

Abstract
Plasminogen activator inhibitor 1 harbors four tryptophan residues at positions 86, 139, 175, and 262. To investigate the contribution of each tryptophan residue to the total fluorescence and to reveal the mutual interactions of the tryptophan residues and interactions with the other amino acids, 15 mutants in which tryptophan residues have been replaced by phenylalanines were constructed, purified, and characterized. Conformational distribution analysis revealed that the tryptophan mutants have a similar conformational distribution pattern as wild-type plasminogen activator inhibitor 1. Mutants in which tryptophan residue 175 was replaced by a phenylalanine displayed an increased functional half-life of the active conformation, whereas the functional half-life of mutants in which tryptophan residue 262 was replaced by a phenylalanine was substantially decreased. Comparative analysis of the fluorescence lifetimes, the extinction coefficients, and the quantum yields of the individual tryptophan residues demonstrates that tryptophan residue 262 gives the highest contribution to the total fluorescence. The other tryptophan residues have a very low quantum yield. In the wild-type protein, the fluorescence of all tryptophan residues is partially quenched as compared to the mutants that contain single tryptophan residues, due to conformational effects. The fluorescence of tryptophan residue 262 is very likely also partially quenched by energy transfer to tryptophan residue 175.
INTRODUCTION
Plasminogen activator inhibitor 1 (PAI-1) is the most important physiological inhibitor of tissue-type and urokinase-type plasminogen activator. It is a 50-kDa protein with 379 amino acids belonging to the serpin superfamily (Andreasen et al., 1986; Ginsburg et al., 1986; Ny et al., 1986; Pannekoek et al., 1986). All serpins consist of three β-sheets, nine α-helices (A through I), and a reactive site loop containing residues P16–P10′, which are highly variable (Gils and Declerck, 1998). The reactive site loop, situated 30–40 amino acids from the carboxy-terminal end, harbors a “bait” residue (P1 residue) that mimics the normal substrate of the target proteinase (Laskowski and Kato, 1980). In PAI-1, the Arg346-Met347 bond has been identified as the P1-P1′ bond (Lindahl et al., 1990). The protein plays an important role in the equilibrium between blood clot formation and blood clot lysis (Nilsson et al., 1985; Juhan Vague et al., 1995; Wiman, 1999). PAI-1 is synthesized as an active molecule that spontaneously converts into a latent form that cannot inhibit the target protease. The latent form can partially be reactivated by denaturants such as guanidinium chloride, sodium dodecyl sulfate, or urea (Hekman and Loskutoff, 1985). The spontaneous conversion to the latent form is unique for PAI-1. Even though for two other serpins a latent form has been described (Carrell et al., 1994; Lomas et al., 1995; Chang and Lomas, 1998), these could only be generated under denaturing conditions. This change in conformation has recently been described by computational methods (Krüger et al., 2001). PAI-1 can also occur in a stable noninhibitory cleavable substrate conformation (Declerck et al., 1992; Urano et al., 1992; Munch et al., 1993).
The x-ray structures of PAI-1 in the latent (Mottonen et al., 1992), the cleaved substrate (PAI-1 mutant in which alanine at position 335 is mutated into a proline) (Aertgeerts et al., 1995a), and recently the active conformation are known (Nar et al., 2000). The x-ray structure of the enzyme/inhibitor complex is not yet known. In the latent conformation, the P15–P4 residues insert into β-sheet A and the P4–P10′ residues form an extended loop on the surface of the molecule (Mottonen et al., 1992). Therefore, the latent conformation cannot interact with the target proteinases at the P1-P1′ site.
PAI-1 harbors four tryptophans at positions 86, 139, 175, and 262 (see Fig. 1). Tryptophan residue 86 is located at the turn connecting helix D with strand s2A. Among the serpins, this tryptophan residue is unique for PAI-1. Tryptophan residue 139 is located on the internal side of helix F and is conserved in antithrombin III and in PAI-1. Tryptophan residue 175 is conserved in most of the inhibitory serpins and is located in the turn connecting strand 3A with strand 4C. Tryptophan residue 262 is conserved in α1-antichymotrypsin and antithrombin III and is situated in helix H.
FIGURE 1.
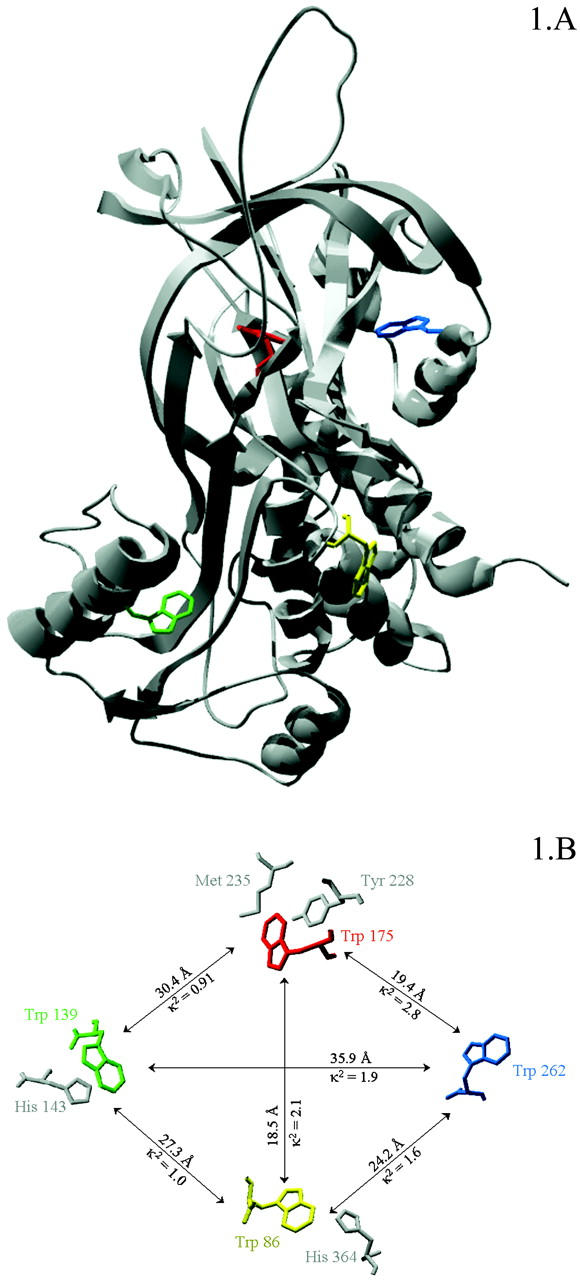
(A) Active conformation of PAI-1 with tryptophan residues 86, 139, 175, and 262, respectively highlighted in yellow, green, red and blue. (B) The different tryptophan residues in PAI-1 with their mutual distances, κ2 values, and possible fluorescence quenchers.
To study the contribution of each of the tryptophans to the total fluorescence of the protein in more detail, we have constructed, purified, and characterized 15 tryptophan mutants in which tryptophans have been replaced by a phenylalanine. This study reveals the importance of the tryptophan side chains on the kinetic stability of the active conformation. It shows that the fluorescence of tryptophan residue 262 gives the highest contribution to the total protein fluorescence, and that the fluorescence of all tryptophan residues is partially quenched in the wild-type protein, as compared to the mutants containing only a single tryptophan residue, whereas the fluorescence of tryptophan residue 262 is very likely also quenched by energy transfer to tryptophan residue 175.
