

Abstract
The cognitive consequences of the apolipoprotein E–ɛ4 (APOE-ɛ4) allele were examined in middle age, before likely onset of symptoms of Alzheimer’s disease. The authors identified 3 cognitive processes—visuospatial attention, spatial working memory, and the effect of visuospatial attention on working memory—and devised “behavioral assays” of the integrity of components of these processes. Redirecting visuospatial attention, retention of memory for location, and attentional modulation of memory of target location were affected by APOE genotype. Visuospatial attention showed additive effects of ɛ4 gene dose; each additional ɛ4 allele inherited further slowed disengagement from invalidly cued space. In contrast, working memory performance was affected only in ɛ4 homozygotes. Effect sizes for the APOE gene were moderate to large, ranging from 14% to 24%. Effects of APOE genotype on component processes of cognition in healthy, middle-aged adults is consistent with the emergence in adulthood of an APOE-ɛ4 cognitive phenotype.
Keywords: attention, memory, APOE, phenotype
Understanding the genetic basis of individual differences in both general and specific cognitive abilities has long been an important goal of research in behavioral genetics (Plomin, DeFries, McClearn, & McGuffin, 2001). Further progress toward this goal has been achieved recently by linking specific polymorphisms to information-processing components underlying cognitive performance (Egan et al., 2001; Greenwood, Sunderland, Friz, & Parasuraman, 2000; Swanson et al., 2000). Rather than use broadly conceived, standardized neuropsychological tests, some investigators have developed more sensitive “behavioral assays” of component processes of cognition that can be linked to the activation of different brain networks. Notably, Posner and colleagues have hypothesized the existence of three attentional networks that can be distinguished anatomically and neurochemically and have developed a set of tasks aimed at behavioral assessment of both the integrity (Fan, McCandliss, Sommer, Raz, & Posner, 2002) and the genetic mediation (Fan, Wu, Fossella, & Posner, 2001; Fossella et al., 2002) of each network.
Using a similar approach, we have sought to understand the effects of the apolipoprotein E (APOE) gene on the component processes of cognition (Greenwood et al., 2000). This gene is known to be related to the risk of late-onset Alzheimer’s disease (AD) in older adults (Corder et al., 1993; Henderson et al., 1995). However, there is also evidence that this gene may influence cognition in individuals without dementia prior to old age (Greenwood et al., 2000; Parasuraman, Greenwood, & Sunderland, 2002). Therefore, we were interested in determining which components of cognition are affected by normal variation of this gene in healthy adults.
The APOE gene is inherited as one of three alleles, termed ɛ2, ɛ3, and ɛ4, with mean frequencies in the general population of about 8%, 78%, and 14%, respectively (Utermann, Langenbeck, Beisiegel, & Weber, 1980). The degree of risk of AD conferred by APOE-ɛ4 rises in a “gene dose” manner, increasing with the number of ɛ4 alleles inherited, from zero (noncarriers), to one (heterozygotes), to two (homozygotes; Corder et al., 1993). Because AD usually appears late in life and progresses slowly, the APOE-ɛ4 allele may exert its effects prior to the clinical diagnosis of AD. Consistent with this view is research showing that older carriers of the ɛ4 allele who do not have dementia nevertheless do show cognitive deficits. For example, men over age 70 declined in performance on the Mini-Mental State Examination (MMSE; Folstein, Folstein, & McHugh, 1975) over a 3-year period in a manner related to ɛ4 gene dose: greatest in homozygotes, intermediate in heterozygotes, and least in noncarriers (Feskens et al., 1994). Nor are deficits seen only on general function tests such as the MMSE. Specific cognitive processes, such as episodic memory, have also been found to be impaired in older ɛ4 carriers compared with noncarriers (Bondi et al., 1995).
The most common type of AD—sporadic and nonfamilial—is primarily a disorder of old age, and its prevalence increases with age (Evans et al., 1989). As a result, any large group of adults over age 65 is likely to include some individuals who are in an early stage of AD (Sliwinski, Lipton, Buschke, & Stewart, 1996). Therefore, the presence of cognitive decline in a group of older adult ɛ4 carriers may simply reflect an increased proportion of persons with undiagnosed, preclinical AD. That otherwise healthy older individuals possessing an ɛ4 allele are cognitively impaired relative to noncarriers suggests the existence of a prodromal stage of AD (Daffner & Scinto, 2000). Researchers have estimated that cognitive dysfunction precedes diagnosis of AD by 4–6 years in those who develop the disease (Fox, Warrington, Seiffer, Agnew, & Rossor, 1998; Linn et al., 1995). Confirming this finding, Small, Fratiglioni, Viitanen, Winblad, and Backman (2000) observed that individuals eventually diagnosed with AD showed rates of cognitive change from 6 to 3 years prior to diagnosis that were similar to those in individuals who did not develop AD. In contrast, there was a precipitous decline beginning about 3 years prior to diagnosis.
Cognitive deficits associated with APOE-ɛ4 are not confined to older adults. They may also occur as early as midlife, a decade or more before the likely onset of symptoms of AD in those destined to get the disease. A group of individuals with a mean age of 46 (range = 24–60) and with at least one ɛ4 allele showed cognitive deficits relative to noncarriers on tasks of verbal learning, visual memory, and attention span (Flory, Manuck, Ferrell, Ryan, & Muldoon, 2000). Greenwood et al. (2000) found that a medically screened group of middle-aged ɛ4 homozygotes without dementia (mean age = 58) who were unimpaired on standard neuropsychological tests nevertheless showed deficiencies in components of tasks of spatial attention and visual search. The results of the Greenwood et al. study are of particular interest because the pattern of attentional impairment in their sample of APOE-ɛ4 carriers without dementia was qualitatively (but not quantitatively) the same as those observed in older, clinically diagnosed AD patients (Parasuraman, Greenwood, Haxby, & Grady, 1992).
Moreover, even when frank neuropsychological deficits are not seen (Plassman et al., 1997; Reiman et al., 1996), there are subtle but distinct brain changes in middle-aged ɛ4 carriers compared with age-matched noncarriers. Reduced hippocampal volume has been observed in APOE-ɛ4 heterozygotes with mean ages of 55 (R. M. Cohen, Small, Lalonde, Friz, & Sunderland, 2001) and 63 (Plassman et al., 1997). In addition to finding brain structural variation, researchers using positron emission tomography have also found evidence of hypometabolism of association cortex in ɛ4 homozygotes with a mean age of 55.4 (Reiman et al., 1996) and, surprisingly, in ɛ4 heterozygotes with a mean age of 30.7 (Reiman et al., 2004). (For a review of cognitive and neural changes associated with APOE-ɛ4 in healthy adults, see Parasuraman et al., 2002.)
Such findings suggest that a cognitive phenotype of APOE could exist independently of the eventual development of AD. Possession of the ɛ4 allele is far from a guarantee of later dementia, given that only about 50% of ɛ4 homozygotes have developed AD by age 80 (Meyer et al., 1998) or 90 (Henderson et al., 1995). Thus, although not all ɛ4 carriers will develop AD, as a group they exhibit altered cognition prior to old age. This suggests either brain development or age-related brain change phenotypic of the allele. There is some evidence that in youth a cognitive phenotype exists in those individuals destined to develop AD late in life. Individuals diagnosed with AD in old age had lower IQs at age 11 (Whalley et al., 2000) and lower “idea density” in written work at age 20 (Snowdon et al., 1996), compared with individuals who did not later develop AD. Carriers of a tau mutation on Chromosome 17 linked to an inherited dementia have specific cognitive deficits in the third decade, compared with noncarriers in the same family (Geschwind et al., 2001). Like the tau mutation, the APOE gene might influence brain functioning only in adulthood. The APOE-ɛ4 allele does not affect IQ in childhood (Turic, Fisher, Plomin, & Owen, 2001), although it is associated with greater cognitive decline from youth to old age (Deary et al., 2002) and greater memory and verbal comprehension impairments in AD (Smith et al., 1998). This evidence suggests that the APOE-ɛ4 cognitive phenotype detected in midlife is related to maintenance of brain integrity in adulthood.
Consistent with an APOE-ɛ4-related reduction in brain and cognitive integrity beginning in midlife is evidence that the protein product of the APOE gene—termed apoE—plays a role in neuronal plasticity and repair (Arendt et al., 1997; Fagan et al., 1998; Krzywkowski et al., 1999). A complex of apoE and cholesterol is a strong promotor of synapse development (Mauch et al., 2001). Further, apoE is upregulated in tandem with clearance of cholesterol and lipid debris from the site of the injury following lesions to the entorhinal cortex in mice. This may reflect the redistribution of lipid in the service of neurite extension (White, Nicoll, & Horsburgh, 2001). Such processes appear to be reduced in effectiveness when the ɛ4 and not the ɛ3 allele directs apoE production (White, Nicoll, Roses, & Horsburgh, 2001). That greater APOE-ɛ3 allele expression was associated with increased sprouting of hippocampal granule cell mossy fibers, whereas greater ɛ4 allele expression was associated with decreased sprouting after injury, has been interpreted to indicate that the lipoprotein produced by the APOE-ɛ4 allele actively suppresses regeneration (Teter et al., 2002). This is evidence for a role for apoE in repair and recovery following brain injury.
Consistent with that evidence are findings that the APOE genotype affects prognosis in several neurologic disorders, namely, amyotrophic lateral sclerosis (Drory, Birnbaum, Korczyn, & Chapman, 2001) and multiple sclerosis (Fazekas et al., 2001), and following head injury (Crawford et al., 2002). This evidence suggests that the APOE gene may modulate the response to brain insult generally. If so, then effects of the APOE genotype may not be evident until midlife—after development is complete and adult aging has begun. It is in midlife that there emerges AD-like neuropathology (Braak & Braak, 1996; Reiman et al., 1998) and subtle cognitive change (Flory et al., 2000; Greenwood et al., 2000). On the basis of these findings, we sought further evidence for a cognitive phenotype of APOE in midlife.
