
TABLE 2.
Conformational analysis
| Peptide sequence* | Zimmerman code of the lowest energy conformation† | Conformation of pyrrolidine ring‡ | Peptide bond conformation§ | PPII content¶ (%) |
|---|---|---|---|---|
| PPP∥ | FFFFFFADD | DDD D DDD | TTC C TCT | 66.7 [66.7] (66.0 ± 2) |
| PPP** | FFFFFFADA | DDD D DDD | TTT T TTT | 68.0 [66.7] (66.0 ± 2) |
| PPP†† | FFFFFFAAD | DDD D DDU | CTC T CTT | 66.7 [66.7] (66.0 ± 2) |
| PAP∥ | FFCDFFADD | UUD - DDD | TTT - TCT | 44.4 [44.4] (63.0 ± 2) |
| PAP** | FFADFFADA | DUD - DDD | TTT - TTT | 46.2 [44.4] (63.0 ± 2) |
| PAP†† | FFFF*FFAAF | DDU - DDU | CTT - TTT | 66.7 [66.7] (63.0 ± 2) |
| PAAP∥ | FFAAFFFCD*D | DDD - - DDU | TTT - - CTT | 50.6 [50.0] (54.0 ± 2) |
| PAAP** | FFAADFFADA | DDD - - DDD | TTT - - TTT | 40.5 [40.0] (54.0 ± 2) |
| PAAP†† | FFAA*A*FFACG | DUD - - DDU | CTT - - CTT | 40.0 [40.0] (54.0 ± 2) |
| PQP∥ | FFAEFFAA*D | DUD - DDD | TTT - TCT | 44.4 [44.5] (65.0 ± 2) |
| PQP** | FFADFFADA | DUD - DDD | TTT - TTT | 46.6 [44.4] (65.0 ± 2) |
| PQP†† | FFAA*FFFAF | DDU - UUU | TTT - CTT | 66.7 [66.7] (65.0 ± 2) |
| PGP∥ | FFFD*FFAGF | DDU - DDD | TTT - TCT | 54.3 [66.5] (58.0 ± 2) |
| PGP** | FFADFFADA | DUD - DDD | TTT - TTT | 46.6 [44.4] (58.0 ± 2) |
| PGP†† | FFADFFAAA | UUU - DDU | TTT - CTT | 44.4 [44.4] (58.0 ± 2) |
| PVP∥ | FFCDFFADD | DUD - DDD | TTT - TCT | 44.8 [44.6] (49.0 ± 1) |
| PVP** | FFADFFADA | DUD - DDU | TTT - TTT | 46.0 [44.4] (49.0 ± 1) |
| PVP†† | FFAAFFACA | UDU - UDD | TTT - TCT | 33.3 [33.3] (49.0 ± 1) |
PXP and PXXP represent the X residue in the sequence Ac-PPPXPPPGY-NH2 (for X = Pro, Ala, Gln, Gly, and Val) and Ac-PPPXXPPPGY-NH2 (for XX = AlaAla), respectively. A dash is used in columns 3 and 4 to denote that the guest residue is not proline, and hence no results are displayed for this residue in these columns.
Conformations are classified in terms of the regions of the φ−ψ Ramachandran (Ramachandran et al., 1963) map in which they occur (Zimmerman et al., 1977). On the left-hand half of the map (φ < 0°), the regions are defined as A–G; on the right-hand half of the map (φ ≥ 0°), the regions are defined by inversion of the left-hand half around the center of the map, and an asterisk is appended to the letters.
The designation of U (up) and D (down) pertain to the (φ = −53.0° and χ1 = −28.1°) and (φ = −68.8° and χ1 = 27.4°) positions, respectively, of the Cγ atom.
C and T are used to designate the cis and trans states of the peptide bond only for prolines. All the non-prolines are trans.
For each peptide in this table, we show 1), the Boltzmann-averaged PPII helix content computed using all accepted conformations listed in the third column of Table 1; 2), within brackets, the Boltzmann-averaged PPII helix content computed by using only the leading members, weighted by the population, of the corresponding cluster listed in the fifth column of Table 1; and 3), in parentheses, the corresponding experimental values (Kelly et al., 2001). Very good agreement for the computed Boltzmann-averaged PPII helix content exists between both procedures 1 and 2, i.e., with a correlation coefficient of R = 0.96 and slope of 1.02 for the correlation line. In these calculations, it is assumed that only residues in the F region contribute to the PPII helix content.
The calculations were carried out by using a GP potential, as explained in Methods.
The calculations were carried by using a GPSAS potential, as explained in Methods.
The calculations were carried out at pH 7 at t = 25°C by using the GPSP potential, as described in Methods. The value of 10.10 was adopted as the 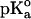 for the ionizable group of the Tyr residue, as an average from the data of Perrin (1972).
for the ionizable group of the Tyr residue, as an average from the data of Perrin (1972).