

Abstract
Of the several hundred proteins induced by interferon (IFN) α/β, the ubiquitin-like ISG15 protein is one of the most predominant. We demonstrate the novel way in which the function of the ISG15 protein is inhibited by influenza B virus, which strongly induces the ISG15 protein: a specific region of the influenza B virus NS1 protein, which includes part of its effector domain, blocks the covalent linkage of ISG15 to its target proteins both in vitro and in infected cells. We identify UBE1L as the E1 enzyme that catalyzes the first activation step in the conjugation of ISG15, and show that the NS1B protein inhibits this activation step in vitro. Influenza A virus employs a different strategy: its NS1 protein does not bind the ISG15 protein, but little or no ISG15 protein is produced during infection. We discuss the likely basis for these different strategies.
Keywords: E1 activating enzyme/influenza virus NS1 protein/interferon-induced proteins/ISG15 protein/ubiquitin-like proteins
Introduction
The cellular proteins that are induced by interferon (IFN α/β) constitute one of the initial host defenses against virus infection (Welsh and Sen, 1997; Stark et al., 1998). Analysis by oligonucleotide arrays indicates that IFN α/β stimulates the transcription of several hundred genes (Der et al., 1998), resulting in at least an equivalent number of IFN-induced proteins, many of which probably participate in the antiviral response (Der et al., 1998; Stark et al., 1998).
The mechanisms by which most IFN-induced proteins carry out their antiviral actions, and the viral countermeasures against these actions, have not been established (Der et al., 1998; Stark et al., 1998). Many investigations have focused on two IFN-induced antiviral pathways, those involving the protein kinase PKR and 2′-5′ oligoadenylate synthetase (2-5 OAS)/RNase L (reviewed in Welsh and Sen, 1997; Stark et al., 1998). These two IFN-induced pathways are directly activated by viral double-stranded RNA (dsRNA) molecules, which are produced during infection by either DNA or RNA viruses. Activated PKR phosphorylates the α-subunit of the translation initiation factor eIF2 (eIF-2α), resulting in the inhibition of protein synthesis, and the products synthesized by activated 2-5 OAS activate RNase L, which efficiently degrades viral and cellular RNAs. There are several mechanisms by which viruses mount counterattacks against these two IFN-induced pathways. The activation of PKR and 2-5 OAS is inhibited by the sequestering of dsRNA by proteins encoded by several viruses (reviewed in Welsh and Sen, 1997). In addition, several viruses employ alternative strategies to thwart the antiviral activity of PKR (Beattie et al., 1991; Matthews and Shenk, 1991).
Little is known about the many other IFN-induced antiviral proteins and the measures by which viruses counter the actions of these proteins (Der et al., 1998; Stark et al., 1998). Here we report the novel mechanism by which the activity of one of these other IFN-induced proteins, the ubiquitin-like ISG15 protein, is counteracted during infection by influenza B virus. ISG15 is one of the most strongly induced proteins after IFN treatment (Farrell et al., 1979; Der et al., 1998), and is also strongly induced in cells infected by influenza B virus (present study). As is the case for other ubiquitin-like proteins such as SUMO-1 and Rub1 (Jentsch and Pyrowolakis, 2000; Yeh et al., 2000), the ISG15 protein is covalently attached via its C-terminal glycine residue to particular target proteins (Loeb and Haas, 1992). It is not known whether ISG15 modification, like ubiquitin modification, results in degradation of the modified target proteins (Loeb and Haas, 1992, 1994; Jentsch and Pyrowolakis, 2000). The specific target proteins that are modified by ISG15 conjugation have not been identified (Loeb and Haas, 1994; Jentsch and Pyrowolakis, 2000).
In this study we demonstrate that influenza B virus, which strongly induces the ISG15 protein during infection, specifically blocks ISG15 protein conjugation. This inhibition is mediated by the viral NS1 protein (NS1B protein): a specific region of this protein blocks the covalent linkage of ISG15 to its target proteins both in vitro and in infected cells. We demonstrate that the first activation step in the conjugation of ISG15 is catalyzed by a previously identified protein, UBE1L, whose function was not known (Kok et al., 1993, 1995). The region of the NS1B protein that is required for the inhibition of this activation step includes both of its functional domains: the RNA-binding domain (the N-terminal 93 amino acids), which binds dsRNA (Wang and Krug, 1996), and part of the adjacent effector domain. Influenza A virus employs a different strategy: its NS1 protein (NS1A protein) does not bind the ISG15 protein, but little or no ISG15 protein is produced during infection. We discuss the likely basis for these different strategies.
Results
Identification of a cellular protein, ISG15, that binds specifically to the influenza virus NS1B protein and not to the influenza NS1A protein
Using a yeast two-hybrid interaction trap, we screened a human cDNA library for human proteins that specifically interact with the NS1B protein of influenza B virus and not with the NS1A protein of influenza A virus. Yeast colonies containing cDNAs encoding such proteins were selected as described in Materials and methods. Most of the identified cDNAs encode an IFN-induced protein, ISG15, a human ubiquitin-like protein that contains a tandem repeat of two ubiquitin-like domains (Haas et al., 1987).
To determine whether ISG15 binds directly to the NS1B protein, we carried out glutathione S-transferase (GST)-pulldown experiments in vitro. The GST–ISG15 fusion protein was immobilized on glutathione–agarose beads, and the ability of a 35S-labeled NS1B or NS1A protein to bind to this fusion protein was determined (Figure 1). The labeled NS1B and NS1A proteins were synthesized in vitro, and the extracts were treated with micrococcal nuclease for 30 min at 37°C prior to the GST-pulldown. The NS1B protein binds efficiently to the GST–ISG15 protein on the glutathione–agarose beads (lanes 1, 3 and 5). In contrast, no binding of the NS1A protein to the GST–ISG15 protein was detected (lanes 2, 4 and 6). These results suggest that the ISG15 protein binds directly to the NS1B protein (see below), and indicate that ISG15 does not bind to the NS1A protein.
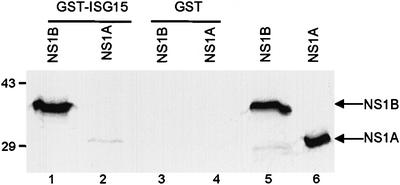
Fig. 1. The NS1B, but not the NS1A protein binds to the ISG15 protein in vitro. GST–ISG15 protein (1 µg) (lanes 1 and 2) or GST (1 µg) (lanes 3 and 4) was incubated with 20 µl of 35S-labeled NS1B protein (lanes 1 and 3) and NS1A protein (lanes 2 and 4) in the presence of 20 µl of glutathione–Sepharose 4B beads. The labeled proteins eluted from the beads were analyzed by electrophoresis on a 12% SDS–polyacrylamide gel. Lanes 5 and 6: 2 µl of the indicated 35S-labeled NS1 protein. The mobilities of molecular weight markers are shown on the left.
