proposed that the distal Arg43 has two functions. It both stabilizes the developing negative charge on the peroxide OH− leaving group, and once the peroxidic O O bond is cleaved, the distal Arg43 forms a hydrogen bond with the ferryl oxygen atom, thus stabilizing LiP Compound I. His47, on the other hand, is a proton acceptor for the bound peroxide (Edwards et al., 1993). Both Arg43 and His47 form a strong network of hydrogen bonds: the former with the propionate group of pyrrole A (as already illustrated) and with the peptide carbonyl oxygen atom of Ile85. The Nδ1 atom of His47 forms a hydrogen bond with the side-chain oxygen of Asn84, whose side-chain nitrogen donates a hydrogen bond to the peptide carbonyl oxygen atom of Glu78.
O bond is cleaved, the distal Arg43 forms a hydrogen bond with the ferryl oxygen atom, thus stabilizing LiP Compound I. His47, on the other hand, is a proton acceptor for the bound peroxide (Edwards et al., 1993). Both Arg43 and His47 form a strong network of hydrogen bonds: the former with the propionate group of pyrrole A (as already illustrated) and with the peptide carbonyl oxygen atom of Ile85. The Nδ1 atom of His47 forms a hydrogen bond with the side-chain oxygen of Asn84, whose side-chain nitrogen donates a hydrogen bond to the peptide carbonyl oxygen atom of Glu78.
The analysis of the MD simulation revealed that in the active site these key residues essentially maintained the conformation they have in the crystal structure; in fact, the RMSD of the active site residues is 0.13 ± 0.01 nm. In particular their hydrogen-bond network is retained and persisted during the simulation (data not shown). The superimposition shown in Fig. 4 of the active site in the x-ray LiP crystal structure (bold line) with different frames from the HTR simulation (gray lines) displays a very similar orientation of all the amino-acidic residues in the active site considered important for LiP activity, confirming that this protein area is very rigid.
FIGURE 4.

Stereo view of the superimposition of 10 conformations sampled along the last 1 ns of HTR simulation. (Bold solid line) The crystallographic structure. The active site, the distal (upper) and proximal (lower) calcium binding sites and the HTR171 region are reported.
Calcium ions
One of the structural elements shared by class II and III peroxidases is the calcium-binding sites proximal and distal to the heme. It seems that the calcium ions play an essential role in maintaining the integrity of the active site and thus the enzymatic activity (Howes et al., 2001). Thermal (Nie and Aust, 1997) and alkaline (George et al., 1999) LiP inactivation has been correlated with the loss of calcium ions and formation of an inactive bis-histidyl hexacoordinate low-spin heme state.
Each LiP calcium ion coordinates to groups attached to helices that form the active site: the proximal side calcium ligand, Ser177, is at the C-terminal end of the proximal helix, and immediately follows the heme ligand, His176, whereas the distal side calcium ligand, Asp48, immediately follows the distal His47. The distal calcium ion has less affinity for the enzyme inasmuch as it is nearer to the surface than the proximal, and has two water molecules as ligands; its binding site is formed by the residues from Gly66 to Ser70. The peptide oxygen of Gly66, the carboxylate group of Asp68, the side-chain oxygen of Ser70, and the carboxylate group of Asp48 bind the distal calcium ion. The proximal calcium, the most tightly bound, has a binding site formed primarily by one loop from Asp194 to Asp201. The carboxylate group of Asp194, both the oxygen atoms of Thr196 and Ser177, the main chain oxygen of Gln199, and the carbonyl oxygen of Asp201 are coordinated to it. The proximal calcium ion has been proposed to fine-tune the topology of the active site, in particular the heme iron-His176 distance, which is thought to influence the enzyme redox potential. Accordingly, it has been suggested that the higher redox potential of LiP, as compared to that of other peroxidases such as cytochrome C peroxidase and horseradish peroxidase, can be due to a lesser imidazolate character of the proximal histidine (Banci et al., 1991). Thus, this structural feature is considered to be in part responsible for LiP's ability to oxidize nonphenolic aromatic substrates with a redox potential up to 1.4 V versus NHE.