EXPERIMENTAL PROCEDURES
Materials
Restriction enzymes were obtained from Amersham Biosciences (Uppsala, Sweden) or from Boehringer Mannheim (Brussels, Belgium). T4 DNA ligase, the Klenow fragment of Escherichia coli DNA polymerase I, and alkaline phosphatase were purchased from Boehringer Mannheim (Brussels, Belgium). The oligonucleotide-directed mutagenesis system (the pMa/c plasmids; (Stanssens et al., 1989)) was kindly provided by Corvas (Ghent, Belgium). The oligonucleotides for mutagenesis were purchased from Amersham Biosciences. M13KO7 helper phage was obtained from Promega (Leiden, The Netherlands). The expression vector pIGE20 was kindly provided by Innogenetics (Ghent, Belgium), together with the bacterial strains E. coli DH1λ for cloning and E. coli MC1061 for expression as well as the pAcI plasmid encoding the thermolabile repressor. The chromogenic substrate S-2403 was obtained from Chromogenix (Mölndal, Sweden). T-PA (predominantly single chain) was a kind gift from Boehringer Ingelheim (Brussels, Belgium).
Construction, expression and purification of PAI-1 and PAI-1 mutants
In vitro site-directed mutagenesis of PAI-1 was performed as described earlier (Audenaert et al., 1994) using the pMa/c system and the following synthetic oligonucleotides to obtain the desired mutation:
PrimW86F: 5′ CGTCTGTAGTACTGATCTCGTCCTTGTTAAATGGC 3′
PrimW139F: 5′ GTGTGTCTTCACAAAGTCATTGATGATG 3′
PrimW175F: 5′ GGGGAAGGGAGTTTTAAACTGGCCG 3′
PrimW262F: 5′ GGTCATGTTGCCTTTAAAGTGGCTGATGAGC 3′.
Except for primer PrimW139F, specific restriction sites were introduced (AGTACT ScaI-site or TTTAAA DraI-site) to allow confirmation of the induced mutation. SacI-XbaI fragments from the pMa-PAI-1-mutants were recovered and substituted for the SacI-XbaI in pIGE20-wild-type (wt)-PAI-1. Transformations of E. coli were performed using the calcium chloride method (Sambrook et al., 1989). Nucleotide sequencing using an Automatic Laser Fluorescence Detection Unit (ALF sequencer, Amersham Biosciences) was used to confirm the desired mutation. Expression and purification of wt-PAI-1 and PAI-1 mutants was performed in E. coli as described earlier (Gils et al., 1996). Extinction coefficients at 280 nm were determined as described by Mach et al. (1992). The extinction coefficients at 295 nm were determined by taking the proportion of the absorbance at 280 nm and 295 nm into account, and corrected as described by Mach et al. (1992).
Conformational distribution of PAI-1 mutants
PAI-1 samples were diluted with phosphate buffered saline (140 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4) to a concentration of 0.3 mg/ml and incubated for 30 min at 37°C with a fivefold molar excess of t-PA. The reaction was terminated by adding sodium dodecyl sulfate (final concentration 1%) and heating for 30 s at 100°C. Reaction products were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis using 10–15% gradient gels under nonreducing conditions with the Phast system (Amersham Biosciences). Proteins were visualized by staining with Coomassie brilliant blue. Reaction products were quantified with the Imagemaster using densitometric scanning (Amersham Biosciences).
Determination of the kinetic stability of purified wt-PAI-1 and PAI-1 variants at 37°C
Purified PAI-1 samples were diluted to a final concentration of 50–120 μg/ml to obtain a buffered solution with 45 mM phosphate pH 7.4 and 70 mM NaCl. Samples were incubated at 37°C and aliquots were removed at various times and assayed for inhibitory activity toward t-PA.
Fluorescence lifetime measurements at 10°C
Isolation of the reactive form of wt-PAI-1 and PAI-1 variants from the latent form was performed as described previously (Declerck et al., 1992). In brief, reactive wt-PAI-1 or PAI-1 variants were applied to immobilized t-PA-S478A, the nonbinding fraction (=latent form) was collected, and bound PAI-1 (=reactive form) was eluted with 1.5 M NaCl in 0.1 M sodium acetate pH 5.5.
All fluorescence measurements were done at 10°C to hinder the conversion of the reactive form into latent. Fluorescence lifetimes were measured using automatic multifrequency phase fluorometry between 1.6 MHz and 1 GHz as described previously (Sillen et al., 2000). N-acetyltryptophanamide in aqueous solution was used as a reference with a lifetime of 3.789 ns at 10°C. The measured phase shifts, φ, at a modulation frequency, ω, of the exciting light are related to the fluorescence decay in the time domain I(t),
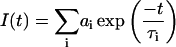 |
(1) |
where ai is the amplitude of the fluorescence signal of the component with lifetime τi, by means of the following equations (Weber, 1981):
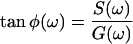 |
(2) |
where S(ω) and G(ω) are the sine and cosine Fourier transforms of I(t)
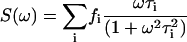 |
(3) |
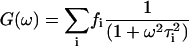 |
(4) |
and fi is the steady-state contribution of the component with phase shift φi
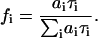 |
(5) |
Quantum yield measurements at 10°C
Fluorescence measurements were performed on a SPEX spectrofluorometer (Fluorolog-1691, Spex Industries, Edison, NJ) with excitation and emission slits at 7.2 and 3.6 nm, respectively. For selective observation of the tryptophan residues, an excitation wavelength of 295 nm was used. All measurements were done at a temperature of 10°C. All fluorescence spectra were corrected for the buffer and the wavelength dependence of the emission monochromator and the photomultiplier. The quantum yields were determined relative to N-acetyltryptophanamide in aqueous solution according to the method of Parker and Rees (1960) using a quantum yield for N-acetyltryptophanamide in water of 0.177 at 10°C:
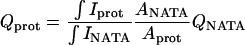 |
(6) |
where ∫I is the integrated intensity over the wavelength region 300–450 nm, Q is the quantum yield, and A is the absorbance at 295 nm.
Calculation of κ2
The geometric orientation factor (κ) has been calculated from:
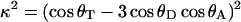 |
(7) |
where θT is the angle between the emission dipole of the donor and the absorption dipole of the acceptor, and θD and θA are the angles between these dipoles and the vector joining the midpoints of the CE2/CD2 bond of the donor and the acceptor, respectively. Indole has two excited states termed 1La and 1Lb (Valeur and Weber, 1977). The direction of transition moment of the 1La state was defined according to Ichiye and Karplus (1983). That of 1Lb was assumed to be at 88° relative to 1La. The values found are shown in Fig. 1.
RESULTS
Conformational distribution of wt-PAI-1 and PAI-1 mutants at 37°C
Despite the fact that all mutations were successfully constructed in the pIGE20-wt-PAI-1 vector, sequenced and transformed in E. coli MC1061 cells, the expression level of PAI-1-W86-139F and PAI-1-W175-262F was very low. As a consequence, further experiments could not be done with these mutants.
