
TABLE 1.
Summary of the EDMC runs
| Peptide sequence* | Number of energy-minimized conformations† | Number of accepted conformations‡ | Zimmerman code of the lowest energy conformation§ | Number of cluster families¶ |
|---|---|---|---|---|
| PPP∥ | 135,222 | 2049 | FFFFFFADD | 77 |
| PPP** | 35,360 | 1000 | FFFFFFADA | 16 |
| PPP†† | 54,871 | 1091 | FFFFFFAAD | 23 (8) |
| PAP∥ | 129,533 | 1798 | FFCDFFADD | 80 |
| PAP** | 124,383 | 2000 | FFADFFADA | 26 |
| PAP†† | 53,675 | 1243 | FFFF*FFAAF | 45 (4) |
| PAAP∥ | 154,277 | 1875 | FFAAFFFCD*D | 99 |
| PAAP** | 147,092 | 2000 | FFAADFFADA | 46 |
| PAAP†† | 107,148 | 2363 | FFAA*A*FFACG | 74 (3) |
| PQP∥ | 178,308 | 2154 | FFAEFFAA*D | 90 |
| PQP** | 122,601 | 2078 | FFADFFADA | 23 |
| PQP†† | 66,747 | 1774 | FFAA*FFFAF | 72 (7) |
| PGP∥ | 162,930 | 2345 | FFFD*FFAGF | 105 |
| PGP** | 145,665 | 3000 | FFADFFADA | 30 |
| PGP†† | 64,634 | 1478 | FFADFFAAA | 46 (5) |
| PVP∥ | 184,106 | 1921 | FFCDFFADD | 86 |
| PVP** | 127,073 | 2629 | FFADFFADA | 29 |
| PVP†† | 114,613 | 3128 | FFAAFFACA | 45 (9) |
PXP and PXXP represent the X residue in the sequence Ac-PPPXPPPGY-NH2 (for X = Pro, Ala, Gln, Gly, and Val) and Ac-PPPXXPPPGY-NH2 (for XX = AlaAla), respectively.
These values correspond to the total number of generated conformations for the runs with three different force fields as explained in Methods and footnotes ∥, **, and ††, using the procedure described in Methods.
According to the Metropolis criterion.
For the leading member of family 1. Conformations are classified in terms of the regions of the φ−ψ Ramachandran (Ramachandran et al., 1963) map in which they occur (Zimmerman et al., 1977). On the left-hand half of the map (φ < 0°), the regions are defined as A–G; on the right-hand half of the map (φ ≥ 0°), the regions are defined by inversion of the left-hand half around the center of the map, and an asterisk is appended to the letters. The conformation of the guest residues are underlined. One of the alanines of PAAP in the PPII region (Zimmerman code F) is shown in boldface.
Total number of families after a minimal-tree cluster analysis of all accepted conformations, i.e., those listed in column 3 within a cutoff of 2 Å RMSD over all heavy atoms in the PXP and PAAP peptides, with no cutoff in energy. The number of cluster families containing >95% of the total accepted conformations is given in parentheses only for those peptides for which the Boltzmann-averaged 13C chemical shifts were computed as explained in Methods.
The calculations were carried out by using a GP potential, as explained in Methods.
The calculations were carried out by using a GPSAS potential, as explained in Methods.
The calculations were carried out at pH 7 at t = 25°C by using the GPSP potential, as described in Methods. The value of 10.10 was adopted as the 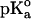 for the ionizable group of the Tyr residue, as an average from the data of Perrin (1972).
for the ionizable group of the Tyr residue, as an average from the data of Perrin (1972).