We used this GST-pulldown assay to determine which region of the NS1B protein binds to ISG15 (Figure 2). The N-terminal, 93-amino acid fragment, NS1B(1–93), which constitutes the RNA-binding domain (Wang and Krug, 1996), does not bind to the ISG15 protein (Figure 2B, lane 2). Surprisingly, a slightly larger N-terminal fragment, NS1B(1–103), binds ISG15 as efficiently as the full-length NS1B protein (compare lane 3 with lane 1). A larger N-terminal fragment, NS1B(1–145), binds with an efficiency similar to that of NS1B(1–103) (lane 4). Deletion of either the 94–103 region (lane 5), or the RNA-binding domain, i.e. the N-terminal 91 amino acids (lane 6), from the full-length NS1B protein abolishes binding to ISG15, verifying that both of these regions of the NS1B protein are required for ISG15 binding. Consequently, ISG15 binding to the NS1B protein requires the N-terminal 103 amino acids of NS1B.
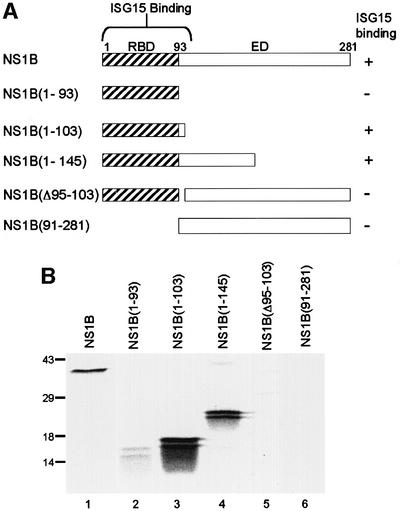
Fig. 2. Identification of the ISG15 binding site on the NS1B protein. (A) Schematic diagram of the fragments of the NS1B protein that were used in the binding assay. RBD, RNA-binding domain; ED, effector domain. (B) Binding assay. GST–ISG15 protein (1 µg) was incubated with 104 c.p.m. of 35S-labeled full-length NS1B protein (lane 1), or the indicated fragment of the NS1B protein (lanes 2–6) in the presence of 20 µl of glutathione–Sepharose 4B beads. The same amount of each radiolabeled NS1B fragment was added to the binding assay. The labeled proteins eluted from the beads were analyzed by electrophoresis on a 12% SDS–polyacrylamide gel. The doublets seen with the NS1B fragments (lanes 2–4) were present both before and after the binding assay. The mobilities of molecular weight markers are shown on the left.
The NS1B protein inhibits ISG15 function in vitro
The first step of the pathway by which ubiquitin is conjugated to its target proteins is the formation of a thioester bond between ubiquitin and the ubiquitin-activating enzyme, E1 (reviewed in Hershko and Ciechanover, 1998). A similar, but distinct pathway for ISG15 activation has been proposed (Narasimhan et al., 1996; Jentsch and Pyrowolakis, 2000). We developed an in vitro assay for an E1-like enzyme specific for ISG15 using extracts from IFN-treated A549 cells, a human lung cancer cell line (Lieber et al., 1976). The substrate for this assay was recombinant ISG15 containing a kinase domain that was labeled with 32P (Figure 3A). Incubation of this substrate with extracts from IFN-treated A549 cells for 2 h at 37°C results in the formation of two labeled protein species that are not formed by extracts from A549 cells not treated with IFN (Figure 3B, lanes 1 and 2). Hence the activities that form these two species, which are candidates for ISG15–E1 and ISG15–E2 adducts, are induced by IFN treatment, as is the case for the ISG15 substrate. We postulated that the species of larger molecular weight is the E1 adduct, based on analogy with the corresponding E1 and E2 enzymes in ubiquitin conjugation (Hershko and Ciechanover, 1998). The formation of these two protein species is inefficient in vitro, as shown by the presence of a large amount of unreacted ISG15.
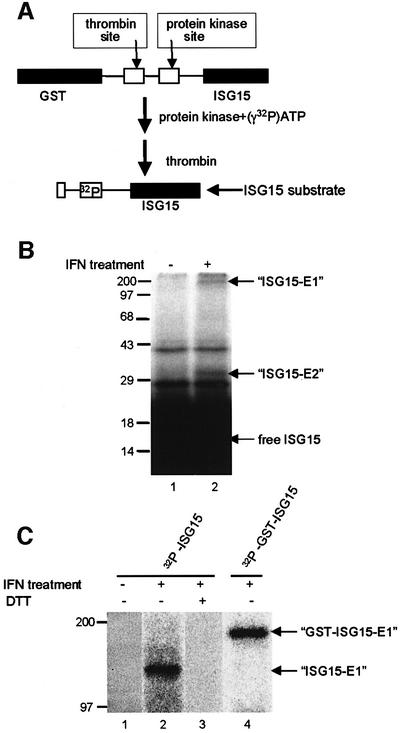
Fig. 3. In vitro assay for the E1-like enzyme that activates ISG15. (A) Schematic diagram of the strategy for preparing the 32P-labeled ISG15 substrate. (B) An extract (40 µg) from IFN-treated A549 cells (lane 2) or from untreated A549 cells (lane 1) was incubated with 106 c.p.m. (70 pmol) of 32P-labeled ISG15 protein in a 50 µl reaction, as described in Materials and methods. After a 2 h incubation at 37°C, the reactions were terminated by adding SDS sample buffer lacking DTT, and subjected to electrophoresis on a 12.5% SDS–PAGE gel in the absence of a reducing agent. The putative ISG15–E1 and ISG15–E2 adducts are denoted by arrows, and the mobilities of molecular weight markers are shown on the left. (C) An extract (40 µl) from IFN-treated A549 cells (lanes 2–4) or from untreated A549 cells (lane 1) was incubated for 2 h with 106 c.p.m. of 32P-labeled ISG15 protein (lanes 1–3) or 32P-labeled GST–ISG15 protein (lane 4) in a 50 µl reaction. The reactions were terminated by adding SDS sample buffer lacking DTT (lanes 1, 2 and 4) or containing DTT (lane 3), and analyzed on a 7.5% SDS–PAGE gel. The putative GST–ISG15–E1 and ISG15–E1 adducts are denoted by arrows.
In the present study we focused on the putative ISG15–E1 adduct, and analyzed the in vitro products on gels that resolve species of >90 kDa in molecular weight (Figure 3C). The putative ISG15–E1 adduct, which is formed by extracts from IFN-treated but not from untreated A549 cells (lanes 1 and 2), has a molecular weight of ∼140 kDa. Treatment of the reaction products of lane 2 with dithiothreitol (DTT) causes the disappearance of the labeled 140 kDa species (lane 3), consistent with the formation of a thioester bond between ISG15 and its E1-like enzyme. Formation of the 140 kDa species requires ATP (data not shown), as is the case for the conjugation of ubiquitin to its E1 activating enzyme (Hershko and Ciechanover, 1998). When the size of the ISG15 substrate was increased by the addition of N-terminal GST, the labeled protein species also increases in size (∼200 kDa) (lane 4), confirming that this E1-like protein forms a specific bond with the ISG15 protein.
To determine whether the NS1B protein inhibits the formation of this ISG15–E1-like intermediate, increasing amounts of GST–NS1B were added to the in vitro reaction (Figure 4A). GST–NS1B dramatically inhibits the formation of this intermediate (lanes 2–4): 30% inhibition in the presence of 20 pmol of NS1B, and >95% inhibition in the presence of either 80 or 160 pmol of NS1B. In contrast, the GST–NS1A protein, even at 160 pmol, has no detectable effect on the formation of the ISG15–E1-like intermediate (lanes 5–7). Thus, the NS1B protein and not the NS1A protein inhibits the coupling of ISG15 to its cognate E1-like enzyme.