Incubation of wt-PAI-1 and the tryptophan mutants with a fivefold molar excess of t-PA all revealed the formation of t-PA/PAI-1 complexes originating from the active conformation—small amounts of cleaved derivative originating from the substrate form and residual nonreactive material being the latent form. The relative fractions of each mutant are displayed in Table 1.
TABLE 1.
Conformational distribution (expressed in %) of wt-PAI-1 and PAI-1 variants upon incubation with a fivefold molar excess of t-PA
| PAI-1 variants | Active (%) | Substrate (%) | Latent (%) |
|---|---|---|---|
| wt-PAI-1 | 53 | 11 | 36 |
| PAI-1-W86F | 59 | 17 | 24 |
| PAI-1-W139F | 57 | 10 | 33 |
| PAI-1-W175F | 68 | 15 | 17 |
| PAI-1-W262F | 52 | 9 | 39 |
| PAI-1-W86-139F | N.D.* | N.D. | N.D. |
| PAI-1-W86-175F | 71 | 13 | 16 |
| PAI-1-W86-262F | 76 | 7 | 17 |
| PAI-1-W139-175F | 86 | 11 | 3 |
| PAI-1-W139-262F | 73 | 15 | 12 |
| PAI-1-W175-262F | N.D. | N.D. | N.D. |
| PAI-1-W86-139-175F | 77 | 19 | 4 |
| PAI-1-W86-139-262F | 69 | 14 | 17 |
| PAI-1-W86-175-262F | 80 | 17 | 3 |
| PAI-1-W139-175-262F | 69 | 28 | 3 |
| PAI-1-W86-139-175-262F | 53 | 44 | 3 |
N.D., not determined.
Functional (kinetic) stability of PAI-1 mutants at 37°C
Wt-PAI-1 and PAI-1 mutants were I ncubated at 37°C and the remaining PAI-1 activity was determined at various time intervals. Table 2 shows the functional half-life of wt-PAI-1 and PAI-1-mutants. This half-life characterizes the loss of active fraction as a function of time at 37°C.
TABLE 2.
Functional half-life at 37°C (expressed in minutes) of wt-PAI-1 and PAI-1 variants
| PAI-1 variants | Functional half-life (min) |
|---|---|
| wt-PAI-1 | 108 ± 18* |
| PAI-1-W86F | 121 ± 19 |
| PAI-1-W139F | 111 ± 14 |
| PAI-1-W175F | 282 ± 17 |
| PAI-1-W262F | 17 ± 2 |
| PAI-1-W86-139F | N.D.† |
| PAI-1-W86-175F | 200 ± 32 |
| PAI-1-W86-262F | 15 ± 2 |
| PAI-1-W139-175F | 291 ± 29 |
| PAI-1-W139-262F | 15 ± 1 |
| PAI-1-W175-262F | N.D. |
| PAI-1-W86-139-175F | 320 ± 33 |
| PAI-1-W86-139-262F | 22 ± 5 |
| PAI-1-W86-175-262F | 17 ± 2 |
| PAI-1-W139-175-262F | 18 ± 3 |
| PAI-1-W86-139-175-262F | 20 ± 3 |
Mean ± SD, n = 3.
N.D., not determined.
In comparison with the half-life of wt-PAI-1 (108 ± 18 min), the mutants in which tryptophan residue 262 is replaced by a phenylalanine have a short half-life and the mutants in which tryptophan residue 175 is replaced by a phenylalanine have a longer half-life. Mutating either tryptophan residue 86 or tryptophan residue 139 into phenylalanine has no effect on the functional stability of PAI-1. When both tryptophan residues 262 and 175 are replaced by a phenylalanine, the functional half-life is shortened.
Fluorescence spectrophotometry of the active form at 10°C
After separation of the reactive forms from the latent forms, the quantum yields and fluorescence lifetimes were measured of the reactive forms. The fluorescence decay parameters were determined at emission wavelengths ranging from 320 to 380 nm in 10 nm intervals. Depending on the mutant, best fit (lowest χ2—no systematic deviation in the autocorrelation function or the weighted residuals) was obtained with a double- or triple-exponential fit.
Fig. 2 displays the phase shifts, the fitted curves, and the residuals values as a function of the frequency, together with the autocorrelation of the residuals for (A) PAI-1-W139-175-262F and (B) PAI-1-W86-139-175F. Both measurements were fitted with a two-exponential decay function. Fittings with a three-exponential decay function resulted in three lifetimes, two of which were the same.
FIGURE 2.

Representation of a phase measurement with the fit against their two-exponential function, the respective residuals, and autocorrelation functions of (A) PAI-1-W139-175-262F and (B) PAI-1-W86-139-175F.
Fig. 3 on the other hand displays the phase shifts, fitted curve, and residuals together with the autocorrelation of the residuals for PAI-1-W86-139-262F. This mutant was fitted with a three-exponential function (A) and a two-exponential function (B). The graphs clearly show that fitting with a three-exponential decay function gives the best result. To increase the accuracy of the recovery of the lifetimes and the fractional intensities, a global analysis of all the phase measurements at the different wavelengths was performed. This means that all the phase data were fitted with a single set of two or three lifetimes and at each wavelength a separate set of three amplitude fractions. There were no significant differences observed between the amplitude fractions at different wavelengths. The different quantum yields and fluorescence lifetimes are shown in Tables 3 and 4, respectively.
FIGURE 3.

Representation of a phase measurement of PAI-1-W86-139-262F fitted with (A) a three-exponential function and (B) a two-exponential function, the respective residuals, and autocorrelation functions.
TABLE 3.
Experimental and calculated quantum yields at 10°C of the reactive conformations of wt-PAI-1 and PAI-1 variants
| PAI-1 variants | Quantum yield | Calculated Q* |
|---|---|---|
| wt-PAI-1 | 0.085 ± 0.008† | 0.159 and 0.083‡ |
| PAI-1-W86F | 0.098 ± 0.007 | 0.185 |
| PAI-1-W139F | 0.126 ± 0.015 | 0.192 |
| PAI-1-W175F | 0.155 ± 0.009 | 0.182 |
| PAI-1-W262F | 0.060 ± 0.007 | 0.058 |
| PAI-1-W86-139F | N.D.§ | – |
| PAI-1-W86-175F | 0.223 ± 0.021 | 0.225 |
| PAI-1-W86-262F | 0.043 ± 0.007 | 0.052 |
| PAI-1-W139-175F | 0.250 ± 0.021 | 0.233 |
| PAI-1-W139-262F | 0.092 ± 0.006 | 0.071 |
| PAI-1-W175-262F | N.D. | – |
| PAI-1-W86-139-175F | 0.331 ± 0.033 | 0.127¶ |
| PAI-1-W86-139-262F | 0.075 ± 0.009 | −0.179¶ |
| PAI-1-W86-175-262F | 0.029 ± 0.011 | −0.078¶ |
| PAI-1-W139-175-262F | 0.067 ± 0.007 | 0.0383¶ |
| PAI-1-W86-139-175-262F | 0.009 ± 0.002 | – |
Calculated additively from data of the single tryptophans.