The term cognitive phenotype has been used to refer to a characteristic cognitive profile in a number of heritable disorders, such as Williams syndrome (Donnai & Karmiloff-Smith, 2000), autism (Hughes, Plumet, & Leboyer, 1999), and Fragile X syndrome (Kaufmann, Abrams, Chen, & Reiss, 1999). A cognitive phenotype characterized by memory and verbal comprehension deficits has been claimed for APOE-ɛ4 homozygotes with AD as well as those with mild cognitive impairment, but not in individuals without dementia (Smith et al., 1998).
We hypothesize that the negative consequences of inheriting the APOE-ɛ4 allele for brain integrity in the face of a range of insults (reviewed above) leads to subtle cognitive deficits by midlife or earlier. Because most APOE-ɛ4 carriers will not develop AD (Henderson et al., 1995), deficits in those individuals could be viewed as a cognitive phenotype evident regardless of the eventual development of AD. In this view, the APOE-ɛ4 allele would not cause AD but could allow its development through weak or inefficient repair mechanisms. This view is in contrast to one that considers all deficits in ɛ4 carriers to be related to the AD prodrome. In either view, cognitive consequences would be related to ɛ4 gene dose, much as risk of AD is related to gene dose (Corder et al., 1993). In the present study, we tested this hypothesis by examining the influence of the ɛ4 allele on spatial attention, working memory, and the effect of spatial attention on working memory in middle age.
Cognitive deficits in healthy, middle-aged adults are likely to be small and subtle, given that an early stage of aging is being probed. Therefore, whole-task performance, particularly on general neuropsychological tests (such as the Mattis Dementia Rating Scale; Mattis, 1976), may be lacking in specificity and hence, in sensitivity, when compared with information-processing tests in which components of cognition can be isolated (Greenwood et al., 2000; Parasuraman et al., 2002). Accordingly, in three experiments we used well-developed information-processing tests to examine three aspects of cognition: (a) spatial attention, (b) spatial working memory, and (c) the interaction between spatial attention and working memory. Evaluation of not only two different domains of cognitive functioning, attention and memory, but also the influence of one on the other provided a test of the generality of the APOE genotype effect and its consequences for the ability to integrate functioning across cognitive systems.
Experiment 1: Spatial Attention
Visuospatial attention is a basic aspect of cognition (Posner, 1980) that is fundamental for effective visual functioning in everyday life. This function may be particularly important for older adults, as it appears to be relatively resistant to age-related decline (Gottlob & Madden, 1999; Hartley, Kieley, & McKenzie, 1992). Alterations in the efficiency of visuospatial attention appear only very late in life and then only in certain conditions (Greenwood & Parasuraman, 1994; Greenwood, Parasuraman, & Haxby, 1993). However, in contrast to that of healthy older adults, the ability to redirect spatial attention in the visual field is impaired in AD patients with mild dementia (Parasuraman et al., 1992). The efficiency of reorienting attention is also diminished in middle-aged individuals without dementia who are APOE-ɛ4 allele carriers, although to a lesser extent (Greenwood et al., 2000). In Experiment 1, we sought to both confirm and extend this latter finding. A larger sample size of healthy, middle-aged APOE-ɛ4 carriers was used to allow an examination of the gene dose (zero, one, or two copies of the ɛ4 allele) effect of the APOE-ɛ4 allele on spatial attention.
A cued discrimination task was used to direct visuospatial attention to regions of space containing letter targets (illustrated in supplemental Figure 1 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp). Cues were valid, invalid, or neutral in predicting the location of the target letter, which they preceded by a cue–target stimulus onset asynchrony (SOA) of 200, 500, or 2,000 ms.
Method
Participants
The sample consisted of healthy individuals (N = 177), aged 41 to 85, who volunteered to participate in the National Institute of Mental Health’s Prospective Study of Biomarkers for Older Controls at Risk for Alzheimer’s Disease (known as BIOCARD), which is double-blind and longitudinal. Individuals were excluded from participation if their performance did not fall within the normal age-related range of scores on a battery of standardized neuropsychological test batteries, including the Mattis Dementia Rating Scale, the Buschke Selective Reminding Test (Buschke, 1973), and the Wechsler Memory Scale—Revised (WMS–R; Wechsler, 1987). Other criteria for exclusion were significant medical problems, including diabetes mellitus, hypertension, cerebrovascular disorder, autoimmune disorder, vitamin B-12 deficiency, or thyroid disorder. Exclusion of individuals for cerebrovascular disease was made on the basis of history of strokes and hypertension and on review of MRI scans. Of the 177 individuals tested, 80% had first-degree relatives diagnosed with AD. Demographic characteristics are given in Table 1. Apolipoprotein E genotypes were determined by restriction endonuclease digestion of polymerase chain reaction amplified genomic DNA (performed by Athena Diagnostics, Worcester, Massachusetts). All procedures were approved by institutional review, and informed consent was obtained from all participants.
Table 1.
Means and Standard Deviations of Group Demographic Characteristics and Selected Neuropsychological Tests
| No. of APOE-ɛ4 alleles
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0
|
1
|
2
|
|||||||
| Variable | n | M | SD | n | M | SD | n | M | SD |
| Demographic | |||||||||
| Participants | 113 | 52 | 12 | ||||||
| Gender | |||||||||
| Female | 66 | 33 | 6 | ||||||
| Male | 47 | 19 | 6 | ||||||
| Age (years) | 59.9 | 0.9 | 58.9 | 1.0 | 57.6 | 2.2 | |||
| Education (years) | 16.7 | 2.5 | 16.9 | 2.7 | 17.4 | 2.5 | |||
| Test | |||||||||
| Mattis | 141.2 | 2.2 | 140.6 | 2.9 | 140.8 | 1.8 | |||
| Buschke | |||||||||
| Consistency | 9.2 | 1.2 | 8.9 | 1.6 | 8.6 | 1.2 | |||
| Delayed recall | 8.6 | 2.7 | 8.2 | 2.6 | 7.4 | 2.7 | |||
| WMS | |||||||||
| Logical Memory II subtest (delayed) | 25.6 | 7.3 | 23.5 | 6.7 | 19.9 | 9.7 | |||
Note. APOE-ɛ4 = apolipoprotein E–ɛ4; Mattis = Mattis Dementia Rating Scale; Buschke = Buschke Selective Reminding Test; WMS = Wechsler Memory Scale.
Because the APOE gene increases the risk of AD according to the gene dose of the ɛ4 allele (Corder et al., 1993), the number of ɛ4 alleles (zero, one, or two) was the basis for grouping participants. The groups were designated as follows: zero ɛ4 alleles (noncarriers), one ɛ4 allele (heterozygotes), and two ɛ4 alleles (homozygotes). Table 1 reports only a subset of an extensive neuropsychological battery administered to these individuals. For those participants common to all experiments, separate one-way analyses of variance (ANOVAs) were used to assess gene dose group differences on each measure. There were no statistical differences on age, on years of education, or on a large number of standardized neuropsychological tests (results of which will be reported in an upcoming paper). Of these comparisons, the only significant difference occurred between the gene dose groups on the delayed measure of the Logical Memory II subtest of the WMS–R, F(2, 165) = 3.70. However, after a Bonferroni correction for number of comparisons, it was no longer significant.
Stimuli and procedures
A spatially cued letter discrimination task, developed in our previous study of APOE-ɛ4 carriers (Greenwood et al., 2000), was used. Following a fixation point (displayed for 500 ms), a centered location cue (an arrow pointing to the left, to the right, or in both directions) appeared (see supplemental Figure 1 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp). The cue was valid in predicting the subsequent target location on 62.5% of the trials, invalid on 18.75%, and neutral on 18.75%. The centered location cue appeared for a variable cue–target SOA of 200, 500, or 2,000 ms, following which a letter target appeared 6.7° to the right or left of fixation. Participants were required to make a speeded categorization of the target as either a consonant or a vowel by pressing one of two response buttons. The intertrial interval was varied randomly (2,200, 2,500 or 2,800 ms).
Results
All statistical tests were performed at the .05 level of significance.
Accuracy
Accuracy of target categorization as consonant or vowel was analyzed as ratios (number correct:number presented) for each condition. These were subjected to a repeated measures ANOVA, with APOE-ɛ4 gene dose as the between-subjects factor and cue validity and cue–target SOA as within-subjects factors. Accuracies ranged from .944 to .993. Accuracy varied with cue validity, F(2, 348) = 4.04, in a manner that interacted with SOA (Validity × SOA), F(4, 696) = 3.15. The only effect of APOE gene dose on accuracy was seen in an SOA × Gene Dose interaction, F(4, 696) = 2.43 (see supplemental Table 1 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp). Accuracy can be seen to decline as reaction time (RT) increased in the ɛ4 homozygotes from neutral to invalid cues. However, across SOA it was a small decline from a high level—from .984 to .968.
