

Abstract
Objectives
Previous work defined two flavin-containing monooxygenase 2 (FMO2) alleles. The major allele, FMO2*2 (g.23,238C> T), encodes truncated inactive protein (p.X472) whereas the minor allele, FMO2*1, present in African- and Hispanic-American populations, encodes active protein (p.Q472). Recently, four common (27 to 51% incidence) FMO2 single nucleotide polymorphisms (SNPs) were detected in African-Americans (N= 50); they encode the following protein variants: p.71Ddup, p.V113fs, p.S195L and p.N413 K. Our objectives were to: (1) determine the incidence of these SNPs in 29 Hispanic individuals previously genotyped as g.23,238C (p.Q472) and 124 previously genotyped as homozygous g.23,238 T (p.X472); (2) determine FMO2 haplotypes in this population; and (3) assess the functional impact of SNPs in expressed proteins.
Methods
SNPs were detected via allele-specific oligonucleotide amplification coupled with real-time or electrophoretic product detection, or single strand conformation polymorphism.
Results
The g.7,700_7,702dupGAC SNP (p.71Ddup) was absent. The remaining SNPs were present but, except for g.13,732C > T (p.S195L), were less common in the current Hispanic study population versus the previously described African-Americans. Only expressed p.N413K was as active as p.Q472, as determined by methimazole- and ethylenethiourea-dependent oxidation. Haplotype determination demonstrated that the g.10,951delG (p.V113fs), g.13,732C > T (p.S195L) and g.22,060T >G (p.N413 K) variants segregated with g.23,238C> T (p.X472).
Conclusions
SNPs would not alter FMO2 activity in individuals possessing at least one FMO2*1 allele. It is likely that these SNPs will segregate similarly in African-American populations. Therefore, estimates that 26% of African-Americans and 2–7% of Hispanic- Americans have at least one FMO2*1 allele should closely reflect the percentages producing active FMO2 protein.
Keywords: SNPs, haplotype, drug metabolism, ethnic polymorphism, human, pulmonary, baculovirus expression, flavin-containing monooxygenase
Introduction
Flavin-containing monooxygenase (FMO) metabolizes a wide range of drugs and xenobiotics, as well as some endogenous substrates [1,2]. FMO contains a single flavin adenine dinucleotide (FAD), which following reduction by reduced nicotinamide adenine dinucleotide phosphate (NADPH) and reaction with oxygen, forms a stable C4a-peroxyflavin (FADOOH) intermediate. The FADOOH reacts with any soft nucleophile gaining access to the flavin. Most substrates characterized to date contain a sulfur or nitrogen susceptible to oxidation. Endogenous substrates include cysteamine, lipoic acid, trimethylamine and biogenic amines. FMO metabolizes many of the same drugs as the cytochrome P450 (CYP) monooxygenase system, but in many cases the products are distinct (e.g., tertiary amines are N-dealkylated by CYP and N-oxygenated by FMO) [3]. Examples of drugs for which FMO-dependent N-oxygenation plays a major role in metabolism include chlor- and bromo-pheniramine, zimeldine, ranitidine, benzydamine, olopatadine, xanomeline, pargyline and itopride. Sulfur-containing drugs for which FMO plays a major role in metabolism include cimetidine, albendazole, sulindac sulfide, methimazole and ethionamide [4].
Eleven distinct FMO genes exist in humans, encoding five active enzymes (FMO1–5) and six pseudogenes [5–7]. FMO2 is not expressed in significant amounts in liver and although mammalian FMO2 is expressed in a number of extrahepatic tissues, including kidney, nasal mucosa and intestine, it is not the predominant FMO isoform in these tissues [8–10]. However, FMO2 is the major isoform expressed in human lung [11,12] and if gene expression studies are correct may also be the predominant heart FMO isoform in humans [13].
Compared to the CYPs, there are few examples of FMOmediated adverse drug–drug interactions or genetic polymorphisms impacting drug or toxicant disposition. This is due in part to the fewer number of FMO enzymes involved in drug metabolism (FMOs 1, 2 and 3) and in part to the fact that FMO activity is not inducible and predominantly produces non-toxic metabolites [14].
A genetic polymorphism (g.23,238C>T) (dbSNP# rs6661174) in FMO2 exon 9, present in all Caucasians and Asians genotyped to date [11,15], converts a glutamine to a stop codon, p.Q472X. This change produces a non-functional truncated FMO2.2 protein from the FMO2*2 allele [11,12]. The FMO2*1 allele (p.Q472) encodes a full-length functional FMO2.1 enzyme. FMO2*1 is present in individuals from African (26%) and Hispanic (2 to 7%) populations [11,15–18]. A second documented SNP present in exon 9, g.23,412dupT (p.C530fs#rs2234889), appears to always segregate with the g.23,238C>T variant and as such, has no functional impact [11,15,16,18].