We have analyzed in detail the behavior of the calcium ions and their binding residues during the MD to determine if all these interactions are maintained or not in solution. The distances between each calcium ion and their closer residues are constant during the simulation and their values are very similar to those reported for LiP crystal structure. Moreover, both the calcium ions retained their positions even with respect to other constituents of the enzyme—in particular, their distances from the heme iron during the MD simulation are similar to the crystallographic ones (for the proximal calcium ion the distance is 1.34 ± 0.02 nm compared to 1.34 nm in the crystal structure and for the distal calcium ion is 1.68 ± 0.02 nm compared to 1.57 nm in the crystal structure). In Fig. 4 a superimposition of the proximal (lower) and distal (upper) calcium ion binding sites in the crystal structure (bold line) with several snapshots from the HTR simulation (gray lines) is reported. All the residues coordinated to both calcium ions are shown and they do not deviate from their crystal orientations, thus indicating that this microenvironment is firmly conserved in solution. These results are in accordance with the expectation that the interactions between the calcium ions and the protein are important to maintain the structure of the active site and, therefore, the enzymatic catalytic activity.
Access channel to the heme
The most interesting and surprising results came from the MD data analysis for the residues forming the substrate access channel to the heme. LiP heme is not in direct contact with the solvent, but is located at the bottom of a fissure, recessed from the protein surface. A cleft, found in heme peroxidases but with variable sizes, provides access to it from the surface. Compared to other peroxidases, like cytochrome C peroxidase and Coprinus cinereus peroxidase, LiP has bulkier residues whose side chains occupy, in part, the entrance, so that a direct interaction of large substrates with the heme in the active site appears highly unlikely (English and Tsaprailis, 1995). Thus, one of the still-open questions regarding LiP activity is how a substrate is oxidized by this enzyme.
In Fig. 3 b the root mean square fluctuation (RMSF) per residue is reported (full line for the HTR model) with the vertical lines indicating the residues forming the access channel to the heme (His82, Pro83, Asn84, Ile85, Glu146, Pro147, Phe148, Asp183, Val184, Ile221, and Gln222). Among these, Pro147, Phe148, Val184, Ile221, and Gln222 exhibit the largest displacements.
A similar indication came out from the analysis of the Rg of the amino-acid residues forming the access channel, the solvent-accessible surface of this area and the number of contacts (NC) between these residues, reported in Fig. 5 (black line for the HTR model). The variations of these properties during the time of the simulation are correlated. Accordingly, to an increase in Rg corresponds an increase in the solvent-accessible surface and a decrease in the NC, indicating that the access channel is increasing its opening. Thus, this analysis pointed out the occurrence of conformational changes of the protein access channel, suggesting that there is the possibility for a substrate to enter in the channel in solution. The enlargement of the heme access channel is pictorially represented in Fig. 6, where the x-ray crystal structure (0 ps) and different frames extracted from the HTR simulation are reported. Hence, a relaxation of the protein occurs in solution, demonstrating that the picture obtained from the x-ray crystal structure represents a lower limit. Therefore, these results indicate that the previous hypothesis (Baciocchi et al., 2000, 2001) of an interaction between the enzyme and the substrate (or its radical cation) in the active site is reliable.
FIGURE 5.

(a) Radius of gyration, (b) total accessible surface area, (c) and number of contacts (NC) within 0.6 nm of the access channel residues along the HTR (black line) and W171A (gray line) simulations. (Dashed lines) The crystallographic values.
FIGURE 6.

LiP x-ray crystal structure (0 ps) and snapshots extracted from the HTR simulation. The residues forming the access channel (see text) are represented as Connolly surfaces. (Blue sticks) The heme.