Mean ± SD, n = 3.
Calculated additively from data of single tryptophan residues W86, W139, and W175 + Q from W262 as present in wt (=0.127).
N.D., not determined.
Calculated subtractively from data of wt and −1W-mutants.
TABLE 4.
Different fluorescence lifetimes (τi) together with their amplitude fractions (ai), χ2 value, mean amplitude lifetime 〈τ〉, and Q/〈τ〉 values of the reactive conformations of wt-PAI-1 and PAI-1 variants measured at 10°C
| PAI-1 variants | a1 | τ1 | a2 | τ2 | a3 | τ3 | χ2 | 〈τ〉 | Q/〈τ〉 |
|---|---|---|---|---|---|---|---|---|---|
| wt-PAI-1 | 0.307* (0.044) | 0.624 (0.095) | 0.394 (0.026) | 2.158 (0.247) | 0.299 (0.047) | 5.349 (0.274) | 2.780 | 2.641 | 0.031 |
| PAI-1-W86F | 0.288 (0.032) | 0.495 (0.087) | 0.265 (0.022) | 2.180 (0.452) | 0.446 (0.074) | 5.532 (0.198) | 1.804 | 3.188 | 0.029 |
| PAI-1-W139F | 0.348 (0.027) | 0.833 (0.087) | 0.262 (0.030) | 2.522 (0.389) | 0.390 (0.054) | 5.416 (0.195) | 2.568 | 3.063 | 0.039 |
| PAI-1-W175F | – | – | 0.246 (0.085) | 1.921 (0.411) | 0.754 (0.231) | 5.355 (0.333) | 3.566 | 4.510 | 0.032 |
| PAI-1-W262F | 0.449 (0.028) | 0.543 (0.048) | 0.498 (0.021) | 1.938 (0.092) | 0.053 (0.004) | 5.344 (0.284) | 2.658 | 1.492 | 0.0375 |
| PAI-1-W86-139F | N.D.† | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| PAI-1-W86-175F | – | – | 0.251 (0.068) | 1.912 (0.324) | 0.749 (0.124) | 5.289 (0.276) | 3.654 | 4.441 | 0.046 |
| PAI-1-W86-262F | 0.561 (0.019) | 0.487 (0.026) | 0.389 (0.018) | 2.050 (0.087) | 0.050 (0.003) | 5.550 (0.208) | 2.842 | 1.348 | 0.029 |
| PAI-1-W139-175F | – | – | 0.278 (0.016) | 2.121 (0.094) | 0.722 (0.042) | 5.425 (0.111) | 1.806 | 4.506 | 0.051 |
| PAI-1-W139-262F | 0.247 (0.022) | 0.455 (0.083) | 0.602 (0.018) | 1.919 (0.081) | 0.151 (0.009) | 5.285 (0.184) | 1.908 | 2.065 | 0.040 |
| PAI-W175-262F | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| PAI-1-W86-139-175F | – | – | 0.122 (0.023) | 2.512 (0.335) | 0.878 (0.166) | 5.596 (0.140) | 1.808 | 5.220 | 0.056 |
| PAI-1-W86-139-262F | 0.398 (0.032) | 0.481 (0.057) | 0.510 (0.023) | 1.886 (0.126) | 0.091 (0.011) | 5.180 (0.241) | 2.356 | 1.625 | 0.038 |
| PAI-1-W86-175-262F | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| PAI-1-W139-175-262F | – | – | 0.668 (0.058) | 2.157 (0.120) | 0.332 (0.029) | 5.523 (0.289) | 1.766 | 3.275 | 0.017 |
Mean (SD), n = 3.
N.D., not determined.
The mutants in which tryptophan residue 262 is replaced by a phenylalanine have a lower quantum yield compared to wt-PAI-1 except for PAI-1-W139-262F. In contrast, the mutants in which tryptophan residues 139 and 175 are replaced by a phenylalanine have a higher quantum yield except for those mutants in which tryptophan residue 262 is also replaced by a phenylalanine.
There are three fluorescence lifetimes present in wt-PAI-1 around 0.6 ns, 2.2 ns, and 5.3 ns. The shortest lifetime only appears when tryptophan residue 175 is present. The other two lifetimes are more or less the same for all mutants. Besides the mutants with a low expression level, the fluorescence lifetimes of PAI-1-W86-175-262F and PAI-1-W86-139-175-262F could not be measured because of very low fluorescence intensities.
DISCUSSION
Conformational properties of the tryptophan residues
The primary intention of making the tryptophan mutants was to study the contribution of each individual tryptophan residue to the total fluorescence of wt-PAI-1 and to map the dissipation of the excited-state energy, and in this way learn more about protein fluorescence in general. This includes the energy transfer between the tryptophan residues as well as the interactions with the surrounding residues. In this study, the replacement of tryptophan by phenylalanine residues was chosen since both residues are aromatic. We didn't choose for replacement by tyrosine since a high number of tyrosine residues could interfere with our fluorescence measurements despite the fact that an excitation wavelength of 295 nm was used. The chosen mutations were expected to have little influence on the stability of PAI-1. However, comparing the functional half-life of wt-PAI-1 (108 ± 18 min) with the functional half-lives of the tryptophan mutants, the importance of tryptophan residues at positions 262 and 175 in the kinetics of the conversion of active to latent PAI-1 appears clearly.
During the conversion from the active to the latent form, the C-terminal portion of the reactive center loop (RCL) has to move through a gate formed between two loops, designated “loop 1” (residues 185–200) and “loop 2” (residues 242–246) (Aertgeerts et al., 1995b), resulting in i), the disruption of s1C (strand 1 of β-sheet C) (Mottonen et al., 1992) and ii), the concomitant insertion of the N-terminal portion (P14-P4) of the RCL into β-sheet A, thus forming an additional strand s4A (strand 4 of β-sheet A). The transition pad of this conversion was recently described at an atomic level using simulation techniques (Krüger et al., 2001).
The present study reveals that replacement of the tryptophan residue at position 175 strongly increases the kinetic stability of PAI-1. This tryptophan residue is positioned in the turn between strand 3A and strand 4C. Visual inspection of the structure reveals that tryptophan residue 175 could influence i), the movement of the RCL because it gets in close contact during the transition, ii), the insertion of the N-terminal portion of the RCL into β-sheet A, and iii), the mobility of “loop 1” thus again affecting the movement of the RCL during the transition.
Replacement of tryptophan residue 262 results in a decrease in functional (kinetic) stability indicating that a tryptophan at this position hinders the conformational transition. Tryptophan residue 262 is positioned at the end of helix H near turn thHs2C (the turn between helix H and strand 2C). We assume that a mutation at position 262 will influence the mobility of “loop 2”, thereby affecting the movement of the C-terminal portion of the RCL through the gate formed by “loop 1” and “loop 2”. The turn at which tryptophan residue 262 is positioned is also in close proximity with “loop 1” so this can also influence the movement of the RCL.