Human FMO2 (Genbank sequence AL021026) spans approximately 25.3 kb of genomic DNA. The translated coding sequence (from p.M1_X536) begins in exon 2 and extends through part of exon 9, encompassing 93% of the genomic distance. While most genotyping studies have focused on the variant already described in exon 9, recent single strand conformation polymorphism (SSCP) studies of DNA from 50 African-American individuals reported a number of previously unidentified FMO2 variants [17]. Of these, the most common (allele frequency of ≥ 27%) polymorphisms encode p.71Ddup (g.7,700_7,702dupGAC), p.V113fs (g.10,951delG), p.S195L (g.13,732C>T; dbSNP#rs2020862) and p.N413K (g.22,060T>G; dbSNP#rs2020865), present in exons 3, 4, 5 and 8, respectively. Whether these polymorphisms segregated with the previously defined FMO2*2 variant was not determined, nor was their catalytic activity determined in a protein expression system.
The present study extends our genotyping results for the FMO2*1 allele in Hispanic-Americans [16,18] to the common single nucleotide polymorphisms (SNPs) reported by Furnes et al. [17], and reports the initial characterization of mutant proteins produced by baculovirus infection of Sf9, Spodoptera frugiperda, cells. We have developed mutation screening procedures for SNP detection that combine single-tube allele-specific polymerase chain reaction (PCR) amplification [19] with either real-time SYBR green discrimination of allelespecific products by their respective melting temperatures (Tm) [20] or by electrophoretic discrimination based on allele-specific product size. SSCP detection was utilized when we were unable to develop successful allele-specific primers. Of the expressed SNP proteins, only p.N413 K was as active as FMO2.1; however, haplotype analysis indicated segregation of the variants with the previously described g.23,238C>T FMO2*2 allele. As such, they will have a minimal impact on existing estimates for the occurrence of individuals producing active FMO2 protein.
Materials and methods
Subjects and sample collection
Genomic DNA used for genotyping studies was obtained, without identifiers, from four sources. Hispanic samples were obtained as donor organs (n=1) (Tissue Transformation Technologies, Edison, New Jersey, USA), from living donors as blood (n=4) [21] or DNA (n=148) (Genomics Collaborative, Inc. (Cambridge, Massachusetts, USA) and from subjects who are participants in the New York Cancer Project [22], Academic Medicine Development Corporation, New York, New York, USA). Ethnicity was self-reported by living donors and included one Honduran, four Mexicans and 147 Puerto Ricans. The single organ donor in the study was a Hispanic of unknown country of origin. The appropriate Institutional Review Boards approved studies prior to initiation of experiments. Informed consent was obtained for all study subjects. DNA was isolated from donor samples as described elsewhere [16,21]. DNA was available from several African-American individuals studied previously [12,15,16] and was used for methods development. All Hispanic individuals in this study are a subset of individuals utilized in previous studies [16,18]; subjects included one FMO2*1/*1 individual (Puerto Rican ancestry), 28 FMO2*1/*2 individuals (one Honduran, four Mexican, 22 Puerto Rican and one of unknown Hispanic ancestry), and 124 FMO2*2/*2 individuals (Puerto Rican ancestry).
Primers and PCR
Primers were designed with the aid of Oligo 6.0. design software (Molecular Biology Insights, Cascade, Colorado, USA) and were synthesized by Invitrogen (Carlsbad, California, USA). Primers were used for cloning and mutagenesis (Table 1) and for genotyping (Table 2). Primers were designed to reclone FMO2*1 and FMO2*2 cDNA from pFastBac1 [12] into pENTR/SD/D-TOPO(Invitrogen) using topoisomerase mediated directional cloning. Pfu turbo (Stratagene, La Jolla, California, USA) was used to amplify the cDNA under the following conditions: 1× Pfu buffer; 1.0 unit Pfu Turbo; 2.0mm magnesium; 0.2mm each dNTP; 0.2 μm each hF2–33 and hF2–18r primer; and 5 ng cDNA template in a reaction volume of 20 μl. A 30-cycle PCR program was used: 1min at 95°C; 1min at 60°C; 2 min 15 s at 72°C. The resulting FMO2*1 vector (pENTR·hF2.1) was the template for synthesis of individual mutations. QuikChange mutagenesis (Stratagene) was used following the manufacturer’s recommendations with the primer pairs indicated in Table 1; the annealing temperature was 55°C for all of the mutagenesis reactions. The fidelity of the resulting plasmid constructs (pENTR·hF2.1, pENTR·hF2.2, pENTR·71Ddup, pENTR·V113fs, pENTR·S195L and pENTR·N413 K) was verified by DNA sequence analysis performed by the Central Services Laboratory (Oregon State University) using BigDye chemistry (Applied Biosystems, Foster City, California, USA).
Table 1.