HTR171 site
The β-hydroxytryptophan (HTR171) area has been recently suggested to be an alternative substrate binding site and HTR171 is considered to be part of an electron transfer pathway (vide infra) (Blodig et al., 1998); Choinowski et al., 1999; Blodig et al., 2001). Thus, taking this suggestion in mind, we have analyzed the MD simulation with respect to the residues around HTR171 and connecting the β-hydroxytryptophan to the heme. Such analysis reveals that HTR171 and all the amino-acid residues in its proximity essentially maintained the conformation they have in the x-ray crystal structure. In Fig. 4, we report the superimposition of the amino-acid residues in this area for the x-ray crystal structure and for 10 frames sampled in the last ns of HTR simulation, which shows a very similar orientation of the amino-acidic residues connecting HTR171 to the heme. Accordingly, the RMSD for the HTR171 residue and its four closer residues (169–173) is 0.08 ± 0.01 nm. This result indicates this to be is a rigid area of the protein.
Essential dynamics (ED) analysis
ED analysis on the Cα atoms of the HTR simulation was performed to obtain information on the collective motions of the native protein. From the obtained eigenvalues it results that ∼50% of the overall protein fluctuations is described by the first eight eigenvectors. In Fig. 7 displacements along the first eight eigenvectors of the Cα are reported. For each Cα atom, the value 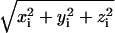 is given, where xi, yi, and zi are the Cartesian components of the ith atom in the respective eigenvectors. The arrows indicate the residues forming the access channel. The protein regions having the largest correlated displacements are defined by the following N- and C-terminal residues, 55–65, 146–150, 182–190, and 220–224. Interestingly, the access channel residues Glu146, Pro147, Phe148, Asp183, Val184, Ile221, and Gln222 are located in the last three regions, and residues 148, 184, 221, and 222, in particular, are the most involved in the correlated protein motion (see Fig. 7, vec 1 and vec 2; and Fig. 7, vec 6, vec 7, and vec 8). HTR171, on the contrary, does not participate in the correlated motion defined by these eigenvectors. Thus, the ED study confirms the results obtained from the standard MD analysis regarding both the flexibility of the access channel residues and the rigidity of the HTR171 region. Furthermore, the finding that the access channel movements are correlated strongly indicates that these residues cooperatively fluctuate, with an in-phase opening, facilitating the access of a substrate to the enzyme active site.
is given, where xi, yi, and zi are the Cartesian components of the ith atom in the respective eigenvectors. The arrows indicate the residues forming the access channel. The protein regions having the largest correlated displacements are defined by the following N- and C-terminal residues, 55–65, 146–150, 182–190, and 220–224. Interestingly, the access channel residues Glu146, Pro147, Phe148, Asp183, Val184, Ile221, and Gln222 are located in the last three regions, and residues 148, 184, 221, and 222, in particular, are the most involved in the correlated protein motion (see Fig. 7, vec 1 and vec 2; and Fig. 7, vec 6, vec 7, and vec 8). HTR171, on the contrary, does not participate in the correlated motion defined by these eigenvectors. Thus, the ED study confirms the results obtained from the standard MD analysis regarding both the flexibility of the access channel residues and the rigidity of the HTR171 region. Furthermore, the finding that the access channel movements are correlated strongly indicates that these residues cooperatively fluctuate, with an in-phase opening, facilitating the access of a substrate to the enzyme active site.
FIGURE 7.

Components of the first eight eigenvectors for the HTR (black line) and W171A (gray line) simulations. (Dashed vertical lines) The position of the access channel residues (see text).
Steered molecular dynamics docking simulations
It has been suggested (Poulos et al., 1993; Schoemaker and Piontek, 1996) that VA can approach the LiP heme edge. Unfortunately no experimental data are reported to support this hypothesis. The entrance of VA into the LiP active site access channel would require a timescale too long to be observed by a free MD simulation. For this reason, we have used the SMD method to accelerate this process. It is important to point out that SMD provides only qualitative information (Izrailev et al., 1998) on the docking process that is, however, sufficient to assess the presence of energy barriers. Quantitative results would require the calculation of the potential mean force along the docking pathway (Meulenhoff, 1999), but this is computationally very expensive, and it is beyond the scope of this article.