RT
Means of median RTs were subjected to a mixed-effects ANOVA, with ɛ4 gene dose as the between-subjects factor and cue validity (valid, neutral, or invalid) and cue–target SOA (200, 500, and 2,000) as within-subjects factors. As can be seen in Figure 1, RT was fastest following valid cues, slowest following invalid cues, and intermediate following neutral cues, F(2, 348) = 50.58. The cue validity effect developed with time following cue onset, as indicated by the significant main effect of SOA, F(2, 348) = 35.09, and the Cue Validity × SOA interaction, F(4, 696) = 32.06. There was no overall effect of ɛ4 gene dose on RT, but there was a significant interaction of gene dose with cue validity, F(4, 348) = 2.50. Figure 1 shows that as ɛ4 gene dose increased, RT on invalidly cued trials was increasingly slowed. In contrast, there was little effect of ɛ4 gene dose on valid cue RT. This does not appear to be due to a speed–accuracy trade-off. In supplemental Table 1 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp, it can be seen that although accuracy decreased as RT slowed from neutral to invalid cues in ɛ4 homozygotes, the change was very small (e.g., from .983 to .969) in the context of overall high accuracy.
Figure 1.
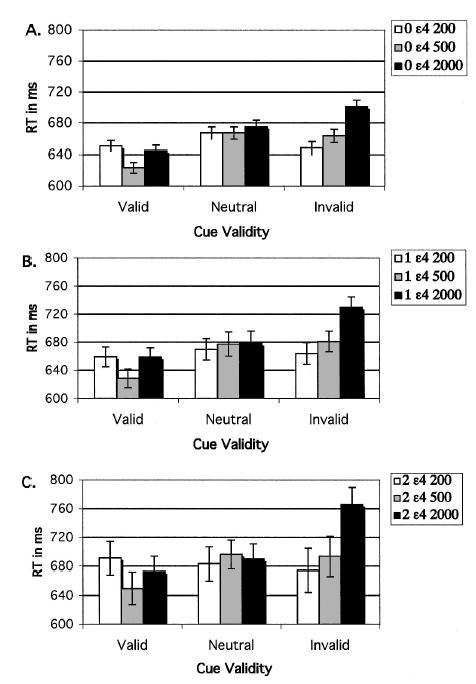
Experiment 1: Results of reaction time (RT) data from the ɛ4 gene dose groups, plotted as a function of cue validity and cue–target stimulus onset asynchrony. A: Zero ɛ4 group (noncarriers). B: One ɛ4 group (ɛ4 heterozygotes). C: Two ɛ4 group (ɛ4 homozygotes). Error bars represent standard errors.
This selective effect on invalid cue RT was confirmed by analysis of the calculated costs of invalid cues (invalid cue – neutral cue RT) and benefits of valid cues (neutral cue – valid cue RT). Costs increased with ɛ4 gene dose, F(2, 169) = 4.27, and as SOA lengthened, F(2, 338) = 27.49 (see Figures 1 and 2 and supplemental Table 2 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp). A post hoc test of costs at the longest SOA revealed that the noncarriers (zero ɛ4 alleles, 22.1 ± 58.6) differed significantly from both heterozygotes (one ɛ4 allele, 39.3 ± 50.8) and homozygotes (two ɛ4 alleles, 81.6 ± 63.9), and these two groups differed significantly from each other (Fisher’s protected least significant difference [PLSD] test, p < .05). Moreover, the main effect of gene dose on cost remained significant even when accuracy was included as a covariate, F(2, 169) = 4.02. In comparison, although the benefits of valid cues varied with SOA, F(2, 338) = 16.16, the gene dose groups did not differ on this component (see Figures 1 and 2). Therefore, the interaction of cue validity and gene dose could be attributed to greater costs of invalid cues (slowed disengagement) rather than to a change in benefits of valid cues.
Figure 2.
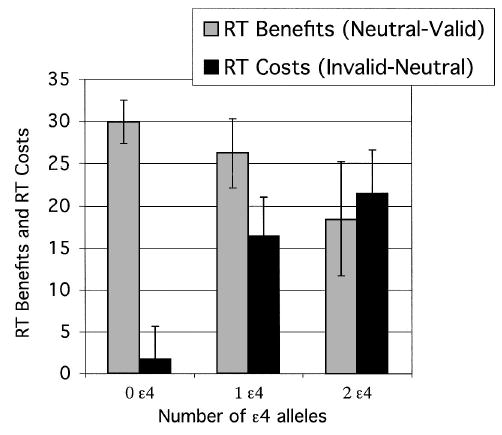
Experiment 1: Calculated benefits of valid cues (neutral cue RT – valid cue RT) and costs of invalid cues (invalid cue RT – neutral cue RT), both plotted as a function of ɛ4 gene dose group. RT = reaction time. Error bars represent standard errors.
Examination of effect size is important in single-gene studies, given that such studies have sometimes been criticized for small effect sizes, low replicability, and Type I error (Ioannidis, Ntzani, Trikalinos, & Contopoulos-Ioannidis, 2001). Accordingly, we calculated effect size for the APOE gene, corrected for unequal sample size (J. Cohen, 1988). The effect size for the significant APOE gene dose effect reported above for RT costs at the long SOA was .24.
Discussion
We previously reported that inheritance of at least one allele of the APOE-ɛ4 gene was associated with a specific impairment in redirecting visuospatial attention to a location in the visual field following an invalid cue (Greenwood et al., 2000). There were two main outcomes of this follow-up experiment. First, the initial results reported by Greenwood et al. (2000) were confirmed. Specifically, healthy, middle-aged adults without dementia who were APOE-ɛ4 allele carriers exhibited an impairment in the disengagement of spatial attention while showing normal overall performance on a spatially cued letter discrimination task. This selective deficit in attentional shifting is qualitatively similar to, though quantitatively smaller than, the deficit found in previous studies of clinically diagnosed AD patients (Greenwood & Parasuraman, 1999; Parasuraman et al., 1992; see Parasuraman et al., 2002, for a recent review). Second, this experiment revealed a selective, systematic effect of the dose of the risky APOE-ɛ4 allele on this component of spatial attention. The greater the number of ɛ4 alleles carried, the slower was the speed of redirection of spatial attention following an invalid location cue. At the same time, RT benefits of valid spatial cues and overall accuracy on the letter discrimination task were unaffected by APOE-ɛ4 gene dose.
It is important to note not only the significance of the APOE gene dose effect on the spatial redirection of attention but also its effect size of .24. This moderate-sized effect in a study of cognitive performance is in marked contrast to single-gene studies of disease category, which often are very low (about 1%–5%; Ioannidis et al., 2001).
This orderly pattern of results suggests that the disengagement of visuospatial attention may be particularly sensitive to the neural consequences of inheritance of the APOE ɛ4 gene (Parasuraman et al., 2002). However, other cognitive functions may be affected as well. Furthermore, variations in the efficiency of the attentional system may affect the integrity of memory and other functions. Investigation of memory functioning would be particularly cogent given that memory dysfunction is a cardinal feature of AD (McKhann et al., 1984) and given the association between APOE and AD. For these reasons, we sought to examine the effect of the ɛ4 allele on a cognitive system other than attention—working memory.
Experiment 2: Working Memory
A decline in working memory is one of the major consequences of normal aging (Salthouse, Mitchell, Skovronek, & Babcock, 1989). In addition, impairments in memory have been defined as the earliest symptom of AD (McKhann et al., 1984). Persons without dementia who are genetically at risk of AD because they possess the ɛ4 allele have been found to exhibit deficits on measures of delayed recall, even when no other neuropsychological deficits are observed (Bondi, Galasko, Salmon, & Thomas, 1999).
If cognitive change in APOE-ɛ4 carriers is prodromal—that is, a consequence of AD-driven pathologic change—then memory should be affected. Of the two pathognomonic neuropathologies of AD, neurofibrillary tangles and amyloid plaques, only tangles appear in mesial temporal regions in a selective and orderly manner. Because tangles develop in mesial temporal (entorhinal) cortex of unselected brains prior to midlife (Braak & Braak, 1995), individuals destined to develop AD might be predicted to have some tangle development by the fifth decade. A number of studies have shown a relation between tangle counts and cognition (Arriagada, Marzloff, & Hyman, 1992; Davis & Chisholm, 1997). Given that memory encoding is known to be dependent on mesial temporal activity (Brewer, Zhao, Desmond, Glover, & Gabrieli, 1998), the entanglement of that region could compromise memory function. Indeed, memory loss is a requirement for diagnosis of probable AD (McKhann et al., 1984). On the other hand, if cognitive decline is phenotypic of the ɛ4 allele, reflecting native adult cognitive integrity, then memory may be deficient, but not selectively so. Other functions, such as attention and spatial ability, would be predicted to be vulnerable as well.
In Experiment 2, a spatial working memory task was used to assess the effect of APOE-ɛ4 gene dose on the ability to form and retain a memory of up to three locations over a delay.
Method
Participants
See Experiment 1.
Stimuli and procedures
The task was a spatial working memory task, illustrated in supplemental Figure 2 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp. Following a fixation cross by 1 s, one, two, or three black dots (.67° in diameter, each indicating a target location) appeared at randomly chosen screen locations for 500 ms. Simultaneously with dot offset, the fixation cross reappeared for a 3-s delay. At the end of the delay, a single red test dot appeared alone on the screen. This test dot appeared either at the same location as one of the target dots (match condition) or at a different location (nonmatch condition). On nonmatch trials, the distance between the correct location and the test dot was varied over three levels, being about 2°, 4°, or 8° of visual angle. Participants had 2 s to decide whether the test dot location matched one of the target dots. RT and accuracy were measured.