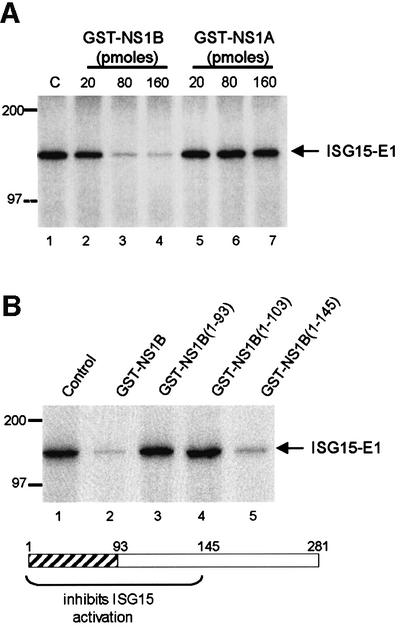
Fig. 4. Identification of the region of the NS1B protein that is required for inhibition of the in vitro ISG15-coupling reaction. (A) The NS1B protein, and not the NS1A protein, inhibits the formation of the ISG15–E1-like intermediate in vitro. 32P-labeled ISG15 protein (106 c.p.m., 70 pmol) was pre-incubated with the indicated amounts of GST–NS1B (lanes 2–4) or GST–NS1A (lanes 5–7) prior to the incubation with the extract from IFN-treated A549 cells. C (lane 1), pre-incubation of ISG15 in the absence of an NS1 protein. (B) The N-terminal 145 amino acid region of the NS1B protein is required for inhibition of the coupling of ISG15 to the E1-like enzyme. 32P-labeled ISG15 protein (106 c.p.m., 70 pmol) was pre-incubated with 160 pmol of either full-length NS1B protein (lane 2), or the indicated fragment of the NS1B protein (lanes 3–5) prior to incubation with the extract from IFN-treated A549 cells. Control (lane 1): pre-incubation of ISG15 in the absence of an NS1 protein. The hatched region in the NS1B protein is the RNA-binding domain.
To identify the region of the NS1B protein that is required for the inhibition of this in vitro ISG15-coupling reaction, the same N-terminal fragments that were used to map the ISG15 binding site on the NS1B protein (Figure 2) were added to the in vitro coupling reaction (Figure 4B). The NS1B fragment containing the first 103 N-terminal amino acids, GST–NS1B(1–103), which binds efficiently to ISG15, does not inhibit the coupling reaction (lane 4). In contrast, the longer N-terminal NS1B fragment containing the first 145 N-terminal amino acids, GST–NS1B(1–145), inhibits the coupling reaction almost as efficiently as the full-length NS1B protein (compare lanes 5 and 2). Consequently, inhibition of the coupling of ISG15 to the E1-like enzyme is mediated by the N-terminal 145 amino acid region of the NS1B protein.
To establish that the 140 kDa species is formed by an ISG15-specific E1 activating enzyme, we purified and sequenced the protein that is linked to ISG15 in this adduct. Extracts from IFN-treated A549 cells were bound to GST–ISG15 immobilized on glutathione–agarose beads, and the eluted proteins were analyzed by electrophoresis on a denaturing gel containing a reducing agent (Figure 5, lane 2). A single protein band of ∼110 kDa was detected by Coomassie Blue staining. This protein was absent from extracts that were eluted from glutathione– agarose beads containing GST alone (lane 1), indicating that the 110 kDa protein was retained by, and eluted from the immobilized ISG15 and not GST. Analysis of the 110 kDa protein by matrix assisted laser deionization-mass spectrometry (MALDI-MS) indicated a match to a protein in the database, the UBE1L protein or ubiquitin E1-like protein, whose function was not known (Kok et al., 1993, 1995). In addition, one of the trypsin-derived peptides of the 110 kDa protein was sequenced by Edman degradation; this sequence exactly matches amino acids 972–986 in the UBE1L protein (Figure 5). We conclude that the 110 kDa protein is the previously identified UBE1L protein.
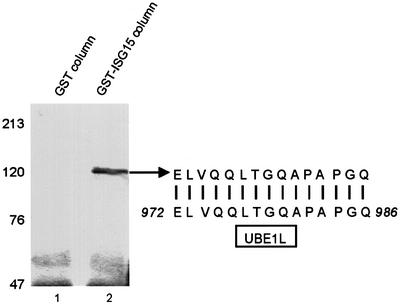
Fig. 5. Identification of the E1 enzyme that activates ISG15. (A) Cell extracts from IFN-treated A549 cells were subjected to affinity selection on a glutathione–Sepharose 4B column containing either GST (lane 1) or the GST–ISG15 fusion protein (lane 2). The eluate was resolved by 7.5% SDS–PAGE, followed by staining with Coomassie Blue. The 110 kDa protein species from lane 2 was analyzed by MALDI-MS, and one of the tryptic peptides was microsequenced by automated Edman degradation. The sequence of this peptide is compared to the amino acid sequence of UBE1L (amino acids 972–986).
To definitively establish that the UBE1L protein functions as the E1 activating enzyme for the ISG15 protein, the UBE1L protein was expressed as a GST fusion protein using a baculovirus vector (Figure 6A). The extracts from cells infected by this recombinant baculovirus catalyze the formation of an adduct with 32P-labeled GST–ISG15 which has a molecular weight of ∼170 kDa (Figure 6B, lanes 1 and 2) and is DTT sensitive (data not shown), indicating that the GST–UBE1L protein in this extract functions as the ISG15-specific E1 activating enzyme. A less abundant, faster migrating species, which varies in amount in different experiments, probably represents a proteolytic cleavage product. The formation of the 170 kDa species was blocked by 160 pmol of the NS1B protein (lane 3), but not by the same amount of the NS1A protein (lane 4). These results provide further evidence that the direct interaction between the NS1B and ISG15 proteins blocks the activation of ISG15 by UBE1L. The GST–UBE1L protein does not form an adduct with 32P-labeled ubiquitin (Ub) (lane 5), indicating that GST–UBE1L specifically activates ISG15 but not ubiquitin. Confirmation of this specificity was obtained using the GST–UBE1L purified on glutathione–Sepharose 4B beads: the purified enzyme activates ISG15, but not ubiquitin (Figure 6C).