As described in Methods, a 180-ps-long SMD docking simulation was performed, starting from a Fe-VA benzylic C atom (C14) distance of 1.4 nm (Fig. 8 a). After 180 ps this distance decreases to ≈0.75 nm. The curve in Fig. 8 a shows two steps in correspondence of 25 ps and 100 ps. The second step corresponds to a situation in which VA is close to the heme edge, as shown in Fig. 8 b where the number of contacts of VA with heme atoms (within 0.4 nm) is reported. The value of the constraint force, Fig. 8 d, fluctuates around 0, indicating the presence of favorable interactions across the access channel. Finally, the lack of channel residues' perturbation by the incoming VA molecule is shown in Fig. 8 c, where the number of contacts among the residues forming the access channel is reported. It is evident that the number of contacts remains similar to that observed in the free HTR simulation. Thus, it would appear that VA could easily approach the heme edge up to a distance of ∼0.4 nm without disturbing the protein. In Fig. 9 the stereo views of the starting and final VA positions in the access channel for the SMD docking simulation are reported.
FIGURE 8.

(Bottom to top) Behavior of different parameters along the SMD docking simulation are reported: (a) distance Fe-C14; (b) number of contacts (<0.4 nm) between VA and the heme atoms; (c) number of contacts (<0.6 nm) among the access channel residues (see text); and (d) constraint force along the Fe-C14 direction.
FIGURE 9.

Stereo views of the starting conformation for the SMD docking simulation (bottom) and the conformation after 180 ps of the SMD docking simulation (top). In this figure, only the access channel residues (see in the text), the heme, and the VA molecule are reported.
Mutant enzyme simulation
Very recently, the high-resolution crystal structure of LiP has shown the presence of a Cβ hydroxylation of Trp171 (HTR) (Blodig et al., 1998; Choinowski et al., 1999; Blodig et al., 2001). Site-directed mutagenesis studies indicated that removal of this residue, and substitution with Phe (Doyle et al., 1998), Ser (Doyle et al., 1998), or Ala (Sollewijn Gelpke et al., 2002) led to the loss of enzymatic activity in the oxidation of VA, whereas a substantial maintenance of the catalytic activity was observed in the oxidation of two dye substrates, 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) and 4-[(3,5-difluoro-4-hydroxyphenyl)azo]benzenesulfonic acid (DFAD), which are larger than VA but characterized by a lower redox potential. Thus, it has been suggested that there might be a second substrate binding site, namely in close proximity of HTR171, which is located on the LiP surface—opposite to the heme access channel and ∼1.1 nm distant from the plane defined by the heme. A long-range ET pathway has been proposed to exist within the protein leading from the heme to HTR171 (Doyle et al., 1998; Blodig et al., 1998; Choinowski et al., 1999).
To get a better insight into this intriguing problem, we have carried out an MD simulation of one of LiP mutants, namely W171A. The structural influence of the mutation on the dynamical behavior of the enzyme can be obtained by the comparison between the simulations of the wild-type enzyme and its mutant.
General structural properties
The time evolution of the backbone RMSD of W171A relative to the crystal structure (of the wild-type enzyme) and the radius of gyration are shown respectively in Fig. 2, a and b (gray line). Both these properties are very similar to the corresponding ones of the wild-type enzyme. The secondary structure features observed in the x-ray crystal structure are maintained during the W171A simulation (Table 2). In Fig. 3 the backbone RMSD and the RMSF per residue are reported (gray line), indicating a similar behavior for the two simulations. The analysis of both the position and the interactions of the two calcium ions (data not shown) reveals that they maintained all their structural characteristics in this simulation as well.
Active site: hydrogen bonds and structure
Both the extensive network of hydrogen bonds considered important for the enzymatic activity and the position of the active site key residues are maintained in this simulation (data not shown), confirming that the W171A mutation does not produce any relevant structural change.
Access channel to the heme
In Fig. 5 (gray line), the structural properties of the residues forming the access channel are reported. They all show a similar behavior compared to that observed in the HTR simulation, indicating that an enlargement of the channel also occurs in the mutant. The ED analysis reported in Fig. 7 (gray line) confirms these observations.