The mutants in which both tryptophan residues 262 and 175 are replaced by a phenylalanine show a half-life comparable to the functional half-life of the mutants in which only tryptophan residue 262 is replaced by a phenylalanine. So both mutations together do not neutralize one another. In contrast, the replacement of tryptophan residue 262 by a phenylalanine is not at all influenced by the other mutations, indicating the importance of a tryptophan residue at position 262.
Although we can clearly see the effect of these mutations on the kinetics of the conformational change, it cannot be automatically assumed that these mutations will alter the reactive conformation in a dramatic way. The difference in equilibrium fractions are rather small and even the kinetics suggest rather small effects. We base this conclusion on the fact that the maximal effect of the mutations is an increase of the rate constant for deactivation with a factor of 10. This corresponds to a change in ΔG* at room temperature of ∼1.38 kCal/mol or 5.5 kJ/mol. This is not a big change. Removing one hydrogen bridge would be sufficient to destabilize the starting state with this value (Pace et al., 1991). The same result could be obtained by stabilizing the transition state with one hydrogen bridge. Therefore we conclude that changes in the overall conformation of the active state cannot be very dramatic.
Fluorescence properties of the individual tryptophan residues
Fluorescence spectra
To calculate the quantum yields, the steady-state spectra had to be measured. Therefore, we were able to assign the four tryptophans of PAI-1 to one of the five classes of tryptophan residues according to Burstein and coworkers (Reshetnyak et al., 2001). Both tryptophan residues 139 (λmax = 324 nm) and 175 (λmax = 324 nm) belong to Class S and tryptophan residues 86 (λmax = 328 nm) and 262 (λmax = 330 nm) can be classified as Class I.
Quantum yields
The experimental quantum yields of wt-PAI-1 and the PAI-1 variants are shown in Table 3. It should be realized that these quantum yields are the average quantum yields of the individual tryptophan residues present in the mutants. It is remarkable that removing tryptophan residues 86, 139, and 175 individually results in an increased average quantum yield compared to the wild-type reference, whereas removing tryptophan residue 262 results in a decreased value. This already indicates that the individual quantum yields of tryptophan residues 86, 139, and 175 must be lower than the average value in the wild-type, and the quantum yield of tryptophan residue 262 must be higher.
This is confirmed by looking at the quantum yields of the individual tryptophan residues.
In a multi-tryptophan protein, the fluorescence quantum yield of an individual tryptophan can be obtained in two ways (De Beuckeleer et al., 1999). It can be obtained experimentally from the single-tryptophan containing protein where all the other tryptophan residues have been replaced by a phenylalanine (Qi,exp). It can also be calculated by subtraction (Qi,calc) from the quantum yield of the wild-type and of the mutant protein where only the concerned tryptophan has been removed (−1W-mutants). The two values do not have to be the same. In the first case, the tryptophan is not influenced by other tryptophan residues; in the second case, the quantum yield of a tryptophan can be influenced by energy transfer to or from the other tryptophan residues. In both cases the results can also be influenced by conformational changes induced by the mutations. These are more likely to occur in the mutants where more than one tryptophan has been replaced.
To calculate the quantum yield of the concerned tryptophan, the calculated fluorescence intensity of the mutant protein is subtracted from the calculated intensity of the wild-type and divided by its own extinction coefficient at 295 nm. The intensities mentioned are always calculated from the quantum yields and the appropriate extinction coefficients (ɛi) (Table 5):
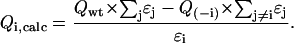 |
(8) |
TABLE 5.
Extinction coefficients at 280 nm of different PAI-1-variants, ratio of the absorbance at 295 nm and 280 nm (corrected according to Mach et al. (1992)), and extinction coefficients of the tryptophan residues at 295 nm, corrected for the contribution of the tyrosine residues at 295 nm (PAI-1-W86-139-175-262F)
| Tryptophan residue | ɛ280 nm (M−1cm−1) PAI-1 variant | A295 nm/A280 nm | ɛ295 nm (M−1cm−1) Trp residue |
|---|---|---|---|
| 86 (PAI-1-W139-175-262F) | 18860 | 0.172 | 2628 |
| 139 (PAI-1-W86-175-262F) | 18860 | 0.161 | 2421 |
| 175 (PAI-1-W86-139-262F) | 18860 | 0.167 | 2534 |
| 262 (PAI-1-W86-139-175F) | 18860 | 0.271 | 4495 |
| PAI-1-W86-139-175-262F | 13050 | 0.047 | – |
Table 3 shows the calculated quantum yields obtained in this way for the single-tryptophan mutants (Q). The results show that the quantum yields of the tryptophan residues 86 and 262 are higher when they are single tryptophan residues than when they are present in the wild-type, indicating that they are losing energy, e.g., by energy transfer or conformational effects. The calculated quantum yields of tryptophan residues 139 and 175 are even negative in the wild-type, indicating that they are decreasing the quantum yield of the other tryptophan residues.
In the additive method the quantum yield of a protein containing more than one tryptophan residue is calculated by averaging the quantum yields of the individual isolated tryptophan residues, taking their extinction coefficient into account.
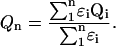 |
(9) |
The results (Table 3) show that for the mutants containing two tryptophan residues, the calculated average quantum yields agree within 10%–20% with the experimental ones. When looking at the calculated quantum yields of the three tryptophan-containing proteins, only the quantum yield of PAI-1-W262F is correctly calculated. Whenever tryptophan residue 262 is present, the experimental quantum yield is a lot smaller than the calculated one. This suggests that the presence of tryptophan residue 262 induces severe quenching of the protein fluorescence compared to the sum of the single tryptophans. It is tempting to attribute this quenching effect to energy transfer from the high quantum yield tryptophan residue 262 to the other residues, e.g., to tryptophan residue 175 at 1.9 nm distance. The calculated efficiency of energy transfer is indeed 63%. We calculated an R0 value of 2.1 nm, which is much bigger than the R0 of 1.1 nm for the tryptophan residues in Barnase (Willaert et al., 1992). The value of κ2 calculated was linked to the most probable 1La-1La transition. The values for the less probable 1La-1Lb transitions are smaller in all cases. The distances between the other tryptophan residues are much larger and their κ2 values are lower, which results in energy transfer efficiencies among the other tryptophan residues always to be lower than 1%. The conclusion is therefore that introducing tryptophan residue 262 brings in a high-intensity tryptophan that is partially quenched by energy transfer to tryptophan residue 175. Taking into account the efficiency of this energy transfer and the quantum yield of the acceptor, we calculate a new quantum yield for tryptophan residue 262 in wt equal to 0.169, which is still higher than the one directly observed. Therefore we must assume that additional quenching is due to a conformational change that leads to quenching of the protein fluorescence, and we cannot exclude that this conformational effect could be responsible for a large part of the quenching effect.