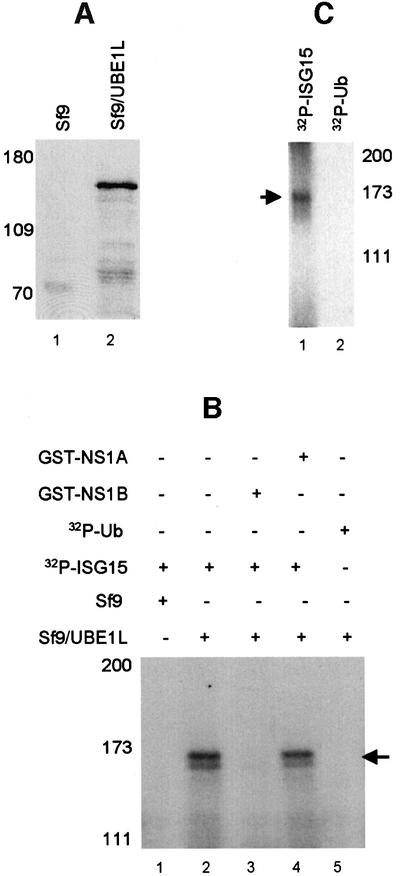
Fig. 6. The recombinant UBE1L protein catalyzes the activation of ISG15. (A) The UBE1L protein is synthesized in insect cells infected with the recombinant virus (lane 2). Lane 1: uninfected Sf9 cells. Extracts from uninfected and infected cells were analyzed by SDS–PAGE followed by western analysis using anti-GST antibody. (B) Extracts from uninfected Sf9 insect cells (lane 1) or Sf9 cells infected with the recombinant baculovirus expressing the GST–UBE1L protein (lane 2) were incubated with 106 c.p.m. (70 pmol) of 32P-labeled ISG15, as described in the legend of Figure 3. Prior to incubation with the extract from cells infected with the recombinant baculovirus, the 32P-labeled ISG15 protein was pre-incubated with 160 pmol of either GST–NS1B (lane 3) or GST–NS1A (lane 4). In lane 5, 32P-labeled ubiquitin (Ub) was incubated with the extract from cells infected with the recombinant baculovirus. Labeled Ub was prepared using a plasmid containing the Ub sequence cloned into pGEX-2TK (kindly provided by Jon Huibregtse). The reaction products were analyzed by gel electrophoresis as described in the legend of Figure 3. The labeled ISG15–GST–UBE1L adduct is denoted by the arrow. (C) The GST–UBE1L protein was purified from the extract of insect cells infected with the recombinant baculovirus using glutathione–Sepharose, and the purified protein was incubated with 106 c.p.m. of either 32P-labeled ISG15 (lane 1) or 32P-labeled ubiquitin (Ub) (lane 2).
The ISG15 protein is synthesized in human cells infected by influenza B virus, but not in cells infected by influenza A virus
To determine whether the ISG15 protein is induced by influenza B virus, A549 human cells were infected with 20 plaque-forming units (p.f.u.) per cell of influenza B/Lee/40 virus. At various times after infection, total cell proteins were resolved by gel electrophoresis, followed by western blotting with ISG15-specific antiserum. For comparison, the proteins of uninfected A549 cells were analyzed for ISG15 production at various times after the addition of IFN. In the presence of IFN, free ISG15 is produced for an extended period of time (for ∼12 h) before conjugation of ISG15 to its target protein begins (Figure 7A), as reported previously by others (Loeb and Haas, 1992). Even after extensive conjugation of ISG15 has occurred at 24 h, a large amount of free (unconjugated) ISG15 is present in the cells. The time course of ISG15 synthesis in influenza virus B-infected cells (lanes 2–5) is similar to that in IFN-treated cells (lanes 6–9). However, although similar amounts of ISG15 are synthesized in both situations, the conjugation of ISG15 to its target proteins occurs in IFN-treated cells but not in cells infected by influenza B virus. The experiments described below establish why ISG15 conjugation products are not produced in influenza B virus-infected cells.
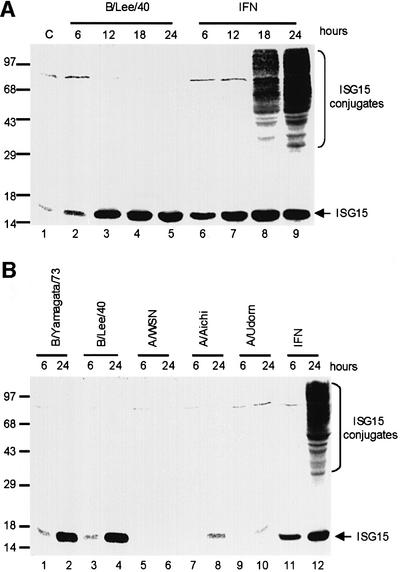
Fig. 7. (A) The ISG15 protein is synthesized in cells infected by influenza B virus, but the conjugation of ISG15 to its target proteins does not occur. A549 cells were infected with 20 p.f.u./cell influenza B/Lee/40 virus (lanes 2–5), or were treated with 1000 U/ml IFN-β (lanes 6–9). At the indicated time points, the cells were collected and proteins were resolved by 12% SDS–PAGE. The proteins were transferred to a BA S-85 filter membrane, which was probed with rabbit anti-ISG15 antiserum. C (lane 1), untreated A549 cells. (B) The ISG15 protein is not synthesized in cells infected by influenza A virus. A549 cells were infected with the indicated influenza B virus (lanes 1–4) or the indicated A virus (lanes 5–10) at 20 p.f.u./cell, or were treated with 1000 U/ml IFN-β (lanes 11 and 12). At the indicated time points, the cells were collected, and proteins were resolved by SDS–PAGE followed by western analysis using rabbit anti-ISG15 antiserum.
The induction of high levels of ISG15 production also occurs in cells infected with other strains of influenza B virus (e.g. B/Yamagata/73) (Figure 7B, lanes 1–4), indicating that this property is probably shared by all influenza B viruses. In contrast, little or no ISG15 protein is synthesized in cells infected with 20 p.f.u./cell of various influenza A virus strains (lanes 6–10) (see Discussion).
The NS1B protein of influenza B virus inhibits the conjugation of ISG15 to its target proteins in infected cells
To determine whether the ISG15 protein is associated with the NS1B protein in influenza B virus-infected cells, extracts from infected cells were immunoprecipitated with anti-ISG15 antiserum or pre-immune serum under conditions in which protein–protein complexes are not dissociated. After gel electrophoresis, the precipitated proteins were analyzed by western blotting using an anti-NS1B antiserum (Figure 8A). The NS1B protein was precipitated by the anti-ISG15 antiserum (lane 2), but not by the pre-immune serum (lane 1), indicating that ISG15 is physically associated with the NS1B protein in influenza B virus-infected cells.
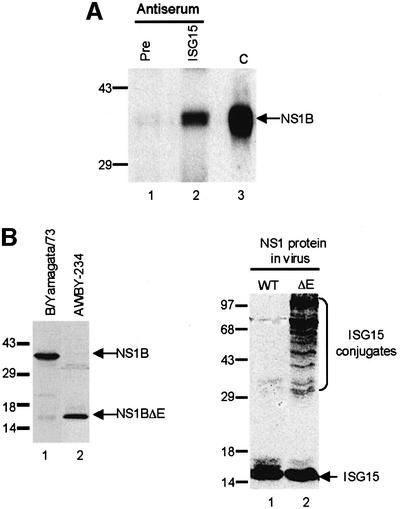
Fig. 8. (A) The NS1B protein is physically associated with the ISG15 protein in influenza B virus-infected cells. A549 cells infected with influenza B/Lee/40 virus were collected at 18 h post-infection, and were immunoprecipitated with either pre-immune mouse serum (lane 1) or mouse anti-ISG15 antiserum (lane 2). The immunoprecipitated proteins were resolved by 12% SDS–PAGE, followed by western blotting using rabbit anti-NS1B antiserum. C (lane 3), purified NS1B protein run on the same gel. (B) The NS1B protein inhibits the coupling of ISG15 to its target proteins in influenza B virus-infected cells. Left panel: the AWBY-234 mutant of B/Yamagata/1/73 synthesizes a truncated NS1B protein. A549 cells were infected with either influenza virus B/Yamagata/1/73 (lane 1) or its AWBY-234 mutant (lane 2) at 20 p.f.u./cell. At 18 h after infection, proteins were resolved by gel electrophoresis, followed by western blotting using rabbit anti-NS1B antiserum. Right panel: conjugation of ISG15 to its target proteins occurs in cells infected with the AWBY-234 mutant (ΔE NS1 protein; lane 2), but not in cells infected with wild-type virus (WT NS1 protein; lane 1). At 18 h after infection, proteins were resolved by gel electrophoresis, followed by western blotting using rabbit anti-ISG15 antiserum.