CONCLUSIONS
The structural and dynamical behavior in aqueous solution of LiP monomer from Phanerochaete chrysosporium has been studied using MD simulations. Different structural and dynamical aspects of the protein have been analyzed. In particular, the conformational changes in the access channel region have been studied to investigate possible variations of its size, which is very important in determining the substrate accessibility to the enzymatic active site.
The simulation of the enzyme has been shown that the overall backbone structure as well as the interactions and the positions of those amino-acidic residues considered important for the catalytic activity are maintained. Interestingly, the residues defining the access channel to the heme show instead large deformations from the crystal structure. Furthermore, large fluctuations of the residues on the rim of the access channel are observed. These variations, as gained from the ED analysis, are correlated with each other, and thus they relax the crystallographic side-chain conformations, opening the rim of the access channel and facilitating the access of small substrates toward the active site. From this simulation it appears that the heme is more exposed and accessible to substrates than what is currently thought on the basis of the x-ray crystal structure analysis. This is also qualitatively evidenced by the docking of VA to the enzyme active site. The SMD simulation has shown an easy accessibility of VA through the access channel; the protein is not perturbed during this docking simulation and, moreover, the value of the constraint force indicates that VA experiences favorable interactions up to a distance of ∼0.4 nm from the heme edge.
On the basis of the enlargement of LiP active site access channel here reported, the accessibility of small LiP substrates to the heme should not be a limiting factor in the enzymatic oxidation. Thus, a direct interaction of a small substrate with the enzyme active site seems possible and this conclusion would be consistent with the results of the kinetic deuterium isotope effects and 18O incorporation studies already mentioned (Baciocchi et al., 2000, 2001).
The simulation with the W171A mutant showed structural features similar to those of the wild-type enzyme. Thus, even in the mutant, the active site access channel seems quite flexible. The actual role of HTR171 remains unclear and it is difficult to understand why a substrate should undergo a long-range ET through HTR171 when it has the possibility to directly interact with the enzyme active site. As a tentative hypothesis, the main HTR171 task might be that of modifying, and namely increasing, the enzyme oxidizing power. Therefore, HTR171 might play a role only in the oxidation of molecules characterized by a relatively high redox potential (e.g., VA). Accordingly, LiP mutants without HTR171 retained catalytic activity toward substrates with a low redox potential, similar to the dyes ABTS and DFAD.
Acknowledgments
We thank Prof. M. Aschi for discussion and helpful suggestions for the ab initio calculations. D.R. is grateful to Prof. Fernando Mazza for his support.
This work was supported by the Italian Ministero dell'Istruzione, Università e Ricerca (as part of national project entitled Structural Biology and Dynamics of Redox Proteins).
References
- Allen, M. P., and D. J. Tildesly. 1989. Computer simulation of liquids. Oxford University Press, Oxford.
- Amadei, A., G. Chillemi, M. A. Ceruso, A. Grottesi, and A. Di Nola. 2000. Molecular dynamics simulations with constrained roto-translational motions: theoretical basis and statistical mechanical consistency. J. Chem. Phys. 112:9–23. [Google Scholar]
- Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Prot. Struct. Funct. Gen. 17:412–425. [DOI] [PubMed] [Google Scholar]
- Andrawis, A., K. A. Johnson, and M. Tien. 1988. Studies on compound I formation of the lignin peroxidase from Phanerochaete chrysosporium. J. Biol. Chem. 263:1195–1198. [PubMed] [Google Scholar]
- Baciocchi, E., M. F. Gerini, P. J. Harvey, O. Lanzalunga, and S. Mancinelli. 2000. Oxidation of aromatic sulfides by lignin peroxidase from Phanerochaete chrysosporium. Eur. J. Biochem. 267:2705–2710. [DOI] [PubMed] [Google Scholar]
- Baciocchi, E., M. F. Gerini, O. Lanzalunga, A. Lapi, M. G. Lo Piparo, and S. Mancinelli. 2001. Isotope-effect profiles in the oxidative n-demethylation of n,n-dimethylanilines catalysed by lignin peroxidase and a chemical model. Eur. J. Org. Chem. 2305–2310.