Fluorescence lifetimes
One would expect that a single tryptophan residue should have only one excited state lifetime. Actually, tryptophan fluorescence in proteins is known to exhibit multiexponential decay of which the origins are not completely understood (Callis, 1999). The existence of multiple fluorescence lifetimes for a single tryptophan residue could be due to excited state reactions (Lakowicz, 2000) or the existence of multiple ground states (e.g., rotamers) (Szabo and Rayner, 1980; Bajzer and Prendergast, 1993; Brown and Royer, 1997; Ababou and Bombarda, 2001). Hudson (1999) proposed a model that involves reversible ionization of the excited tryptophan residue due to collisional transfer of an electron to a neighboring residue. Other groups have also suggested the involvement of an electron transfer process as the principal quenching mechanism in proteins and peptides (Antonini et al., 1997; Chen and Barkley et al., 1998; Ababou et al., 2001). In the absence of quenching side chains, the carbonyl group of the peptide itself is thought to be the responsible quencher (Ricci and Nesta, 1976; Chen et al., 1998; Sillen et al., 2000; Adams et al., 2002). Since no difference is seen in amplitude fractions at different wavelengths, it is safe to assume that in PAI-1, the cause for multiple fluorescence lifetimes could lie in the existence of multiple microstates. Multiple protein micro conformations (Dahms et al., 1995) cause different interactions of the tryptophan side chain with amino acid groups of the surrounding protein matrix (Bajzer and Prendergast, 1993; Chen and Barkley, 1998). The longer lifetimes are thought to originate from conformations with little interaction with the environment or long distances between the indole ring and the peptide carbonyl, whereas the shorter lifetimes are the result of quenching from the solvent or the protein matrix (Chen et al., 1996).
From a first observation of the fluorescence lifetimes, we can conclude that the smallest fluorescence lifetime (∼0.5 ns) originates from tryptophan residue 175, because this fluorescence lifetime doesn't appear in the absence of this tryptophan residue. Together with the analysis of the quantum yields, this is a clear indication that this tryptophan residue is dynamically quenched by its environment. After closer investigation of its environment, two possible quenchers appear: methionine residue 235 (distance ∼0.4 nm) and tyrosine residue 228 (the aromatic rings of the two residues are situated parallel to each other within a distance of ∼0.45 nm).
Tryptophan residue 139 must be quenched, as indicated by the small quantum yield of PAI-1-W86-175-262F. Although the fluorescence lifetimes of PAI-1-W86-175-262F could not be determined, an average lifetime of 〈τ〉 ≅ 1 ns is calculated starting from data of the mutants PAI-1-W86-175F and PAI-1-W86-139-175F. Since there is no clear indication for energy transfer between tryptophan residues 262 and 139, we can calculate the Q/〈τ〉 value (which is the radiative rate constant kr) for PAI-1-W86-175-262F (tryptophan residue 139) starting from the Q/〈τ〉 values of PAI-1-W86-175F (tryptophan residues 139 and 262 present) and PAI-1-W86-139-175F (tryptophan residue 262), taking the extinction coefficients into account (Sillen and Engelborghs, 1998).
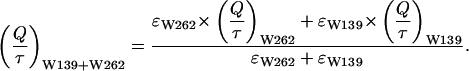 |
(10) |
Since the Q value is known for PAI-1-W86-175-262F, the 〈τ〉 value is easily calculated. This is a clear indication that tryptophan residue 139 is dynamically quenched. This dynamic quenching is possible by the presence of histidine residue 143, which is in close proximity of tryptophan residue 139 (∼0.36 nm).
In the same manner, the kr values for the other double mutants can be calculated, starting from the measured values of the triple tryptophan mutants (only one tryptophan present). The calculated and measured values should not differ, despite the fact that energy transfer or dynamic quenching could occur. Only static quenching or a change of kr will influence these parameters. One must keep in mind that these values can still differ somewhat because the propagation of the errors of the measurements. In case of PAI-1-W86-262F and PAI-1-W139-175F, the measured kr values (0.029 and 0.051, respectively) are close the calculated values (0.033 and 0.044, respectively). There is, however, a larger difference between the measured (0.040) and calculated (0.029) kr values of PAI-1-W139-262F. This can be due to larger errors in the measurements, or a change in conformational distribution.
The quantum yield of W86 is also very low. We assume this is due to static quenching, as clearly indicated by the Q/〈τ〉 (Sillen et al., 1999) value of PAI-1-W139-175-262F. In this case, the value is very low (0.017) in comparison with the Q/〈τ〉 value of wt-PAI-1 (0.031). The Q/〈τ〉 value will be independent of the fact that the fluorophore is dynamically quenched or not. When the quantum yield decreases due to a dynamic quencher, the 〈τ〉 will decrease to the same extent. But in case of static quenching, the quantum yield will decrease, but 〈τ〉 will stay unaffected, so the Q/〈τ〉 value will be small. At a first sight, there is no clear indication why tryptophan residue 86 is quenched. The only quenching residue in the neighborhood is histidine residue 364 at a distance of ∼0.35 nm. Although histidine is basically a dynamic quencher (Vos and Engelborghs, 1994), a very high frequency of interactions between the histidine residue and the tryptophan residue, thereby causing very fast dynamic quenching that escapes lifetime measurements, could be interpreted as static quenching (Webber, 1997).
To check the assumptions made in the last three paragraphs on the possible quenchers of tryptophan residues 86, 139, and 175, three mutants were constructed and purified, i.e., PAI-1-H143N, PAI-1-H364N, and PAI-1-Y228F. The functional half-lives as well as the quantum yields are displayed in Table 6.
TABLE 6.
Functional half-life at 37°C and quantum yields at 10°C of the reactive conformations of PAI-1 variants, constructed to reveal possible quenchers
| PAI-1 variants | Functional half-life (min) | Quantum yield |
|---|---|---|
| PAI-1-H143N | 123 ± 37* | 0.145 ± 0.014* |
| PAI-1-H364N | 12 ± 4 | 0.149 ± 0.029 |
| PAI-1-Y228F | 13 ± 5 | 0.076 ± 0.021 |
Mean ± SD, n = 3.
Because of the low expression levels, there was insufficient material to determine the fluorescence lifetimes of these mutants. To see directly the effect of the possible quenchers on their neighboring tryptophan residues without the influence of the other tryptophan residues, the mutants PAI-1-H143N-W86-175-262F and PAI-1-H364N-W139-175-262F were also constructed, but the expression levels were too low to perform any measurements.
The mutant PAI-1-H143N has a functional half-life of 123 ± 37 min, which is very similar to that of wt-PAI-1. This mutant was constructed to investigate if histidine residue 143 is a quencher of tryptophan residue 139. The quantum yield of this mutant is significantly larger than that of wt-PAI-1 and therefore we can conclude that this histidine residue is a possible quencher of tryptophan residue 139.
The mutant PAI-1-H364N was constructed to investigate the influence of histidine residue 364 on the fluorescence of tryptophan residue 86. This mutant has a significantly larger quantum yield than wt-PAI-1, which suggests that histidine residue 364 is indeed a static quencher of tryptophan residue 86. Unfortunately the mutant has a functional half-life of 12 ± 4 min, which is significantly smaller than wt-PAI-1. Therefore there exists the possibility that introducing the asparagine residue caused a conformational change.
The mutant PAI-1-Y228F was constructed to investigate the influence of tyrosine residue 228 on the fluorescence of residue 175. Again, this mutant has a significant smaller half-life than wt-PAI-1, but more important, the quantum yield is not significantly different from wt-PAI-1. Therefore, we can assume that this tyrosine residue cannot be considered as a quencher of tryptophan residue 175.