To determine whether the binding of the NS1B protein to ISG15 inhibits the coupling of ISG15 to its target proteins in influenza B virus-infected cells, we utilized the AWBY-234 mutant of B/Yamagata/1/73 (Tobita et al., 1990). This mutant virus synthesizes a truncated NS1B protein containing only the 90 N-terminal amino acids, which would not be expected to bind to ISG15. We confirmed that this mutant virus synthesized such a truncated NS1B protein (Figure 8B, left panel): the NS1B protein encoded by the mutant virus has an apparent molecular weight of 15 kDa (lane 2) compared with the 38 kDa NS1B protein encoded by the wild-type virus (lane 1). The mutant and wild-type viruses induce similar amounts of the ISG15 protein in A549 cells (Figure 8B, right panel). However, conjugation of ISG15 to its target proteins, which is not detectable in cells infected with the wild-type virus (lane 1), is greatly enhanced in cells infected by the mutant virus (lane 2). Because ISG15 conjugation occurs in cells infected with the mutant virus, we conclude that: (i) the ISG15 conjugation enzymes, including UBE1L, are produced during infection by influenza B virus; (ii) inhibition of the conjugation of ISG15 to its target proteins in influenza B virus-infected cells is mediated by the NS1B protein; and (iii) this inhibition requires a NS1B sequence longer than 90 amino acids.
Discussion
IFN α/β, which establishes an antiviral state in cells, induces several hundred proteins (Der et al., 1998; Stark et al., 1998). Previous studies have focused on the roles of three IFN-induced proteins during virus infection (PKR, 2–5 OAS and RNase L) and the viral countermeasures against these proteins (Welsh and Sen, 1997; Stark et al., 1998). Here we report on one of the other IFN-induced proteins, the ubiquitin-like ISG15 protein, which is one of the most strongly induced proteins in IFN-treated cells (Farrell et al., 1979; Der et al., 1998). We describe the novel mechanism by which influenza B virus, which strongly induces the ISG15 protein, counteracts its activity: a specific region of the NS1B protein encoded by this virus blocks the conjugation of ISG15 to its target proteins.
The induction of ISG15 and its conjugation products by IFN
ISG15, like other ubiquitin-like proteins, is present either in a free form or, catalyzed by specific enzymes, is covalently attached via its C-terminal glycine residue to target proteins (Narasimhan et al., 1996; Potter et al., 1999). It is not known whether linkage of ISG15 to its target proteins results in their degradation or rather, as is the case for other ubiquitin-like proteins such as SUMO-1 and Rub1 (Jentsch and Pyrowolakis, 2000; Yeh et al., 2000), this linkage modifies the biological activities of the targeted proteins (Loeb and Haas, 1992, 1994). Because the proteins that are targeted by ISG15 have not been identified (Loeb and Haas, 1994), the exact function of ISG15 modification is not known. ISG15, which can be detected at low constitutive levels in cells, is strongly induced by IFN α/β (Loeb and Haas, 1992; present study). In the presence of IFN, free ISG15 is produced for an extended period of time before conjugation of ISG15 to its target protein begins (Loeb and Haas, 1992; present study), indicating that the synthesis of the specific enzymes that couple ISG15 to its target proteins occurs at a later time.
Our results indicate that ISG15-coupling enzymes function like the enzymes that couple ubiquitin and other ubiquitin-like proteins to their target proteins. The first step in the ubiquitin conjugation pathway is the ATP-dependent formation of a thioester bond between ubiquitin and an E1 ubiquitin-activating enzyme (Hershko and Ciechanover, 1998). We demonstrate that IFN treatment of human A549 cells induces an E1 activating enzyme that is specific for ISG15 and identify this enzyme as the UBE1L protein, which was originally identified because its gene is deleted in many lung cancer cells (Carritt et al., 1992). Although UBE1L has 45% homology to the ubiquitin E1 enzyme (UBE1) (Kok et al., 1995), it does not activate ubiquitin. In addition, UBE1L is different from the E1 enzymes for two other ubiquitin-like proteins, SUMO-1 and Rub1, whose E1 activating enzymes are heterodimers (Johnson et al., 1997; Lammer et al., 1998; Liakopoulos et al., 1998). Because the genomic sequence upstream of the UBE1L gene does not appear to contain any of the known IFN-inducible elements (Kok et al., 1995), it is possible that transcription of the UBE1L gene is not induced by IFN, but rather is induced by one or more of the proteins that are directly induced by IFN, thereby explaining the delay in the conjugation of ISG15. Interestingly, although IFN induces ISG15 in all cell types that have been examined, ISG15 conjugation does not always occur, indicating that ISG15-coupling enzymes, including UBE1L, are probably not efficiently induced in all cell types after IFN treatment (Loeb and Haas, 1992).
The NS1B protein of influenza B virus blocks the conjugation of ISG15 to its target proteins both in vitro and in virus-infected human A549 cells
Using both extracts from IFN-treated A549 cells and the recombinant UBE1L enzyme, we established that the NS1B protein prevents ISG15 from being activated by UBE1L in vitro, whereas the NS1A protein of influenza A virus does not have this function. The smallest fragment of the NS1B protein that inhibits ISG15 activation by UBE1L consists of the 145 N-terminal amino acids of NS1B. This fragment of the NS1B protein includes both of its functional domains: the RNA-binding domain (the N-terminal 93 amino acids) (Wang and Krug, 1996) and 52 amino acids of the adjacent effector domain (Figure 4). A smaller N-terminal fragment of the NS1B protein (amino acids 1–103) efficiently binds ISG15 but does not inhibit its conjugation, indicating either that the UBE1L enzyme can effectively compete with this smaller fragment for binding to ISG15, or that the 103–145 region of the effector domain is responsible for inhibiting conjugation.
Two possible reasons why the RNA-binding domain is required for the binding of ISG15 can be postulated: (i) this region is required for the dimerization, and hence the proper folding of the NS1B protein; and/or (ii) specific amino acids in this domain interact directly with the ISG15 protein. Computer alignment of the amino acid sequences of the NS1A and NS1B RNA-binding domains suggests that the structure of the NS1B RNA-binding domain is similar to that of the NS1A protein: the NS1B RNA-binding domain is predicted to be comprised of a dimer of two chains, each of which is comprised of three α-helices, and two basic amino acids in helix 2 of both chains are predicted to be the only amino acids that interact directly with the dsRNA target (Chien et al., 1997; Liu et al., 1997; Wang et al., 1999). Consequently, in the absence of its RNA-binding domain, the NS1B protein would not dimerize, and this lack of dimerization could adversely affect the conformation of the ISG15 binding site in the effector domain. However, the NS1B RNA-binding domain may also have a binding site for ISG15. The NS1B RNA-binding domain apparently differs from that of the NS1A protein in one respect: the NS1B domain is predicted to contain large loops between the three α-helices in each chain (Wang et al., 1999), which are potential sites for ISG15 binding.