Results
The measure of interest was accuracy of memory for target dot location. Ratios were formed (number correct:number presented) for each condition. APOE-ɛ4 gene dose was the between-subjects factor, and memory load (one, two, or three target dots) and trial type (match or nonmatch) were within-subjects factors. Target–test distance could be varied only under nonmatch conditions. Therefore, an omnibus ANOVA was conducted comparing match with nonmatch data, collapsed across target–test distance. This revealed main effects of gene dose, F(2, 128) = 3.47, and memory load, F(2, 256) = 376.03. Memory for target location declined with ɛ4 gene dose in a way that interacted with the other two factors (Gene Dose × Trial Type × Memory Load), F(4, 256) = 3.33. To check for response bias on match versus nonmatch trials, we compared the proportion of false alarms (match responses on nonmatch trials) for trial type and level of memory load. Although the homozygotes had more false alarms overall, F(2, 128) = 3.38, results that are consistent with their lower accuracy, there was no main effect of trial type or a Trial Type × Memory Load interaction. False alarms did increase with memory load, F(2, 256) = 393.80, and this effect interacted with trial type and gene dose, F(4, 256) = 3.10. Examination of Figure 3 indicates that this interaction was not due to biased responding as a function of trial type but rather to relatively greater false alarms of homozygotes under high-load conditions on match trials but under low-load conditions on nonmatch trials.
Figure 3.
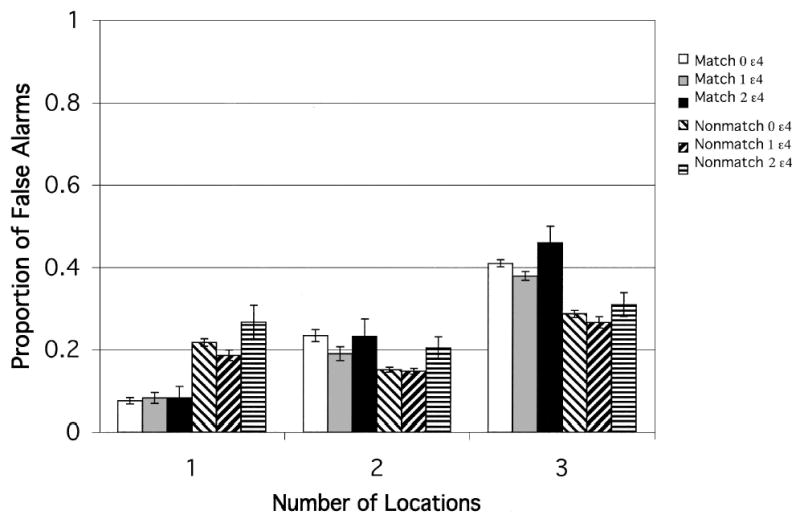
Experiment 2: Match and nonmatch conditions. Proportion of false alarms after a 3-s delay for each ɛ4 gene dose group, plotted as a function of memory load (number of locations to be retained). Error bars represent standard errors.
Because of the different patterns of responding, we analyzed match and nonmatch data separately. Under match conditions in which the test dot appeared at the target location, accuracy declined with memory load (one, two, or three locations to remember), F(2, 256) = 318.21. This effect of memory load varied with ɛ4 gene dose, F(4, 256) = 2.53. Figure 4 shows that the number of ɛ4 alleles exerted the strongest effect when the memory load was greatest. Confirming this result was our finding that the decrease in accuracy as the number of locations increased was greatest in the APOE-ɛ4 homozygotes (two ɛ4 alleles), who differed significantly from both the heterozygotes (one ɛ4 allele) and the noncarriers (PLSD, p < .05). The effect size calculated for unequal sample sizes (J. Cohen, 1988) was .23.
Figure 4.
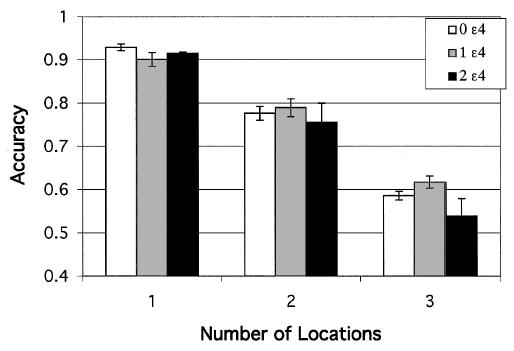
Experiment 2: Match condition. Accuracy of retention of target location after a 3-s delay for each ɛ4 gene dose group, plotted as a function of memory load (number of locations to be retained). Error bars represent standard errors.
On nonmatch trials, accuracy again declined with memory load, F(2, 256) = 83.22, but increased with distance between target and test location, F(2, 256) = 46.85. These two factors interacted, F(4, 512) = 53.19, largely because of effects of target–test distance when the memory load was greatest (three locations). When the test dot was near target location, the presence of more than one dot made it easier to tell if it was in the wrong location. When the test dot was relatively distant from one of the target locations, accuracy declined as the number of locations increased. There was no interaction of this effect with genotype. The effect of gene dose was seen only as a main effect, F(2, 128) = 3.36. The homozygotes differed significantly from only the heterozygotes (PLSD, p < .05).
Discussion
Inheritance of the APOE-ɛ4 allele has previously been shown to modulate attentional shifting between locations (Greenwood et al., 2000). In Experiment 1, this effect was found to vary with APOE-ɛ4 gene dose. The present results extend this finding to spatial working memory. The deleterious effects of the ɛ4 allele on spatial working memory were strongest under conditions of greatest memory load, that is, when three, compared with one or two, locations had to be kept in mind. However, as we discuss again in the General Discussion, the effect of genotype was seen mainly in the homozygotes.
Thus, the ɛ4 homozygotes showed reduced ability to retain memory for location. This was true whether or not the test dot matched the target location. When the test location did not match the target location, despite poorer retention the homozygous ɛ4 group showed effects of target–test distance that were similar to those of the other groups.
Thus, the homozygote group along with the other groups (a) performed the task well and (b) showed effects of the target–test distance manipulation. This indicates that APOE genotype affected memory but not overall task performance. Moreover, effects were somewhat selective, as under match conditions effects of genotype were evident mainly when demands on working memory were substantial. However, as pointed out by a reviewer, neuropsychological assessments of working memory used to diagnose AD and mild cognitive impairment use much longer delays than the 3 s used in this task.
Considered together, the results of Experiments 1 and 2 indicate that the ɛ4 allele alters aspects of both attention and working memory. The effects are subtle, being seen largely under conditions of higher processing demand (under match conditions). However, the effects are also robust and systematic, given that both attention and working memory were influenced by ɛ4 genotype. Moreover, the effect sizes were moderate to large. That both domains were affected is more consistent with the interpretation that APOE is associated with a cognitive phenotype than with a prodromal stage of AD. As discussed above, AD neuropathology is typically seen initially and selectively in the memory-associated mesial temporal regions (Braak & Braak, 1995). If the ɛ4 carriers were in a prodromal stage, then selective effects on memory would be predicted. Instead, both attention and memory were affected. Strengthening this view of an adult cognitive phenotype of APOE-ɛ4 would be evidence that the gene not only affects different cognitive systems but also the integration between systems. We next investigate the effect of this gene on the interaction between working memory and attention.
Experiment 3: Effect of Attention on Working Memory
As discussed above, the lipoprotein apoE appears to have a role in promoting neuronal health and plasticity in response to brain insult (Arendt et al., 1997; Fagan et al., 1998; Teter et al., 2002). Consistent with this evidence is the prediction that the APOE genotype would be expected to affect a number of cognitive processing systems, not just a single domain of cognition. We hypothesized, therefore, that in addition to its influence on integrity of separate cognitive systems, APOE genotype may also influence the ability to integrate across cognitive systems. Consequently, we next sought to determine the effects of APOE-ɛ4 gene dose on a task requiring the integration of attention and working memory. We predicted that the precision with which cues indicate the location of a target would modulate the accuracy of memory for that location. Accordingly, we used a cued working memory task in which a precue varying in size predicted the location of a target that had to be retained over a 3-s delay. After the delay, participants had to indicate whether a test dot was in the same location as the original target dot (see supplemental Figure 3 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp).
Method
Participants
See Experiment 1.
Stimuli and procedures
A task was designed to manipulate the accuracy of memory for location by varying the precision of location precues (see supplemental Figure 3 on the Web at http://dx.doi.org/10.1037/0894-4105.19.2.199.supp). The task was a spatial working memory task similar to that in Experiment 2, but with the target location precued with varying precision. Following a 1-s duration fixation cross, a circular cue appeared for 500 ms in 1 of 12 randomly selected locations on the screen and in one of three sizes (1.6°, 5.2°, and 8.1° of visual angle). At cue offset, one black target dot (.67° in diameter) appeared centered in the cue for 100 ms. Thus, cues were 100% valid in predicting target location. At target offset, a 3-s delay began, during which time only the fixation cross was visible. After the delay, the screen cleared and a red test dot appeared either at the same location as the target dot (match trial) or at a different location (nonmatch trial). On nonmatch trials, the distance between the target location (or nearest target location when there were multiple targets) and the test dot varied, being 1.9°, 3.8°, or 5.7° apart. The red test dot remained visible for 2 s, during which a match–nonmatch decision was required. Both accuracy and RT were measured.
Results
Accuracy
The measure of interest was accuracy of memory for target dot location. Accuracy ratios (defined as number correct: number presented) were subjected to a repeated measures ANOVA. Within-subjects factors were trial type (match or non-match) and target–test distance. Because target–test distance could be varied only under nonmatch conditions, an omnibus ANOVA was conducted comparing trial type by averaging across the target–test distance factor on nonmatch trials. As shown in Figure 5, this analysis revealed that accuracy was lower overall on match trials, F(1, 196) = 57.26, and in homozygotes (two ɛ4 alleles), F(2, 196) = 4.21. The interaction of trial type with cue size, F(2, 392) = 9.42, appears to be due to a benefit on accuracy from small cues on match trials but a cost from small cues on nonmatch trials.
Figure 5.