CONCLUSION
Two issues are clearly illustrated in this paper. First, the importance of tryptophan residues as large rigid amino acids emerges in the kinetics of the conformational change of PAI-1. Although the tryptophan residues in PAI-1 are not directly involved in the movement of the reactive center loop, tryptophan residues 262 and 175 influence the transition from the active to the latent conformation. While this paper was being reviewed, a paper appeared that also demonstrates the role of the tryptophan residues in the kinetics of the conformational changes, based on the results obtained with 7-azatryptophan mutants (Blouse et al., 2002).
We also show that the overall fluorescence properties of PAI-1 are governed mainly by the fluorescence of residue, i.e., tryptophan residue 262. The other residues have a very low quantum yield and the reasons are identified. Additivity of the quantum yields holds as long as tryptophan residue 262 is absent. When tryptophan residue 262 is introduced, a strong quenching appears, probably partially due to energy transfer to tryptophan residue 175 and partially to a conformational effect that leads to quenching of the protein fluorescence.
Acknowledgments
The authors are grateful to Griet Compernolle and Isabelle Knockaert for excellent technical assistance. We also thank Dr. Peter Krüger for discussions during the course of these investigations.
S. Verheyden is a recipient of an Institute for Science & Technology Studies fellowship. Dr. A. Gils is a postdoctoral fellow of the Fund for Scientific Research (FWO-Vlaanderen). This study was supported in part by the Institute for Science & Technology, the Research Fund of the Katholieke Universiteit Leuven (OT/98/37) (OT/97/19), and the FWO (G.0092.01).
References
- Ababou, A., and E. Bombarda. 2001. On the involvement of electron transfer reactions in the fluorescence decay kinetics heterogeneity of proteins. Protein Sci. 10:2102–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, P. D., Y. Chen, K. Ma, M. G. Zagorski, F. D. Sönnichsen, M. L. McLaughin, and M. D. Barkley. 2002. Intramolecular quenching of tryptophan fluorescence by the peptide bond in cyclic hexapeptides. J. Am. Chem. Soc. 124:9278–9286. [DOI] [PubMed] [Google Scholar]
- Aertgeerts, K., H. L. De Bondt, C. De Ranter, and P. J. Declerck. 1995a. Crystallization and X-ray diffraction data of the cleaved form of plasminogen activator inhibitor-1. Proteins. 23:118–121. [DOI] [PubMed] [Google Scholar]
- Aertgeerts, K., H. L. De Bondt, C. J. De Ranter, and P. J. Declerck. 1995b. Mechanisms contributing to the conformational and functional flexibility of plasminogen activator inhibitor-1. Nat. Struct. Biol. 2:891–897. [DOI] [PubMed] [Google Scholar]
- Andreasen, P. A., L. S. Nielsen, P. Kristensen, J. Grondahl Hansen, L. Skriver, and K. Dano. 1986. Plasminogen activator inhibitor from human fibrosarcoma cells binds urokinase-type plasminogen activator, but not its proenzyme. J. Biol. Chem. 261:7644–7651. [PubMed] [Google Scholar]
- Antonini, P. S., W. Hillen, N. Ettner, W. Hinrichs, P. Fantucci, S. M. Doglia, J. A. Bousquet, and M. Chabbert. 1997. Molecular mechanics analysis of Tet repressor Trp43 fluorescence. Biophys. J. 72:1800–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert, A. M., I. Knockaert, D. Collen, and P. J. Declerck. 1994. Conversion of plasminogen activator inhibitor-1 from inhibitor to substrate by point mutations in the reactive-site loop. J. Biol. Chem. 269:19559–19564. [PubMed] [Google Scholar]
- Bajzer, Z., and F. G. Prendergast. 1993. A model for multiexponential tryptophan fluorescence intensity decay in proteins. Biophys. J. 65:2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouse, G. E., M. J. Perron, J. H. Thompson, D. E. Day, C. A. Link, and J. D. Shore. 2002. A concerted structural transition in the plasminogen activator inhibitir-1 mechanism of inhibition. Biochemistry. 41:11997–12009. [DOI] [PubMed] [Google Scholar]
- Brown, M. P., and C. Royer. 1997. Fluorescence spectroscopy as a tool to investigate protein interactions. Curr. Opin. Biotechnol. 8:45–49. [DOI] [PubMed] [Google Scholar]
- Callis, P. R. 1999. Origins of nonexponential tryptophan fluorescence decay in proteins. Biophys. J. 76:A2, SaSG11–2. [DOI] [PubMed] [Google Scholar]
- Carrell, R. W., P. E. Stein, G. Fermi, and M. R. Wardell. 1994. Biological implications of a 3 A structure of dimeric antithrombin. Structure. 2:257–270. [DOI] [PubMed] [Google Scholar]
- Chang, W. S. W., and D. A. Lomas. 1998. Latent alpha(1)-antichymotrypsin. A molecular explanation for the inactivation of alpha(1)-antichymotrypsin in chronic bronchitis and emphysema. J. Biol. Chem. 273:3695–3701. [DOI] [PubMed] [Google Scholar]
- Chen, Y., and M. D. Barkley. 1998. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 37:9976–9982. [DOI] [PubMed] [Google Scholar]
- Chen, Y., B. Liu, H. T. Yu, and M. D. Barkley. 1996. The peptide bond quenches indole fluorescence. J. Am. Chem. Soc. 11:9271–9278. [Google Scholar]
- Dahms, T. E. S., K. J. Willis, and A. G. Szabo. 1995. Conformational heterogeneity of tryptophan in a protein crystal. J. Am. Chem. Soc. 117:2321–2326. [Google Scholar]
- De Beuckeleer, K., G. Volckaert, and Y. Engelborghs. 1999. Time resolved fluorescence and phosphorescence properties of individual tryptophan residues of barnase: evidence for protein-protein interactions. Proteins. 36:42–53. [PubMed] [Google Scholar]
- Declerck, P. J., M. De Mol, D. E. Vaughan, and D. Collen. 1992. Identification of a conformationally distinct form of plasminogen activator inhibitor-1, acting as a noninhibitory substrate for tissue-type plasminogen activator. J. Biol. Chem. 267:11693–11696. [PubMed] [Google Scholar]
- Gils, A., and P. J. Declerck. 1998. Structure-function relationship in serpins: current concepts and controversies. Thromb. Haemost. 80:531–541. [PubMed] [Google Scholar]
- Gils, A., I. Knockaert, and P. J. Declerck. 1996. Substrate behavior of plasminogen activator inhibitor-1 is not associated with a lack of insertion of the reactive site loop. Biochemistry. 35:7474–7481. [DOI] [PubMed] [Google Scholar]
- Ginsburg, D., R. Zeheb, A. Y. Yang, U. M. Rafferty, P. A. Andreasen, L. Nielsen, K. Dano, R. V. Lebo, and T. D. Gelehrter. 1986. cDNA cloning of human plasminogen activator-inhibitor from endothelial cells. J. Clin. Invest. 78:1673–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman, C. M., and D. J. Loskutoff. 1985. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J. Biol. Chem. 260:11581–11587. [PubMed] [Google Scholar]