Previous studies have not identified the function(s) of the effector domain of the NS1B protein of influenza B virus. Unlike the effector domain of the NS1A protein of influenza A virus, the NS1B effector domain does not inhibit the nuclear export of cellular mRNAs (Wang and Krug, 1996). As reported here, the NS1B effector domain has a different function. A portion of the effector domain of the NS1B protein (amino acids 94–145) is required for inhibition of the conjugation of the IFN-induced ISG15 protein to its target proteins in infected cells as well as in vitro. Consequently, it is likely that the function of this portion of the NS1B effector domain is to counter this major IFN-induced pathway in humans. It is conceivable that the effector domain of the NS1B protein also binds to, and inhibits the activity of other major IFN-induced antiviral proteins that are produced in humans, the principal host of this virus (Murphy and Webster, 1996).
The cellular and/or viral proteins that are targeted for ISG15 conjugation have not been identified (Loeb and Haas, 1994; Jentsch and Pyrowolakis, 2000), and it is not known how the conjugation of ISG15 to its target proteins affects the replication of influenza B virus. The only pertinent study regarding the effects on virus replication used MDCK cells infected with the same AWBY-234 mutant of B/Yamagata/1/73 that we employed (Tobita et al., 1990). MDCK cells infected with this mutant virus showed an earlier cytopathic effect and produced more defective viruses than cells infected with the wild-type virus. However, it is not known whether these effects are due to the conjugation of ISG15 to one or more of its target proteins, because it was not determined whether this conjugation occurs efficiently in infected MDCK cells. In addition, the observed effects could also be due to the absence of the region of the NS1B effector domain that does not participate in functional ISG15 binding, i.e. amino acids 146–281.
Comparison of the NS1A and NS1B proteins
Because the induction of ISG15 protein synthesis during infection by influenza B virus is similar to that in IFN-treated cells, it is evident that transcription of the IFN-induced gene encoding the ISG15 protein is efficiently activated in influenza B virus-infected cells. In addition, we have found that synthesis of the IFN-induced P56 protein, which inhibits translation by binding to the eIF3 initiation factor (Guo and Sen, 2000), is efficiently induced in influenza B virus-infected cells (unpublished experiments). It is, therefore, likely that the transcription of many, if not all of the several hundred IFN-induced genes is efficiently activated in cells infected by influenza B virus. Hence, this transcriptional activation, which occurs at early times of infection, is not suppressed by the RNA-binding domain of the NS1B protein via the binding of dsRNA.
The inability of the NS1B RNA-binding domain to sequester dsRNA molecules at early times of infection can be attributed to its extremely low affinity for dsRNA. As is the case for the RNA-binding domain of the NS1A protein, the RNA-binding domain of the NS1B protein is likely to interact with dsRNA solely through electrostatic interactions between four basic amino acids and the phosphate backbone of dsRNA, and has a Kd of ∼1 µM (Wang et al., 1999; C.Chien, Y.Xu, P.V.Sahasrabudhe, R.Xiao, R.M.Krug and G.T.Montelione, in preparation). In contrast, the dsRNA-binding domain (dsRBD), which is found in many cellular proteins in both the nucleus and cytoplasm, has an affinity for dsRNA that is several orders of magnitude higher: the Kd values of cellular dsRBDs range from 0.1 to 1.0 nM (Kim et al., 1994; Lai et al., 1995; Ohman et al., 2000). This high affinity is attributable to the extensive interaction that these cellular dsRBD domains have with dsRNA (Ryter and Schultz, 1998). Therefore, because only a small number of NS1B protein molecules would be present at early times of influenza B virus infection when the transcription of IFN-induced genes is activated, it is understandable why the RNA-binding domain of the NS1B protein does not block this transcriptional activation.
In the present study we show that influenza A virus differs from influenza B virus in two ways: (i) the NS1A protein of influenza A virus does not bind the IFN-induced ISG15 protein; and (ii) little or no ISG15 protein is produced in human A549 cells infected by influenza A virus. In addition, we have found that little or no IFN-induced P56 protein is produced in influenza A virus-infected cells (unpublished experiments).
Two explanations can be proposed for the virtual absence of the ISG15 protein and of other IFN-induced proteins in influenza A virus-infected cells. One hypothesis is that the RNA-binding domain of the NS1A protein, by binding dsRNA, blocks the transcription of IFN-induced genes (Talon et al., 2000). However, it is unclear how the weak RNA-binding domain of the small number of NS1A protein molecules that are present at early times of infection would be able to compete for dsRNA molecules with the much stronger cellular dsRBD domains, particularly as we have shown that the similar RNA-binding domain of the NS1B protein is not able to compete with these cellular domains. An alternative hypothesis is that the inhibition of the synthesis of IFN-induced proteins in influenza A virus-infected cells is at the post-transcriptional level, and is mediated by the known functions of the effector domain of the NS1A protein. Like all poly(A)-containing cellular mRNAs made after infection by influenza A virus (Katze and Krug, 1984; Fortes et al., 1994; Qian et al., 1994; Qiu and Krug, 1994; Nemeroff et al., 1998; Chen et al., 1999; Chen and Krug, 2000), the nuclear export of the mRNAs encoding IFN-induced proteins would be inhibited by the binding of the NS1A effector domain to the 30 kDa subunit of CPSF and to PABII (Nemeroff et al., 1998; Chen et al., 1999; Chen and Krug, 2000). In addition, like other cellular pre-mRNAs and mRNAs made after infection (Katze and Krug, 1984; Krug et al., 1989), newly synthesized IFN-induced pre-mRNAs and mRNAs that are trapped in the nucleus would be almost completely degraded. Consequently, little or no IFN-induced mRNAs should survive, and little or no IFN-induced proteins should be synthesized in influenza A virus-infected cells. Future experiments should resolve this issue.
Materials and methods
Yeast two-hybrid library screening
The NS1B gene of influenza B/Lee/40 was kindly provided by Robert Lamb (Briedis and Lamb, 1982). The open reading frame of the NS1B protein was fused to the DNA-binding domain of Gal4 in the pAS2-1 yeast plasmid. The yeast pACT plasmid containing a human B-cell cDNA library was kindly provided by Stephen Elledge. The two plasmids were co-transfected into yeast strain Y190. The selection of transformants that express β-galactosidase, and the identification of yeast colonies that interact with the NS1B protein and fail to interact with the NS1A protein from A/Udorn/72 virus, were carried out as described previously (Chen et al., 1999). Plasmids were isolated from these yeast colonies and sequenced.
Glutathione–Sepharose affinity selection
To prepare the GST–NS1B fusion protein, the NS1B open reading frame was cloned into pGEX-3X. To prepare GST fusion proteins containing the 93, 103 or 145 N-terminal amino acids of the NS1B protein, two stop codons (TAGTAA) were introduced into the appropriate positions of the NS1B reading frame using PCR. Deletion mutants of the NS1B protein were generated by PCR and cloned into pGEX-3X. GST fusion proteins were expressed and purified as previously described (Qiu and Krug, 1994). The indicated GST fusion protein was combined with the indicated 35S-labeled protein(s) synthesized in vitro, and the mixture was subjected to glutathione–Sepharose affinity selection as previously described (Nemeroff et al., 1995). To prepare the labeled proteins, the DNA encoding the indicated protein was subcloned into pGEM1 and translated using a Promega TNT Coupled Transcription/Translation kit in the presence of [35S]methionine.