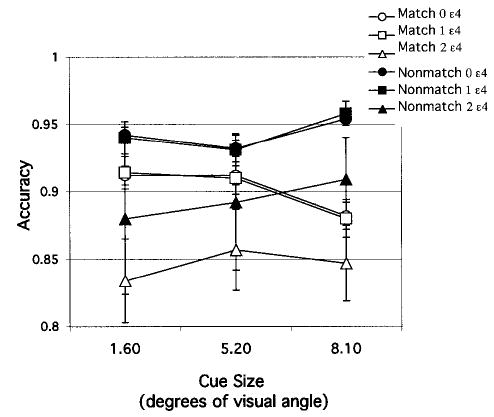
Experiment 3: Match and nonmatch (collapsed across target–test distance) conditions. Accuracy of retention of target location after a 3-s delay for each gene dose group, plotted as a function of cue size. Error bars represent standard errors.
On the basis of finding a significant main effect of trial type on accuracy ratios, we analyzed match and nonmatch conditions separately. Under match conditions, APOE-ɛ4 gene dose was the between-subjects factor, and cue size was the within-subjects factor. Accuracy on match trials, which ranged narrowly from .83 to .95, decreased as cue size increased, F(2, 196) = 3.01. Accuracy also decreased with ɛ4 gene dose, F(2, 196) = 3.37. A post hoc test of the group effect indicated that noncarriers differed significantly from homozygotes, who differed significantly from heterozygotes (PLSD, p < .02). The size of the main effect of ɛ4 gene dose was .14 (formula for unequal sample sizes, J. Cohen, 1988). As can be seen in Figure 5, both noncarriers (zero ɛ4 alleles) and heterozygotes (one ɛ4 allele) tended to decrease in accuracy with increasing cue size. In contrast, the homozygotes (two ɛ4 alleles) showed little change with cue size.
In analyzing nonmatch conditions, we set APOE-ɛ4 gene dose as the between-subjects factor and cue size and target–test distance as within-subjects factors. Accuracy was lower overall in the homozygotes, F(2, 196) = 3.30, and increased with target–test distance, reflecting the easier discrimination, when there was more distance between target and test stimuli, F(2, 392) = 83.89. The cue size effect, F(2, 392) = 12.40, was strongest when target and test locations were close together (Cue Size × Target–Test Distance; see Figure 6), F(4, 784) = 40.62. When the discrimination was more difficult, a benefit was obtained from a larger attentional scale. There were no significant interactions involving APOE-ɛ4 gene dose.
Figure 6.
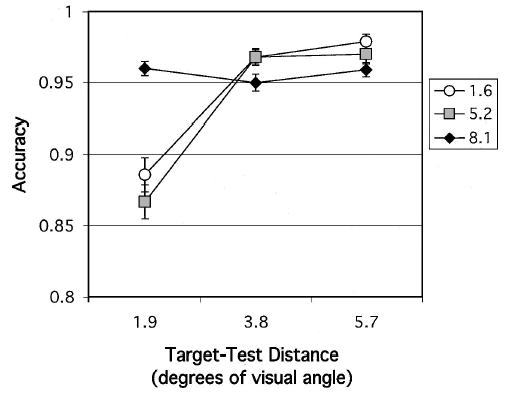
Experiment 3: Nonmatch condition. Accuracy of retention of target location after a 3-s delay for each cue size, plotted as a function of target–test distance. Error bars represent standard errors.
RT
Because decision accuracy was relatively high overall, speed of decision RT was also analyzed for match trials. RT was slowed by increased cue size, F(2, 392) = 4.25. This result complements those of the accuracy results described above, in which accuracy was reduced as cue size increased. Thus, as precue size became larger, which necessarily lessened precue precision, responses became both slower and less accurate. This effect of cue size on RT interacted with APOE-ɛ4 gene dose, F(4, 392) = 2.43. This was due to the different pattern shown by the homozygotes compared with the other gene dose groups (see Figure 7). Although RT in the noncarriers and heterozygotes slowed as cue size was enlarged, RT in the homozygotes showed only a weak effect of cue size. This variation of the homozygotes from the overall trend was also seen in the accuracy results above (see Figure 5).
Figure 7.
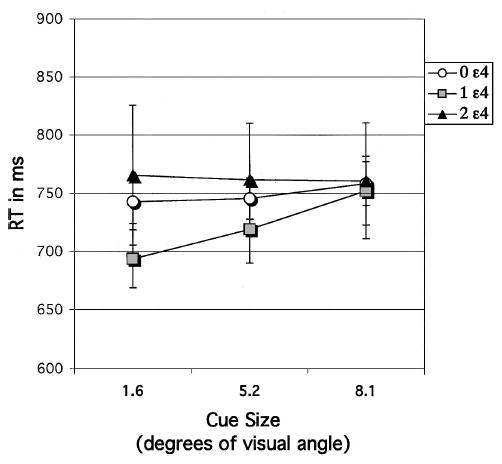
Experiment 3: Match condition. Reaction time (RT) of decision about target location following a 3-s delay, plotted as a function of cue size (1.6, 5.2, or 8.1) to target location for each apolipoprotein E–ɛ4 gene dose group. Error bars represent standard errors.
Discussion
These results show that both memory for spatial location and the effect of attention on memory for spatial location were modulated by APOE genotype. As in Experiment 2, effects of ɛ4 gene dose on memory were not graded—that is, they were seen mainly when ɛ4 gene dose was high (in homozygotes). The performance of heterozygotes was closer to that of noncarriers than of homozygotes, who were both slower and less accurate in responding.
Even though the target was always centered within the cued space—thereby eliminating spatial uncertainty—cue size modulated memory for target location. We interpret this result as reflecting a cue-induced adjustment of the distribution of visuospatial attention at the cued location—narrowly following small cues and broadly following large cues. Thus, scaling of the attentional focus around a location can facilitate encoding and/or retention of the memory for that location, much as previous work has shown that attentional scaling modulates detection (Castiello & Umilta, 1990), discrimination (Eriksen & St. James, 1986), and visual search (Greenwood, Parasuraman, & Alexander, 1997). These effects varied with APOE genotype. Noncarriers and ɛ4 heterozygotes obtained a processing benefit from a small cue, whereas homozygotes experienced little effect.
Researchers have argued that spatial working memory and visuospatial attention are the same process, with visuospatial attention acting as a rehearsal mechanism for spatial working memory (Awh & Jonides, 1998). Offered as evidence is research showing that working memory for a location is impaired when a secondary task inhibits attending to the location (Awh, Jonides, & Reuter-Lorenz, 1998) and manipulations of visuospatial attention and spatial working memory modulate the same early event-related-potential components (Awh, Anllo-Vento, & Hillyard, 2000). However, in the latter study, attention could have been directed to the memorized locations. The current results are not easily interpreted as supporting this view. It is not clear why increased precision of location cuing should affect the efficiency of rehearsal in spatial working memory. Rather, our results are consistent with the view that visuospatial attention and spatial working memory are separate processes, but with attention modulating memory.
As pointed out by a reviewer, the results of this study could be due to genotype-related differences in effects of attention on either encoding or retention. This study was not designed to distinguish between effects on encoding or on retention of memory for location. However, it is possible that heightened target perception led to improved memory for target location, a result that is consistent with research showing that attention modulates perception (Hawkins et al., 1990).
General Discussion
We conducted three experiments to examine the influence of the APOE-ɛ4 allele on cognition in healthy adults without dementia. The results showed that the APOE-ɛ4 allele affects several cognitive systems in middle-age—a decade or more before likely onset of symptoms of AD itself. We had previously shown that inheritance of at least one ɛ4 allele is associated with selective impairment in redirecting attention to a location in the visual field following an invalid cue (Greenwood et al., 2000). Experiment 1 confirmed this result in a larger sample of healthy adults and extended it by showing that the effect varied systematically with APOE-ɛ4 gene dose. The slowing of target discrimination following invalid cues (costs) increased with ɛ4 gene dose: the more alleles, the greater the slowing. Benefits of valid cues were unaffected. Experiment 2 extended the finding of ɛ4-related deficits to spatial working memory. The accuracy of retaining the location of a target in working memory over a delay was reduced in APOE-ɛ4 homozygotes, particularly under conditions of high memory load (three target locations). Finally, Experiment 3 showed that the relation between attention and working memory was also altered by APOE genotype. Memory for spatial location was modulated by the precision of information about future target location, provided by a precue of variable size. However, this effect of cue size on memory accuracy was reduced in APOE-ɛ4 homozygotes. Overall, therefore, the APOE-ɛ4 allele was associated with relative impairment in (a) redirecting visuospatial attention to an unexpected location, (b) retaining a target location in working memory, and (c) using attentional scaling to enhance spatial working memory. Moreover, these effects were seen in healthy, mostly middle-aged individuals with few deficits on standardized cognitive tests.
These findings indicate that this polymorphism in the APOE gene exerts effects on specific components of cognition in midlife. Although the effects are subtle in that only particular components of cognition are affected under certain task conditions, the effect sizes for the APOE gene were moderate to large, ranging from .14 to .24. Effects of the APOE-ɛ4 allele on spatial attention (Greenwood et al., 2000) and memory (Bondi et al., 1995) have been reported previously. However, in the present study we were able to examine effects of the APOE-ɛ4 gene dose, rather than just the presence of the ɛ4 allele. Furthermore, our results show that these genotype effects occur not only over a range of cognitive domains but also in the interaction between cognitive domains, namely, the effect of visuospatial attention on working memory.