- Banci, L. 1997. Structural properties of peroxidases. J. Biotechnol. 1:253–263. [DOI] [PubMed] [Google Scholar]
- Banci, L., I. Bertini, P. Turano, M. Tien, and T. K. Kirk. 1991. Proton NMR investigation into the basis for the relatively high redox potential of lignin peroxidase. Proc. Natl. Acad. Sci. USA. 88:6956–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci, L., P. Carloni, A. Diaz, and G. Gori Savellini. 1996. Molecular dynamics calculations on peroxidases: the effect of calcium ions on protein structure. J. Biol. Inorg. Chem. 1:264–272. [Google Scholar]
- Banci, L., P. Carloni, and G. Gori Savellini. 1994. Molecular dynamics studies on peroxidases: a structural model for horseradish peroxidase and a substrate adduct. Biochemistry. 33:12356–12366. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, the Netherlands. p. 331.
- Blodig, W., W. A. Doyle, A. T. Smith, K. H. Winterhalter, T. Choinowski, and K. Piontek. 1998. Autocatalytic formation of a hydroxy group at Cβ of Trp171 in lignin peroxidase. Biochemistry. 37:8832–8838. [DOI] [PubMed] [Google Scholar]
- Blodig, W., A. T. Smith, W. A. Doyle, and K. Piontek. 2001. Crystal structures of pristine and oxidatively processed lignin peroxidase expressed in Escherichia coli and of the W171F variant that eliminates the redox active tryptophan 171. Implications for the reaction mechanism. J. Mol. Biol. 305:851–861. [DOI] [PubMed] [Google Scholar]
- Breneman, C. M., and K. B. Wiberg. 1990. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comp. Chem. 11:361–397. [Google Scholar]
- Choinowski, T., W. Blodig, K. H. Winterhalter, and K. Piontek. 1999. The crystal structure of lignin peroxidase at 1.70 Å resolution reveals a hydroxy group on the Cβ of tryptophan 171: a novel radical site formed during the redox cycle. J. Mol. Biol. 286:809–827. [DOI] [PubMed] [Google Scholar]
- Doyle, W. A., W. Blodig, N. C. Veitch, K. Piontek, and A. T. Smith. 1998. Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry. 37:15097–15105. [DOI] [PubMed] [Google Scholar]
- Dunford, H. B. 1999. Heme Peroxidases. Wiley-VCH, New York.
- Edwards, S. L., R. Raag, H. Wariishi, M. H. Gold, and T. L. Poulos. 1993. Crystal structure of lignin peroxidase. Proc. Natl. Acad. Sci. USA. 90:750–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, A., and G. Tsaprailis. 1995. Catalytic structure-function relationships in heme peroxidases. Adv. Inorg. Chem. 43:79–125. [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, and J. A. Pople. 1998. Gaussian 98, Rev. A.3. Gaussian, Inc., Pittsburgh, PA.
- George, S. J., M. Kvratskhelia, M. J. Dilworth, and R. N. F. Thorneley. 1999. Reversible alkaline inactivation of lignin peroxidase involves the release of both the distal and proximal site calcium ions and bis-histidine coordination of the heme. Biochem. J. 344:237–244. [PMC free article] [PubMed] [Google Scholar]
- Hess, B., H. Bekker, J. Fraaije, and H. Berendsen. 1997. A linear constraint solver for molecular simulations. J. Comp. Chem. 18:1463–1472. [Google Scholar]
- Howes, B. D., A. Feis, L. Raimondi, C. Indiani, and G. Smulevich. 2001. The critical role of the proximal calcium ion in the structural properties of horseradish peroxidase. J. Biol. Chem. 276:40704–40711. [DOI] [PubMed] [Google Scholar]
- Izrailev, S., S. Stepaniants, B. Isralewitz, D. Kosztin, H. Lu, F. Molnar, W. Wriggers, and K. Schulten. 1998. Steered molecular dynamics. In Computational Molecular Dynamics: Challenges, Methods, Ideas, Vol. 4; Lecture Notes in Computational Science and Engineering. Springer-Verlag, Berlin, Germany. pp. 39–65.