In vitro assay for the E1-like enzyme that activates ISG15
A549 cells were treated, where indicated, with 1000 U/ml IFN-β (Berlex Co.) for 24 h at 37°C. Cell extracts were prepared by Dounce homogenization in a buffer containing 50 mM Tris–HCl pH 7.5, 2 mM ATP, 10 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), followed by sonication for 10 s. To prepare the 32P-labeled ISG15 protein substrate, the DNA encoding ISG15 was cloned into pGEX-2TK, thereby attaching a cAMP-dependent kinase domain onto the N-terminus of a GST–ISG15 fusion protein. The fusion protein was purified using glutathione–Sepharose affinity chromatography. After labeling the kinase domain with 32P, the labeled ISG15 protein was cleaved from GST using thrombin (see Figure 3A). 32P-labeled ISG15 protein (106 c.p.m.) was incubated with 40 µg of the A549 cellular extract at 37°C for 2 h in a 50 µl reaction containing 50 mM Tris–HCl pH 7.5, 3 mM ATP, 10 mM MgCl2, 10 U/ml inorganic pyrophosphatase, 20 mM creatine phosphate and 4 µg/ml creatine phosphokinase. Where indicated, the NS1A protein, the NS1B protein or a specific fragment of the NS1B protein was incubated with the 32P-labeled ISG15 protein for 5 min at room temperature prior to incubation with the A549 cellular extract. After the 2 h incubation at 37°C, the reaction was terminated by adding an equal volume of a buffer containing 100 mM Tris–HCl pH 6.8, 20% glycerol and 4% SDS, and the mixture incubated at 37°C for 10 min. Where indicated, the SDS buffer also contained 200 mM DTT. An aliquot (20 µl) was analyzed by 7.5% SDS–PAGE in the absence of a reducing agent.
Purification, identification and expression of the ISG15 E1 activating enzyme
Cell extracts, prepared from IFN-treated A549 cells as described above, were loaded onto a 1 ml GST–Sepharose 4B column containing 2 mg of either purified GST or the GST–ISG15 fusion protein. After washing with 10 ml of a buffer containing 50 mM Tris–HCl pH 7.5 and 0.5 M NaCl, followed by 10 ml of 20 mM Tris–HCl pH 7.5, the column was eluted with a buffer containing 20 mM Tris–HCl pH 7.5 and 40 mM DTT. The eluate was concentrated with a Microcon filter and subjected to 7.5% SDS–PAGE, followed by staining with Coomassie Blue. The 110 kDa protein species eluted from the column containing GST–ISG15 was cut out and sent to the HHMI Biopolymer Facility and W.M.Keck Foundation Biotechnology Resource Laboratory at Yale University for MALDI-MS and microsequencing by automated Edman degradation. To express the identified UBE1L protein, the full-length cDNA of UBE1L (kindly provided by Charles Buys) (Kok et al., 1993) was PCR cloned into a modified pVL1393 baculovirus transfer vector containing GST upstream of the cloning site (kindly provided by Jon Huibregtse). The resulting plasmid and Autographa californica nuclear polyhedrosis virus DNA were co-transfected into Sf9 insect cells (Frankel et al., 1994), and the recombinant viruses that were produced were then amplified in Sf9 cells. The GST–UBE1L fusion protein was produced by infecting Sf9 cells at a multiplicity of 10 p.f.u./cell for 3 days at 27°C. Cell extracts were prepared by Dounce homogenization as described above, and the GST–UBE1L protein was purified using glutathione–Sepharose affinity chromatography. The in vitro activation assays were carried out as described above.
Identification of ISG15 and its conjugation products in cells
A549 cells were treated with 1000 U/ml IFN-β, or were infected with 20 p.f.u./cell of the indicated influenza A or B virus. Influenza virus B/Yamagata/1/73 and its AWBY-234 mutant were kindly provided by Kiyotake Tobita (Tobita et al., 1990). At the indicated time points, the cells were disrupted in the SDS buffer containing 100 mM DTT, followed by sonication for 10 s. The proteins in the resulting extracts (20 µl) were subjected to 12% SDS–PAGE. The resolved proteins were electrophoretically transferred to a BA S-85 filter membrane, which was probed with 10 µg/ml rabbit anti-ISG15 antiserum kindly provided by Arthur Haas (Loeb and Haas, 1992). The ISG15–antibody complexes were identified using ECL western blotting reagents from Amersham.
Co-immunoprecipitation assay
A549 cells infected with influenza B/Lee/40 virus were collected at 18 h post-infection, and disrupted in a buffer containing 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% NP-40 and 1 mM PMSF (NP-40 buffer) by passage through a 26 gauge needle. After low speed centrifugation, 100 µl of the supernatant were incubated for 1 h at 4°C with 20 µl of pre-immune mouse serum, followed by a 1 h incubation in the presence of 40 µl of protein A–agarose beads that had been blocked by a prior incubation with 1 mg/ml bovine serum albumin (BSA). After another low speed centrifugation, the entire supernatant was incubated at 4°C for 1 h with 20 µl of either pre-immune mouse serum or mouse anti-ISG15 antiserum that was prepared against the GST–ISG15 protein expressed in Escherichia coli. Forty microliters of BSA-blocked protein A–agarose beads were added to each reaction, and the mixtures were incubated at 4°C for 1 h. The beads were washed three times with the NP-40 buffer, and then suspended in 50 µl of the SDS buffer containing 100 mM DTT. The resulting supernatant was subjected to 12% SDS–PAGE, followed by western blotting using rabbit anti-NS1B antiserum.
Acknowledgments
Acknowledgements
We thank Arthur Haas for kindly providing valuable reagents and for helpful discussions and information, and Charles Buys, Kiyotake Tobita and Jon Huibregtse for providing reagents. This investigation was supported by NIH grant AI11772 to R.M.K.