Although significant genotype effects were obtained in both visuospatial attention and working memory performance, there were some differences in the effects observed on these two cognitive systems. For attention, the effect of the APOE-ɛ4 allele was graded and quasilinear, results that are consistent with a gene dose interpretation. For working memory, contrary to our hypothesis, the genotype effect was not graded. In Experiment 2, there was relatively little effect when only one ɛ4 allele was inherited (ɛ4 heterozygotes), but there was a marked effect when two alleles were inherited (ɛ4 homozygotes). A similar result was seen in Experiment 3, which examined the effect of attention on working memory and found the modulation of working memory by APOE genotype to be significant only at the highest gene dose, that is, in ɛ4 homozygotes but not in ɛ4 heterozygotes.
The reason for the observed lack of an effect of ɛ4 heterozygosity on working memory is not fully clear. Evidence of additivity of the APOE-ɛ4 allele, manifested in a gene dose effect, has been shown in a number of studies with regard to risk of AD (e.g., Corder et al., 1993) and with regard to cognitive decline without dementia (Feskens et al., 1994). Therefore, it is interesting that we found an effect of ɛ4 gene dose on attention but an effect of ɛ4 presence or absence on memory. Although memory loss is defined to be the earliest symptom of AD (McKhann et al., 1984), the literature does not agree on the order in which deficits appear in APOE-ɛ4 carriers without dementia. Some investigators have reported selective effects on episodic memory (Bondi et al., 1999; Wilson et al., 2002), whereas others have observed the earliest effects of APOE genotype on spatial ability (Bretsky, Guralnik, Launer, Albert, & Seeman, 2003).
It is possible that we would have obtained significant effects in heterozygotes on the memory tasks if a higher memory load had been used. Alternatively, if confirmed, this finding suggests that the ɛ4 allele exerts additive effects on risk of AD and on integrity of attention systems but not on integrity of memory systems. This possibility would suggest that the brain regions mediating working memory are somehow less vulnerable to the negative consequences of ɛ4 inheritance, perhaps because of greater redundancy or relative invulnerability to weaker neuronal repair mechanisms associated with APOE-ɛ4 (Teter & Ashford, 2002). This is contradicted by the literature. Working memory is at least partly dependent on functioning of the hippocampus (Burgess, Maguire, & O’Keefe, 2002; Mitchell, Macrae, & Gilchrist, 2002), one of the first regions affected in the progression of AD pathology (Braak & Braak, 1996). However, the known plasticity of the hippocampus in response to experience in adulthood (Maguire et al., 2000) might contribute to a reduced vulnerability to deleterious effects of the ɛ4 allele. Regardless of the reason for the difference in APOE-ɛ4 gene dose effects, APOE genotype did affect spatial attention, working memory, and the interaction of spatial attention and working memory.
What do these results imply for the association of APOE-ɛ4 with the existence of (a) an adult cognitive phenotype or (b) a prodromal stage of AD? Although the existence of a prodromal stage of AD cannot be completely ruled out, we argue that the pattern of results we obtained is more consistent with the existence of a cognitive phenotype. In the first place, a prodromal phase in these middle-aged individuals would have to be more than a decade long, as the average age of the homozygote group in the present study was 57.6 (SD = 7.7, range = 48.5 to 73.4), with only 3 individuals over age 61. Average age of onset of AD is 68.4 in homozygotes and 75.5 in heterozygotes (Corder et al., 1993). The prodromal view as described by Daffner and Scinto (2000) assumes that the pathology of AD is initially clinically silent (presymptomatic stage) but progresses over time to the point where symptoms reach clinical significance. Although this view is intuitive, it has not been consistently shown that the existence of pathology in cognitively unimpaired individuals predicts progression to AD. This is not to argue that AD is not accompanied by pathology, but rather to point out that it is hard to predict which cognitively unimpaired individuals will develop AD, given that only about 50% of ɛ4 homozygotes develop AD by age 80 or 90 (Henderson et al., 1995; Meyer et al., 1998). Cognitively unimpaired individuals can and do show the characteristic plaques and tangles of AD (reviewed in Arriagada et al., 1992; Geula, 2000), including a beta-amyloid load equal to or greater than that seen in some individuals with AD (Mufson et al., 1999). Similarly, although some investigators have found that medial temporal atrophy in healthy individuals predicts AD (Killiany et al., 2000), others have not (Jack et al., 2002; Visser et al., 1999). Moreover, although the natural history of the AD prodrome is not known, we are unaware of evidence that it is decades long. Researchers investigating this question appear to agree that symptoms precede diagnosis by 4–6 years (Backman, Laukka, Wahlin, Small, & Fratiglioni, 2002; Linn et al., 1995; Small et al., 2000). Therefore, it cannot be assumed that cognitive deficits shown in the present study by healthy ɛ4 homozygotes in middle age are indicative of an AD prodrome.
Second, given that the neurofibrillary tangles pathognomonic of AD first appear in the hippocampus and related structures (Braak & Braak, 1995), a prodromal explanation would predict selective effects on memory, such as reported recently (Wilson et al., 2002). In contrast to that study, in the present study we obtained little evidence of cognitive selectivity. One difference could be the age of the participants. The use of participants in their mid-70s by Wilson et al. (2002) increases the likelihood of including individuals in the early stages of AD (Sliwinski et al., 1996). Another source of difference could be the tasks used. The standardized tests used by Wilson et al. may be less sensitive to small deficits in component processes of cognition than the more specific and tailored tasks used in the present study.
We acknowledge that the existence of an ɛ4-related AD prodrome could explain the present results if (a) a subset of these middle-aged ɛ4 carriers were in the early stages of the disease and (b) the performance of those individuals was sufficient to affect the group mean. Examining the means for RT costs in Experiment 1 shows that all but 3 of those in the homozygote group had values that exceeded the mean of the noncarriers. In Experiment 2, all but 4 of those in the homozygote group had difference scores that were larger than the mean of the noncarriers. In Experiment 3, all but 3 of the homozygotes had lower accuracy than the noncarriers. Therefore, in each study, most of the individuals in the homozygote group showed the group effect.
We conclude that the present evidence of subtle cognitive deficits in middle-aged ɛ4 carriers without dementia is more consistent with the emergence of an APOE-ɛ4 cognitive phenotype in adulthood rather than with an AD prodrome lasting a decade or longer. Such a cognitive phenotype could arise from suboptimal repair processes related to brain aging and resulting in subtle deficits in a broad range of abilities. Consistent with this view of a cognitive phenotype related to efficiency of neuronal repair in adulthood is the following evidence. First, in childhood, IQ is unrelated to APOE genotype (Turic et al., 2001). Second, in adulthood, the APOE-ɛ4 allele is associated with a range of neurologic conditions: poorer prognosis following head injury (Crawford et al., 2002), incidence of late-onset AD (Corder et al., 1993), rate of progression in multiple sclerosis (Fazekas et al., 2001), and amyotrophic lateral sclerosis (Drory et al., 2001). The APOE-ɛ4 allele has also been linked to cognitive integrity in individuals with diabetes (Ferguson et al., 2003) and folate deficiencies (Mattson, 2003). Third, the protein product of the APOE gene appears to play a role in neuronal plasticity and repair. As discussed previously, the apoE lipoprotein has a role in synapse development (Mauch et al., 2001), clearance of lipid debris from the site of the injury (White, Nicoll, & Horsburgh, 2001), and promotion of granule cell mossy fiber sprouting (Teter et al., 2002). These effects of APOE genotype on neuronal health and plasticity may become particularly important once processes of adult aging have begun. Considered together with results of the present study, this evidence suggests that an APOE-ɛ4 cognitive phenotype reflects reduced integrity of a number of cognitive systems attributable to inefficient mechanisms of neuronal repair and plasticity.
This study has several limitations. The sample is highly educated, and therefore, results may not be generalizable to the population as a whole. Also, no attempt was made to control for effects of gender. Finally, a longitudinal design would have advantages for investigating the natural history of the AD prodrome as a function of APOE genotype, and we are presently conducting such a study.
Acknowledgments
This research was supported in part by National Institute on Aging Grants AG07569 and AG19653 to Raja Parasurman and AG12387 to P. M. Greenwood. We thank the clinical staff of the Geriatric Psychiatry Branch of the National Institute of Mental Health.
Footnotes
P. M. Greenwood and Raja Parasuraman, Cognitive Science Laboratory, Catholic University of America; Chantal Lambert, Cognitive Science Laboratory, Catholic University of America, and Geriatric Psychiatry Branch, National Institute of Mental Health, Bethesda, Maryland; Trey Sunderland, Geriatric Psychiatry Branch, National Institute of Mental Health.
Contributor Information
P. M. Greenwood, Catholic University of America.
Chantal Lambert, Catholic University of America and National Institute of Mental Health.
Trey Sunderland, National Institute of Mental Health.
Raja Parasuraman, Catholic University of America.
References
- Arendt T, Schindler C, Bruckner MK, Eschrich K, Bigl V, Zedlick D, et al. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. Journal of Neuroscience. 1997;17:516–529. doi: 10.1523/JNEUROSCI.17-02-00516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- Awh E, Anllo-Vento L, Hillyard SA. The role of spatial selective attention in working memory for locations: Evidence from event-related potentials. Journal of Cognitive Neuroscience. 2000;12:840–847. doi: 10.1162/089892900562444. [DOI] [PubMed] [Google Scholar]
- Awh, E., & Jonides, J. (1998). Spatial working memory and spatial selective attention. In R. Parasuraman (Ed.), The attentive brain (pp. 353–380). Cambridge, MA: MIT Press.
