
TABLE 4.
Accuracy and coverage of secondary structure prediction by different predictors
| Cut1* | Cut2† | PHD | PSIPRED | SAM-T99 | Overlap‡ | Consensus§ |
|---|---|---|---|---|---|---|
| 0 | 0.23 | 76.6 | 80.8 | 80.1 | 79.2 | 81.3 |
| 1 | 0.33 | 76.6 | 81.0 | 80.1 | 79.6 | 81.0 |
| 2 | 0.37 | 76.9 | 80.8 | 80.1 | 79.9 | 80.7 |
| 3 | 0.41 | 76.7 | 80.6 | 80.2 | 80.4 | 80.0 |
| 4 | 0.45 | 76.2 | 79.9 | 80.1 | 80.8 | 78.9 |
| 5 | 0.49 | 75.8 | 78.7 | 79.5 | 81.1 | 77.0 |
| 6 | 0.53 | 75.1 | 77.3 | 77.6 | 80.4 | 74.4 |
| 7 | 0.57 | 73.4 | 74.6 | 75.5 | 78.9 | 71.2 |
| 0 | 0.23 | 52.1 | 51.6 | 51.7 | 57.3 | 44.9 |
| 1 | 0.33 | 51.9 | 48.9 | 51.7 | 56.2 | 43.4 |
| 2 | 0.37 | 49.0 | 46.5 | 51.2 | 55.1 | 41.9 |
| 3 | 0.41 | 46.0 | 43.7 | 49.4 | 53.3 | 39.4 |
| 4 | 0.45 | 42.7 | 41.2 | 45.9 | 50.3 | 36.6 |
| 5 | 0.49 | 39.3 | 38.4 | 41.7 | 46.5 | 33.5 |
| 6 | 0.53 | 35.8 | 35.1 | 37.2 | 42.4 | 29.9 |
| 7 | 0.57 | 32.0 | 31.0 | 32.5 | 38.1 | 25.5 |
Averaged on 125 test proteins. The upper part of the table is the percentage of accuracy defined as, 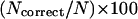 , where
, where 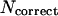 is the number of residues that are correctly assigned to either α-helix, β-strand or loop state, and N is the length of the sequence. The secondary structure elements in native structures are classified according to DSSP (Kabsch and Sander, 1983). The lower part of the table is the average number of residues that are assigned as α-helix or β-strand. The bold and italic bold numbers denote those used in our test runs for the evaluations of the secondary structure predictions in our fold simulations. The italic bold numbers are used in our final fold simulations.
is the number of residues that are correctly assigned to either α-helix, β-strand or loop state, and N is the length of the sequence. The secondary structure elements in native structures are classified according to DSSP (Kabsch and Sander, 1983). The lower part of the table is the average number of residues that are assigned as α-helix or β-strand. The bold and italic bold numbers denote those used in our test runs for the evaluations of the secondary structure predictions in our fold simulations. The italic bold numbers are used in our final fold simulations.
The threshold of confidence level (0 = low, 9 = high) for PHD and PSIPRED predictors.
The threshold of confidence level for SAM-T99. The confidence level for SAM-T99 is defined as the difference of the possibilities of the two highest confident assignments.
The definitions of “overlap” and “consensus” are in Table 5.