- Hudson, B. S. 1999. An ionization/recombination mechanism for complexity of the fluorescence of tryptophan in proteins. Acc. Chem. Res. 32:297–300. [Google Scholar]
- Ichiye, T., and M. Karplus. 1983. Fluorescence depolarization of tryptophan residues in proteins: a molecular dynamics study. Biochemistry. 22:2884–2893. [DOI] [PubMed] [Google Scholar]
- Juhan Vague, I., M. C. Alessi, and P. J. Declerck. 1995. Pathophysiology of fibrinolysis. Baillieres Clin. Haematol. 8:329–343. [DOI] [PubMed] [Google Scholar]
- Krüger, P., S. Verheyden, P. J. Declerck, and Y. Engelborghs. 2001. Extending the capabilities of targeted molecular dynamics: simulation of a large conformational transition in plasminogen activator inhibitor 1. Protein Sci. 10:798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. R. 2000. On spectral relaxation in proteins. Photochem. Photobiol. 72:421–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, M., Jr., and I. Kato. 1980. Protein inhibitors of proteinases. Annu. Rev. Biochem. 49:593–626. [DOI] [PubMed] [Google Scholar]
- Lindahl, T. L., P. I. Ohlsson, and B. Wiman. 1990. The mechanism of the reaction between human plasminogen-activator inhibitor 1 and tissue plasminogen activator. Biochem. J. 265:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomas, D. A., P. R. Elliott, W. S. Chang, M. R. Wardell, and R. W. Carrell. 1995. Preparation and characterization of latent alpha 1-antitrypsin. J. Biol. Chem. 270:5282–5288. [DOI] [PubMed] [Google Scholar]
- Mach, H., C. R. Middaugh, and R. V. Lewis. 1992. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal. Biochem. 200:74–80. [DOI] [PubMed] [Google Scholar]
- Mottonen, J., A. Strand, J. Symersky, R. M. Sweet, D. E. Danley, K. F. Geoghegan, R. D. Gerard, and E. J. Goldsmith. 1992. Structural basis of latency in plasminogen activator inhibitor-1. Nature. 355:270–273. [DOI] [PubMed] [Google Scholar]
- Munch, M., C. W. Heegaard, and P. A. Andreasen. 1993. Interconversions between active, inert and substrate forms of denatured/refolded type-1 plasminogen activator inhibitor. Biochim. Biophys. Acta. 1202:29–37. [DOI] [PubMed] [Google Scholar]
- Nar, H., M. Bauer, J. M. Stassen, D. Lang, A. Gils, and P. J. Declerck. 2000. Plasminogen activator inhibitor 1. Structure of the native serpin, comparison to its other conformers and implications for serpin inactivation. J. Mol. Biol. 297:683–695. [DOI] [PubMed] [Google Scholar]
- Nilsson, I. M., H. Ljungner, and L. Tengborn. 1985. Two different mechanisms in patients with venous thrombosis and defective fibrinolysis: low concentration of plasminogen activator or increased concentration of plasminogen activator inhibitor. Br. Med. J. (Clin. Res. Ed.) 290:1453–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ny, T., M. Sawdey, D. Lawrence, J. L. Millan, and D. J. Loskutoff. 1986. Cloning and sequence of a cDNA coding for the human beta-migrating endothelial-cell-type plasminogen activator inhibitor. Proc. Natl. Acad. Sci. USA. 83:6776–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, N. C., U. Heinemann, U. Hahn, and W. Saenger. 1991. Ribonuclease T1: Structure, function, and stability. Angew. Chem. Int. Ed. Engl. 30:343–360. [Google Scholar]
- Pannekoek, H., H. Veerman, H. Lambers, P. Diergaarde, C. L. Verweij, A. J. van Zonneveld, and J. A. van Mourik. 1986. Endothelial plasminogen activator inhibitor (PAI): a new member of the Serpin gene family. EMBO J. 5:2539–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, C. A., and W. T. Rees. 1960. Corrections of fluorescence spectra and the measurement of fluorescence efficiency. Analyst. 85:587–600. [Google Scholar]
- Reshetnyak, Y. K., Y. Koshevnik, and E. A. Burstein. 2001. Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 81:1735–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci, R. W., and J. M. Nesta. 1976. Inter- and intramolecular quenching of indole fluorescence by carbonyl compounds. J. Phys. Chem. 80:974–980. [Google Scholar]
- Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sillen, A., F. Díaz, and Y. Engelborghs. 2000. A step toward the prediction of the fluorescence lifetimes of tryptophan residues in proteins based on structural and spectral data. Protein Sci. 9:158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillen, A., and Y. Engelborghs. 1998. The correct use of “average” fluorescence parameters. Photochem. Photobiol. 67:475–486. [Google Scholar]
- Sillen, A., J. Hennecke, D. Roethlisberger, R. Glockshuber, and Y. Engelborghs. 1999. Fluorescence quenching in the DsbA protein from Escherichia coli. The complete picture of the excited state energy pathway and evidence for the reshuffling dynamics of the microstates of tryptophan. Proteins. 37:253–263. [DOI] [PubMed] [Google Scholar]
- Stanssens, P., C. Opsomer, Y. M. McKeown, W. Kramer, M. Zabeau, and H. J. Fritz. 1989. Efficient oligonucleotide-directed construction of mutations in expression vectors by the gapped duplex DNA method using alternating selectable markers. Nucleic Acids Res. 17:4441–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, A. G., and D. M. Rayner. 1980. Fluorescence decay of tryptophan conformers in aqueous solution. J. Am. Chem. Soc. 102:554–563. [Google Scholar]
- Urano, T., L. Strandberg, L. B. Johansson, and T. Ny. 1992. A substrate-like form of plasminogen-activator-inhibitor type 1. Conversions between different forms by sodium dodecyl sulphate. Eur. J. Biochem. 209:985–992. [DOI] [PubMed] [Google Scholar]
- Valeur, B., and G. Weber. 1977. Resolution of the fluorescence excitation spectrum of indole into the 1La and 1Lb excitation bands. Photochem. Photobiol. 25:441–444. [DOI] [PubMed] [Google Scholar]
- Vos, R., and Y. Engelborghs. 1994. A fluorescence study of tryptophan-histidine interaction in the peptide anantin and in solution. Photochem. Photobiol. 60:24–32. [DOI] [PubMed] [Google Scholar]
- Webber, S. E. 1997. The role of time-dependent measurements in elucidating static versus dynamic quenching processes. Photochem. Photobiol. 65:33–38. [Google Scholar]
- Weber, G. 1981. Resolution of the fluorescence lifetimes in a heterogeneous system by phase and modulation measurements. J. Phys. Chem. 85:949–953. [Google Scholar]
- Willaert, K., R. Loewenthal, J. Sancho, M. Froeyen, A. R. Fersht, and Y. Engelborghs. 1992. Determination of the excited-state lifetimes of the tryptophan residues in barnase, via multifrequency phase fluorometry of tryptophan mutants. Biochemistry. 31:711–717. [DOI] [PubMed] [Google Scholar]
- Wiman, B. 1999. Predictive value of fibrinolytic factors in coronary heart disease. Scand. J. Clin. Lab. Invest. 59:23–31. [PubMed] [Google Scholar]