- Awh E, Jonides J, Reuter-Lorenz PA. Rehearsal in spatial working memory. Journal of Experimental Psychology: Human Perception and Performance. 1998;24:780–790. doi: 10.1037//0096-1523.24.3.780. [DOI] [PubMed] [Google Scholar]
- Backman L, Laukka EJ, Wahlin A, Small BJ, Fratiglioni L. Influences of preclinical dementia and impending death on the magnitude of age-related cognitive deficits. Psychology and Aging. 2002;17:435–442. doi: 10.1037//0882-7974.17.3.435. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Galasko D, Salmon DP, Thomas RG. Neuropsychological function and apolipoprotein E genotype in the preclinical detection of Alzheimer’s disease. Psychology and Aging. 1999;14:295–303. doi: 10.1037//0882-7974.14.2.295. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Salmon DP, Monsch AU, Galasko D, Butters N, Klauber MR, et al. Episodic memory changes are associated with the APOE-ɛ4 allele in nondemented older adults. Neurology. 1995;45:2203–2206. doi: 10.1212/wnl.45.12.2203. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiology of Aging. 1995;16:271–284. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurologica Scandinavica. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- Bretsky P, Guralnik JM, Launer L, Albert M, Seeman TE. The role of APOE-ɛ4 in longitudinal cognitive decline: MacArthur Studies of Successful Aging. Neurology. 2003;60:1077–1081. doi: 10.1212/01.wnl.0000055875.26908.24. [DOI] [PubMed] [Google Scholar]
- Brewer JB, Zhao Z, Desmond JE, Glover GH, Gabrieli JD. Making memories: Brain activity that predicts how well visual experience will be remembered. Science. 1998 August 21;281:1185–1187. doi: 10.1126/science.281.5380.1185. [DOI] [PubMed] [Google Scholar]
- Burgess N, Maguire EA, O’Keefe J. The human hippocampus and spatial and episodic memory. Neuron. 2002;35:625–641. doi: 10.1016/s0896-6273(02)00830-9. [DOI] [PubMed] [Google Scholar]
- Buschke H. Selective reminding for analysis of memory and learning. Journal of Verbal Learning and Verbal Behavior. 1973;12:543–550. [Google Scholar]
- Castiello U, Umilta C. Size of the attentional focus and efficiency of processing. Acta Psychologica. 1990;73:195–209. doi: 10.1016/0001-6918(90)90022-8. [DOI] [PubMed] [Google Scholar]
- Cohen, J. (1988). Statistical power analysis for the behavioral sciences (2nd ed.). Hillsdale, NJ: Erlbaum.
- Cohen RM, Small C, Lalonde F, Friz J, Sunderland T. Effect of apolipoprotein E genotype on hippocampal volume loss in aging healthy women. Neurology. 2001;57:2223–2228. doi: 10.1212/wnl.57.12.2223. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of Apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993 August 13;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Crawford FC, Vanderploeg RD, Freeman MJ, Singh S, Waisman M, Michaels L, et al. APOE genotype influences acquisition and recall following traumatic brain injury. Neurology. 2002;58:1115–1118. doi: 10.1212/wnl.58.7.1115. [DOI] [PubMed] [Google Scholar]
- Daffner, K. R., & Scinto, L. F. M. (2000). Early diagnosis of Alzheimer’s disease. In L. F. M. Scinto & K. R. Daffner (Eds.), Early diagnosis of Alzheimer’s disease (pp. 1–27). Totowa, NJ: Humana Press.
- Davis JN, II, Chisholm JC. The “amyloid cascade hypothesis” of AD: Decoy or real McCoy? Trends in Neuroscience. 1997;20:558–559. doi: 10.1016/s0166-2236(97)85989-9. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, et al. Cognitive change and the APOE ɛ4 allele. Nature. 2002 August 29;418:932. doi: 10.1038/418932a. [DOI] [PubMed] [Google Scholar]
- Donnai D, Karmiloff-Smith A. Williams syndrome: From genotype through to the cognitive phenotype. American Journal of Medical Genetics. 2000;97:164–171. doi: 10.1002/1096-8628(200022)97:2<164::aid-ajmg8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Drory VE, Birnbaum M, Korczyn AD, Chapman J. Association of APOE ɛ4 allele with survival in amyotrophic lateral sclerosis. Journal of Neurological Science. 2001;190:17–20. doi: 10.1016/s0022-510x(01)00569-x. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proceedings of the National Academy of Sciences, USA. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen CW, St James JD. Visual attention within and around the field of focal attention: A zoom lens model. Perception and Psychophysics. 1986;40:225–240. doi: 10.3758/bf03211502. [DOI] [PubMed] [Google Scholar]
- Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, et al. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. Journal of the American Medical Association. 1989;262:2551–2556. [PubMed] [Google Scholar]
- Fagan AM, Murphy BA, Patel SN, Kilbridge JF, Mobley WC, Bu G, et al. Evidence for normal aging of the septo-hippocampal cholinergic system in apoE (−/−) mice but impaired clearance of axonal degeneration products following injury. Experimental Neurology. 1998;151:314–325. doi: 10.1006/exnr.1998.6818. [DOI] [PubMed] [Google Scholar]
- Fan J, McCandliss BD, Sommer T, Raz A, Posner MI. Testing the efficiency and independence of attentional networks. Journal of Cognitive Neuroscience. 2002;14:340–347. doi: 10.1162/089892902317361886. [DOI] [PubMed] [Google Scholar]
- Fan J, Wu Y, Fossella JA, Posner MI. Assessing the heritability of attentional networks. BMC Neuroscience. 2001;2:14. doi: 10.1186/1471-2202-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazekas F, Strasser-Fuchs S, Kollegger H, Berger T, Kristoferitsch W, Schmidt H, et al. Apolipoprotein E epsilon 4 is associated with rapid progression of multiple sclerosis. Neurology. 2001;57:853–857. doi: 10.1212/wnl.57.5.853. [DOI] [PubMed] [Google Scholar]
- Ferguson SC, Deary IJ, Evans JC, Ellard S, Hattersley AT, Frier BM. Apolipoprotein-e influences aspects of intellectual ability in type 1 diabetes. Diabetes. 2003;52:145–148. doi: 10.2337/diabetes.52.1.145. [DOI] [PubMed] [Google Scholar]
- Feskens EJM, Havekes LM, Kalmijn S, Knijff PD, Launer LJ, Kromhout D. Apolipoprotein ɛ4 allele and cognitive decline in elderly men. British Journal of Medicine. 1994;309:1202–1206. doi: 10.1136/bmj.309.6963.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory JD, Manuck SB, Ferrell RE, Ryan CM, Muldoon MF. Memory performance and the apolipoprotein E polymorphism in a community sample of middle-aged adults. American Journal of Medical Genetics. 2000;96:707–711. doi: 10.1002/1096-8628(20001204)96:6<707::aid-ajmg1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. Mini-mental state: A practical method for grading the state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fossella J, Sommer T, Fan J, Wu Y, Swanson JM, Pfaff DW, et al. Assessing the molecular genetics of attention networks. BMC Neuroscience. 2002;3:14. doi: 10.1186/1471-2202-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NC, Warrington EK, Seiffer AL, Agnew SK, Rossor MN. Presymptomatic cognitive deficits in individuals at risk of familial Alzheimer’s disease. A longitudinal prospective study. Brain. 1998;121:1631–1639. doi: 10.1093/brain/121.9.1631. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Robidoux J, Alarcon M, Miller BL, Wilhelmsen KC, Cummings JL, et al. Dementia and neurodevelopmental predisposition: Cognitive dysfunction in presymptomatic subjects precedes dementia by decades in frontotemporal dementia. Annals of Neurology. 2001;50:741–746. doi: 10.1002/ana.10024. [DOI] [PubMed] [Google Scholar]
- Geula, C. (2000). Pathological diagnosis of Alzheimer’s disease. In L. F. M. Scinto & K. R. Daffner (Eds.), Early diagnosis of Alzheimer’s disease (pp. 65–82). Totowa, NJ: Humana Press.