- Kersten, P. J., B. Kalyanaraman, K. E. Hammel, B. Reinhammar, and T. K. Kirk. 1990. Comparison of lignin peroxidase, horseradish peroxidase and lactase in the oxidation of methoxybenzenes. Biochem. J. 268:475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosztin, D., S. Izrailev, and K. Schulten. 1999. Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophys. J. 76:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labat, G., and B. Meunier. 1990. Recent advances on the enzymic degradation of lignin. Bull. Soc. Chim. Fr. 127:553–564. [Google Scholar]
- MacKerell, A. D., B. Brooks, C. L. Brooks III, L. Nilsson, B. Roux, Y. Won, and M. Karplus. 1998. CHARMM: the energy function and its parameterization with an overview of the program. In Encyclopedia of Computational Chemistry, Vol 1. P. v. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. Kollman, and H. F. Schaeffer, III, editors. J. Wiley & Sons, New York. pp. 271–277.
- Marquez, L., H. Wariishi, H. B. Dunford, and M. H. Gold. 1988. Spectroscopic and kinetic properties of the oxidized intermediates of lignin peroxidase from Phanerochaete chrysosporium. J. Biol. Chem. 263:10549–10552. [PubMed] [Google Scholar]
- Meulenhoff, P. 1999. Interfacial action of phospholipase A2: a molecular dynamics study. University of Groningen, the Netherlands. (Ph.D. thesis.)
- Miyamoto, S., and P. A. Kollman. 1992. SETTLE: an analytical version of the shake and rattle algorithms for rigid water models. J. Comp. Chem. 13:952–962. [Google Scholar]
- Nie, G., and S. D. Aust. 1997. Spectral changes of lignin peroxidase during reversible inactivation. Biochemistry. 36:5113–5119. [DOI] [PubMed] [Google Scholar]
- Piontek, K., T. Glumoff, and K. Winterhalter. 1993. Low ph crystal structure of glycosylated lignin peroxidase from Phanerochaete chrysosporium at 2.5 Å resolution. FEBS Lett. 315:119–124. [DOI] [PubMed] [Google Scholar]
- Poulos, T. L., S. L. Edwards, H. Wariishi, and M. H. Gold. 1993. Crystallographic refinement of lignin peroxidase at 2 Å. J. Biol. Chem. 268:4429–4440. [DOI] [PubMed] [Google Scholar]
- Schoemaker, H. E. 1990. On the chemistry of lignin biodegradation. Recl. Trav. Chim. Pays-Bas. 109:255–272. [Google Scholar]
- Schoemaker, H. E., and K. Piontek. 1996. On the interaction of lignin peroxidase with lignin. Pure Appl. Chem. 68:2089–2096. [Google Scholar]
- Sollewijn Gelpke, M. D., J. Lee, and M. H. Gold. 2002. Lignin peroxidase oxidation of veratryl alcohol: effects of the mutants H82A, Q222A, W171A, and F267L. Biochemistry. 41:3498–3506. [DOI] [PubMed] [Google Scholar]
- ten Have, R., and P. J. M. Teunissen. 2001. Oxidative mechanisms involved in lignin degradation by white-rot fungi. Chem. Rev. 101:3397–3413. [DOI] [PubMed] [Google Scholar]
- van der Spoel, D., R. van Drunen, and H. J. C. Berendsen. 1994. GRoningen MAchine for Chemical Simulations. Department of Biophysical Chemistry, BIOSON Research Institute, Nijenborgh.
- van Gunsteren, W. F., X. Daura, and A. E. Mark. 1998. GROMOS force field. In Encyclopedia of Computational Chemistry, Vol. 2. P. v. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. Kollman, and H. F. Schaeffer, III, editors. J. Wiley & Sons, New York. pp. 1211–1216.