
Fig. 12. Model of trans-10, cis-12 CLA-mediated adipocyte delipidation: metabolic control through adipocytokine-initiated, MEK/ERK-dependent repression of PPAR-γ.
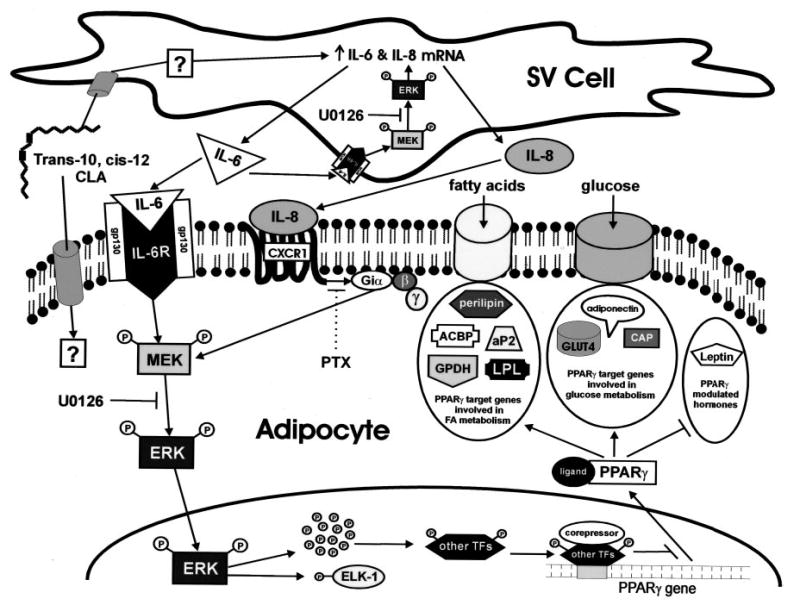
CLA can either enter into the adipocyte, where it may have direct effects or enter into the supporting SV cells to initiate an autocrine/paracrine signaling network. In the SV cell, CLA through a currently unidentified mechanism, increases the mRNA expression and secretion of IL-6 and IL-8. The resulting nascent IL-6 binds to its obligate transmembrane receptor (IL-6R), both on the surface of the adipocyte and the SV cell, where it activates MEK/ERK signaling in both cell populations. In addition, nascent IL-8 binds to its obligate heptahelical receptor (CXCR1), a PTX-sensitive GPCR only expressed in adipocytes, to amplify further MEK/ERK signaling in the adipocyte. The autocrine actions of nacent IL-6 in SV cells result in sustained activation of MEK/ERK signaling, which augments IL-6 and IL-8 mRNA expression, thereby feed-forwarding their synthesis and secretion. The collective paracrine actions of both IL-6 and IL-8 in the adipocyte results in sustained MEK and ERK hyperphosphorylation. Hyperphosphorylated ERK is then shuttled into the nucleus where it can phosphorylate multiple transcription factors (TFs) including ELK-1 and potentially other unidentified transcription factors. The ERK-dependent phosphorylation of other transcription factors may repress the expression of PPAR-γ itself. Collectively, ERK-dependent repression of PPAR-γ gene expression blocks the ability of PPAR-γ to modulate its traditional downstream target genes. The final result is decreased expression of genes involved in FA uptake and metabolism such as perilipin, acyl-CoA binding protein (ACBP), aP2, GPDH, and lipoprotein lipase (LPL) and genes involved in glucose uptake and metabolism such as GLUT4, CAP, and adiponectin. In addition, the ability of ligand-bound PPAR-γ to repress leptin expression is alleviated by CLA, thereby augmenting leptin, a critical regulator of lipid metabolism.