- Gottlob LR, Madden DJ. Age differences in the strategic allocation of visual attention. Journals of Gerontology Series B: Psychological Science & Social Science. 1999;54:165–172. doi: 10.1093/geronb/54b.3.p165. [DOI] [PubMed] [Google Scholar]
- Greenwood PM, Parasuraman R. Attentional disengagement deficit in nondemented elderly over 75 years of age. Aging and Cognition. 1994;1:188–202. [Google Scholar]
- Greenwood PM, Parasuraman R. Scale of attentional focus in visual search. Perception and Psychophysics. 1999;61:837–859. doi: 10.3758/bf03206901. [DOI] [PubMed] [Google Scholar]
- Greenwood PM, Parasuraman R, Alexander GE. Controlling the focus of spatial attention during visual search: Effects of advanced aging and Alzheimer disease. Neuropsychology. 1997;11:3–12. doi: 10.1037//0894-4105.11.1.3. [DOI] [PubMed] [Google Scholar]
- Greenwood PM, Parasuraman R, Haxby JV. Changes in visuospatial attention over the adult lifespan. Neuropsychologia. 1993;31:471–485. doi: 10.1016/0028-3932(93)90061-4. [DOI] [PubMed] [Google Scholar]
- Greenwood PM, Sunderland T, Friz JL, Parasuraman R. Genetics and visual attention: Selective deficits in healthy adult carriers of the ɛ4 allele of the apolipoprotein E gene. Proceedings of the National Academy of Sciences, USA. 2000;97:11661–11666. doi: 10.1073/pnas.97.21.11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley AA, Kieley J, McKenzie CRM. Allocation of visual attention in younger and older adults. Perception and Psychophysics. 1992;52:175–185. doi: 10.3758/bf03206771. [DOI] [PubMed] [Google Scholar]
- Hawkins HL, Hillyard SA, Luck SJ, Mouloua M, Downing CJ, Woodward DP. Visual attention modulates signal detectability. Journal of Experimental Psychology. 1990;16:802–811. doi: 10.1037//0096-1523.16.4.802. [DOI] [PubMed] [Google Scholar]
- Henderson AS, Easteal S, Jorm AF, Mackinnon AJ, Korten AE, Christensen H, et al. Apolipoprotein E allele ɛ4, dementia, and cognitive decline in a population sample. Lancet. 1995;346:1387–1390. doi: 10.1016/s0140-6736(95)92405-1. [DOI] [PubMed] [Google Scholar]
- Hughes C, Plumet MH, Leboyer M. Towards a cognitive phenotype for autism: Increased prevalence of executive dysfunction and superior spatial span amongst siblings of children with autism. Journal of Child Psychology and Psychiatry. 1999;40:705–718. [PubMed] [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nature and Genetics. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Dickson DW, Parisi JE, Xu YC, Cha RH, O’Brien PC, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750–757. doi: 10.1212/wnl.58.5.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Abrams MT, Chen W, Reiss AL. Genotype, molecular phenotype, and cognitive phenotype: Correlations in Fragile X syndrome. American Journal of Medical Genetics. 1999;83:286–295. [PubMed] [Google Scholar]
- Killiany RJ, Gomez-Isla T, Moss M, Kikinis R, Sandor T, Jolesz F, et al. Use of structural magnetic resonance imaging to predict who will get Alzheimer’s disease. Annals of Neurology. 2000;47:430–439. [PubMed] [Google Scholar]
- Krzywkowski P, Ghribi O, Gagne J, Chabot C, Kar S, Rochford J, et al. Cholinergic systems and long-term potentiation in memory-impaired apolipoprotein E-deficient mice. Neuroscience. 1999;92:1273–1286. doi: 10.1016/s0306-4522(99)00061-5. [DOI] [PubMed] [Google Scholar]
- Linn RT, Wolf PA, Bachman DL, Knoefel JE, Cobb JL, Belanger AJ, et al. The “preclinical phase” of probable Alzheimer’s disease. A 13-year prospective study of the Framingham cohort. Archives of Neurology. 1995;52:485–490. doi: 10.1001/archneur.1995.00540290075020. [DOI] [PubMed] [Google Scholar]
- Maguire EA, Gadian DG, Johnsrude IS, Good CD, Ashburner J, Frackowiak RS, et al. Navigation-related structural change in the hippocampi of taxi drivers. Proceedings of the National Academy of Sciences, USA. 2000;97:4398–4403. doi: 10.1073/pnas.070039597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis, S. (1976). Mental status evaluation for organic mental syndrome in the elderly patient. In L. Bellak & T. B. Karasu (Eds.), Geriatric psychiatry (77–121). New York: Grune and Stratton.
- Mattson MP. Gene–diet interactions in brain aging and neuro-degenerative disorders. Annals of Internal Medicine. 2003;139:441–444. doi: 10.7326/0003-4819-139-5_part_2-200309021-00012. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001 November 9;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report on the NINCDS–ADRDA work group under the auspices of the Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Tschanz JT, Norton MC, Welsh-Bohmer KA, Steffens DC, Wyse BW, et al. APOE genotype predicts when—not whether—one is predisposed to develop Alzheimer disease. Nature Genetics. 1998;19:321–322. doi: 10.1038/1206. [DOI] [PubMed] [Google Scholar]
- Mitchell JP, Macrae CN, Gilchrist ID. Working memory and the suppression of reflexive saccades. Journal of Cognitive Neuroscience. 2002;14:95–103. doi: 10.1162/089892902317205357. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, Kordower JH. Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Experimental Neurology. 1999;158:469–490. doi: 10.1006/exnr.1999.7086. [DOI] [PubMed] [Google Scholar]
- Parasuraman R, Greenwood PM, Haxby JV, Grady CL. Visuospatial attention in dementia of the Alzheimer type. Brain. 1992;115:711–733. doi: 10.1093/brain/115.3.711. [DOI] [PubMed] [Google Scholar]
- Parasuraman R, Greenwood PM, Sunderland T. The apolipoprotein E gene, attention, and brain function. Neuropsychology. 2002;16:254–274. doi: 10.1037//0894-4105.16.2.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Welsh-Bohmer KA, Bigler ED, Johnson SC, Anderson CV, Helms MJ, et al. Apolipoprotein E ɛ4 allele and hippocampal volume in twins with normal cognition. Neurology. 1997;48:985–989. doi: 10.1212/wnl.48.4.985. [DOI] [PubMed] [Google Scholar]
- Plomin, R., DeFries, J. C., McClearn, G. E., & McGuffin, P. (2001). Behavioral genetics (4th ed.). New York: Worth.
- Posner MI. Orienting of attention. Quarterly Journal of Experimental Psychology. 1980;32:3–25. doi: 10.1080/00335558008248231. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the ɛ4 allele for apolipoprotein E. New England Journal of Medicine. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proceedings of the National Academy of Sciences, USA. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Uecker A, Caselli RJ, Lewis S, Bandy D, de Leon MJ, et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer’s disease. Annals of Neurology. 1998;44:288–291. doi: 10.1002/ana.410440226. [DOI] [PubMed] [Google Scholar]
- Salthouse TA, Mitchell DRD, Skovronek E, Babcock RL. Effects of adult age and working memory on reasoning and spatial abilities. Journal of Experimental Psychology: Learning, Memory, and Cognition. 1989;15:507–516. doi: 10.1037//0278-7393.15.3.507. [DOI] [PubMed] [Google Scholar]
- Sliwinski M, Lipton RB, Buschke H, Stewart W. The effects of preclinical dementia on estimates of normal cognitive functioning in aging. Journals of Gerontology B. 1996;51:P217–P225. doi: 10.1093/geronb/51b.4.p217. [DOI] [PubMed] [Google Scholar]
- Small BJ, Fratiglioni L, Viitanen M, Winblad B, Backman L. The course of cognitive impairment in preclinical Alzheimer disease: Three- and 6-year follow-up of a population-based sample. Archives of Neurology. 2000;57:839–844. doi: 10.1001/archneur.57.6.839. [DOI] [PubMed] [Google Scholar]
- Smith GE, Bohac DL, Waring SC, Kokmen E, Tangalos EG, Ivnik RJ, et al. Apolipoprotein E genotype influences cognitive “phenotype” in patients with Alzheimer’s disease but not in healthy control subjects. Neurology. 1998;50:355–362. doi: 10.1212/wnl.50.2.355. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Kemper SJ, Mortimer JA, Greiner LH, Wekstein DR, Markesbery WR. Linguistic ability in early life and cognitive function and Alzheimer’s disease in late life. Findings from the Nun Study. Journal of the American Medical Association. 1996;275:528–532. [PubMed] [Google Scholar]
- Swanson JM, Oosterlaan J, Murias M, Schuck S, Flodman P, Spence MA, et al. Attention deficit/hyperactivity disorder children with a 7-repeat allele of the dopamine receptor D4 gene have extreme behavior but normal performance on critical neuropsychological tests of attention. Proceedings of the National Academy of Sciences, USA. 2000;97:4754–4759. doi: 10.1073/pnas.080070897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter B, Ashford JW. Neuroplasticity in Alzheimer’s disease. Journal of Neuroscience Research. 2002;70:402–437. doi: 10.1002/jnr.10441. [DOI] [PubMed] [Google Scholar]
- Teter B, Xu PT, Gilbert JR, Roses AD, Galasko D, Cole GM. Defective neuronal sprouting by human apolipoprotein E4 is a gain-of-negative function. Journal of Neuroscience Research. 2002;68:331–336. doi: 10.1002/jnr.10221. [DOI] [PubMed] [Google Scholar]
- Turic D, Fisher PJ, Plomin R, Owen MJ. No association between apolipoprotein E polymorphisms and general cognitive ability in children. Neuroscience Letters. 2001;299:97–100. doi: 10.1016/s0304-3940(00)01789-4. [DOI] [PubMed] [Google Scholar]
- Utermann G, Langenbeck U, Beisiegel U, Weber W. Genetics of the apolipoprotein E system in man. American Journal of Human Genetics. 1980;32:339–347. [PMC free article] [PubMed] [Google Scholar]
- Visser PJ, Scheltens P, Verhey FR, Schmand B, Launer LJ, Jolles J, et al. Medial temporal lobe atrophy and memory dysfunction as predictors for dementia in subjects with mild cognitive impairment. Journal of Neurology. 1999;246:477–485. doi: 10.1007/s004150050387. [DOI] [PubMed] [Google Scholar]
- Wechsler, D. (1987). Wechsler Memory Scale—Revised. New York: Psychological Corporation.
- Whalley LJ, Starr JM, Athawes R, Hunter D, Pattie A, Deary IJ. Childhood mental ability and dementia. Neurology. 2000;55:1455–1459. doi: 10.1212/wnl.55.10.1455. [DOI] [PubMed] [Google Scholar]
- White F, Nicoll JA, Horsburgh K. Alterations in ApoE and ApoJ in relation to degeneration and regeneration in a mouse model of entorhinal cortex lesion. Experimental Neurology. 2001;169:307–318. doi: 10.1006/exnr.2001.7655. [DOI] [PubMed] [Google Scholar]
- White F, Nicoll JA, Roses AD, Horsburgh K. Impaired neuronal plasticity in transgenic mice expressing human apolipoprotein E4 compared to E3 in a model of entorhinal cortex lesion. Neurobiology of Disease. 2001;8:611–625. doi: 10.1006/nbdi.2001.0401. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Schneider JA, Barnes LL, Beckett LA, Aggarwal NT, Cochran EJ, et al. The apolipoprotein E ɛ4 allele and decline in different cognitive systems during a 6-year period. Archives of Neurology. 2002;59:1154–1160. doi: 10.1001/archneur.59.7.1154. [DOI] [PubMed] [Google Scholar]