References
- Beattie E.J., Tartaglia,J. and Paoletti,E. (1991) Vaccinia virus-encoded eIF-2α homolog abrogates the antiviral effects of interferon. Virology, 183, 419–422. [DOI] [PubMed] [Google Scholar]
- Briedis D. and Lamb,R.A. (1982) Influenza B virus genome: sequences and structural organization of RNA segment 8 and the mRNAs coding for the NS1 and NS2 proteins. J. Virol., 42, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carritt B. et al. (1992) A gene from human chromosome region 3p21 with reduced expression in small cell lung cancer. Cancer Res., 52, 1536–1541. [PubMed] [Google Scholar]
- Chen Z. and Krug,R.M. (2000) Selective nuclear export of viral mRNAs in influenza-virus-infected cells. Trends Microbiol., 8, 376–383. [DOI] [PubMed] [Google Scholar]
- Chen Z., Li,Y. and Krug,R.M. (1999) Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′ end processing machinery. EMBO J., 18, 2273–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien C.Y., Tejero,R., Huang,Y., Zimmerman,D.E., Rios,C.B., Krug,R.M. and Montelione,G.T. (1997) A novel RNA-binding motif in influenza A virus non-structural protein 1. Nature Struct. Biol., 4, 891–895. [DOI] [PubMed] [Google Scholar]
- Der S.D., Zhou,A., Williams,B.R. and Silverman,R.H. (1998) Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl Acad. Sci. USA, 95, 15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell P.J., Broeze,R.J. and Lengyel,P. (1979) Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumor cells. Nature, 279, 523–525. [DOI] [PubMed] [Google Scholar]
- Fortes P., Beloso,A. and Ortin,J. (1994) Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J., 13, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A., Roberts,H., Afrin,L., Vesely,J. and Willingham,M. (1994) Expression of ricin B chain in Spodoptera frugiperda. Biochem. J., 303, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. and Sen,G.C. (2000) Characterization of the interaction between the interferon-induced protein P56 and the Int6 protein encoded by a locus of insertion of the mouse mammary tumor virus. J. Virol., 74, 1892–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A.L., Ahrens,P., Bright,P.M. and Ankel,H. (1987) Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J. Biol. Chem., 262, 11315–11323. [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Jentsch S. and Pyrowolakis,G. (2000) Ubiquitin and its kin: how close are the family ties? Trends Cell Biol., 10, 335–342. [DOI] [PubMed] [Google Scholar]
- Johnson S.E., Schwienhorst,I., Dohmen,R.J. and Blobel,G. (1997) The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J., 16, 5509–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze M.G. and Krug,R.M. (1984) Metabolism and expression of RNA polymerase II transcripts in influenza virus-infected cells. Mol. Cell. Biol., 4, 2198–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U., Garner,T.L., Sanford,T., Speicher,D., Murray,J.M. and Nishikura,K. (1994) Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J. Biol. Chem., 269, 13480–13489. [PubMed] [Google Scholar]
- Kok K., Hofstr,R., Pilz,A., van den Berg,A., Terpstra,P., Buys,C.H. and Carritt,B. (1993). A gene in the chromosomal region 3p21 with greatly reduced expression in lung cancer is similar to the gene for ubiquitin-activating enzyme. Proc. Natl Acad. Sci. USA, 90, 6071–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok K., Berg,A.V., Veldhuis,P.M.J.F., Franke,M., Terpstra,P. and Buy,C.H.C.M. (1995) The genomic structure of the human UBE1L gene. Gene Expr., 4, 163–175. [PMC free article] [PubMed] [Google Scholar]
- Krug R.M., Alonso-Caplen,F.V., Julkunen,I. and Katze,M. (1989) Expression and replication of the influenza virus genome. In Krug,R.M. (ed.), The Influenza Viruses. Plenum Press, New York and London, pp. 89–152.
- Lai F., Drakas,R. and Nishikura,K. (1995) Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J. Biol. Chem., 270, 17098–17105. [DOI] [PubMed] [Google Scholar]
- Lammer D., Mathias,N., Laplaza,J.M., Jiang,W., Liu,Y., Callis,J., Goebl,M. and Estelle,M. (1998) Modification of yeast Cdc53p by the ubiquitin-related protein Rub1p affects function of the SCFCdc4 complex. Genes Dev., 12, 914–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liakopoulos D., Doenges,G., Matuschewski,K. and Jentsch,S. (1998) A novel protein modification pathway related to the ubiquitin system. EMBO J., 17, 2208–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M., Smith,B., Szakal,A., Nelson-Rees,W. and Todaro,G. (1976) A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer, 17, 62–70. [DOI] [PubMed] [Google Scholar]
- Liu J., Lynch,P.A., Chien,C., Montelione,G.T., Krug,R.M. and Berman,H.M. (1997) Crystal structure of the unique multifunctional RNA-binding domain of the influenza virus NS1 protein. Nature Struct. Biol., 4, 896–899. [DOI] [PubMed] [Google Scholar]
- Loeb K.R. and Haas,A.L. (1992) The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J. Biol. Chem., 267, 7806–7813. [PubMed] [Google Scholar]
- Loeb K.R. and Haas,A.L. (1994) Conjugates of ubiquitin cross-reactive protein distribute in a cytoskeletal pattern. Mol. Cell. Biol., 14, 8408–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews M.B. and Shenk,T. (1991) Adenovirus-associated RNA and translation control. J. Virol., 65, 5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B.R. and Webster,R.G. (1996) Orthomyxoviruses. In Fields,B.N., Knipe,D.M. and Howley,P.M. (eds), Fields Virology. Lippincott-Raven, Philadelphia, PA, pp. 1397–1446.
- Narasimhan J., Potter,J.L. and Haas,A.L. (1996) Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J. Biol. Chem., 271, 324–330. [DOI] [PubMed] [Google Scholar]
- Nemeroff M.E., Qian,X.-Y. and Krug,R.M. (1995) The influenza virus NS1 protein forms multimers in vitro and in vivo. Virology, 212, 422–428. [DOI] [PubMed] [Google Scholar]
- Nemeroff M., Barabino,S.M.L., Keller,W. and Krug,R.M. (1998) Influenza virus NS1 protein interacts with the 30 kD subunit of cleavage and specificity factor and inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell, 1, 991–1000. [DOI] [PubMed] [Google Scholar]
- Ohman M., Kallman,A.M. and Bass,B.L. (2000) In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA, 6, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter J.L., Narasimhan,J., Mende-Mueller,L. and Haas,A.L. (1999) Precursor processing of pro-ISG15/UCRP, an interferon-β-induced ubiquitin-like protein. J. Biol. Chem., 274, 25061–25068. [DOI] [PubMed] [Google Scholar]
- Qian X.Y., Alonso-Caplen,F. and Krug,R.M. (1994) Two functional domains of the influenza virus NS1 protein are required for regulation of nuclear export of mRNA. J. Virol., 68, 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y. and Krug,R.M. (1994) The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol., 68, 2425–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter J.M. and Schultz,S.C. (1998) Molecular basis of double-stranded RNA–protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J., 17, 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G.R., Kerr,I.M., Williams,B.R., Silverman,R.H. and Schreiber,R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Talon J., Horvath,C.M., Polley,R., Basler,C.F., Muster,T., Palese,P. and Garcia-Sastre,A. (2000) Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol., 74, 7989–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobita K., Tanaka,T., Odagiri,T., Tashiro,M. and Feng,S.Y. (1990) Nucleotide sequence and some biological properties of the NS gene of a newly isolated influenza B virus mutant which has a long carboxyl terminal deletion in the NS1 protein. Virology, 174, 314–319. [DOI] [PubMed] [Google Scholar]
- Wang W. and Krug,R.W. (1996) The RNA-binding and effector domains are conserved to different extents among influenza A and B viruses. Virology, 223, 41–50. [DOI] [PubMed] [Google Scholar]
- Wang W., Riedel,K., Lynch,K., Chien,C., Montelione,G.T. and Krug,R.M. (1999) RNA-binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA, 5, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh R.M. and Sen,G.C. (1997) Nonspecific host response to viral infections. In Nathanson,N. (ed.), Viral Pathogenesis. Lippincott-Raven, Philadelphia, PA, pp. 109–141.
- Yeh E.T.H., Gong,L. and Kamintini,T. (2000) Ubiquitin-like proteins: new wines in new bottles. Gene, 248, 1–14. [DOI] [PubMed] [Google Scholar]