Primers used in cloning and mutagenesis reactions to transfer FMO2*1 and FMO2*2 cDNA into pENTR, primers used to create mutant cDNA expression vectors and primers used to clone genomic DNA for haplotype analysis
| Primera | Location | Sequence (5′ to 3′)b |
|---|---|---|
| Primers used to transfer FMO2*1 and FMO2*2 cDNA into pENTR | ||
| hF2–33 | FMO2*1/*2 | cac cAT GGC AAA GAA GGT AGC TGT |
| hF2–18r | FMO2*1/*2 | caa agc TTA TGC TGA CTA GGc |
| Primers used to create mutations in the FMO2*1 pENTR construct | ||
| hF2–36 | g.7,700_7,702dupGAC | G TCC TGT TTC AGT GAC GAC TTT CCA ATG C |
| hF2–37r | g.7,700_7,702dupGAC | G CAT TGG AAA GTC GTC ACT GAA ACA GGA C |
| hF2–56 | g.10,951delG | CTG TGG TTA GT_ TGA GAA AAT GTC |
| hF2–57r | g.10,951delG | GAC ATT TTC TCA_AC TAA GGA CAG |
| hF2–38 | g.13,732C > T | GAA TGG GAA ACT TAG GCT CAG ATA TC |
| hF2–39r | g.13,732C > T | GAT ATC TGA GCC TAA GTT TCC CAT TC |
| hF2–40 | g.22,060T > G | ATC AAA AGG AAG GAA AAA AGA ATT GAC C |
| hF2–41r | g.22,060T > G | GGT CAA TTC TTT TTT CCT TCC TTT TGA T |
| Primers used for long PCR and cloning of genomic DNA | ||
| hF2–98 | Intron 3 | P-ACT GCT GAG TCA GGC CTC TTG CAT GAAd |
| hF2–99r | Exon 9 | P-TAA CTT TGG TGA ACT GAG GGA AGC GTG GAAd |
Lower case ‘r’ indicates a reverse primer; absence of an ‘r’ indicates a forward primer.
All primers are shown from 5′ to 3′. Lower case indicates intentional sequence mismatches; bold/underlining indicates mutation creating change(s).
This primer has been previously described [16].
Primers are phosphorylated at their 5′ ends.
Table 2.
Primers used for allele-specific and SSCP genotyping
| Primera | Description | Sequence (5′ to 3′)b |
|---|---|---|
| Exon 3: g.7,700_7,702dupGAC polymorphism (p.71Ddup) | ||
| hF2–44 | wild-type forward | AGA AAT GTC CTG TTT CAG TGc CT |
| hF2–45 | 71Ddup forward | gcc ccg ggg gCA Aga AAA TGT CCT GTT TCA GTc ACG |
| hF2–46r | common reverse | TTA TGC AGG AAG TTT GG |
| Exon 4: g.10,951delG polymorphism (p.V113fs) | ||
| hF2–83 | common forward | TTC AAG ACA ACT GTC CTT A |
| hF2–49r | common reverse | AAC CAT AAC TGC GTC AAA G |
| Exon 5: g.13,732C > T polymorphism (p.S195L) | ||
| hF2–50 | wild-type forward | ggg ggg ggg ggc CCT caT GAT TGG AAT GGG AAC cTC |
| hF2–79 | S195L forward | CTG GTG ATT GGt ATG GGA AAg TT |
| hF2–52r | common reverse | TGA GCA GCA TTC TTA CTC AG |
| Exon 8: g.22,060T > G polymorphism (p.N413 K) | ||
| hF2–59 | common forward | TTG TGT AGC CTG CCC TCA GA |
| hF2–60r | wild-type reverse | ccc ccc ccc cgt GGT CAA TTC TTT TgT CA |
| hF2–61r | N413 K reverse | CAc GTC AAT TCT TTT TaC C |
| Exon 9: g.23,238C > T polymorphism (p.Q472X) | ||
| hF2–42 | FMO2*1 forward | CGG ACC CTG CAA CTC CTt TC |
| hF2–20 | FMO2*2 forward | caa gct tat aTA TTT CGG ACC CTG CAA gTC CTA TTc |
| hF2–18r | Common reverse | caa agc TTA TGC TGA CTA GGc |
Lower case ‘r’ indicates a reverse primer, while absence of an ‘r’ indicates a forward primer.
Lower case indicates intentional sequence mismatches; bold/underlining indicates the polymorphism discriminating nucleotide.
These primers have been described previously [16].
Long PCR was used to separate variant polymorphisms to their source chromosomes for haplotype analysis. Primers, hF2–98 and hF2–99r, were designed to clone genomic DNA encompassing segregating polymorphisms into pSMART-LC (Lucigen, Middleton, Wisconsin, USA); these primers were purchased 5′ phosphorylated. The 12.8 kb genomic DNA insert was amplified using Herculase hotstart polymerase (Stratagene) in a 75 μl reaction containing: 1 × Herculase buffer; 3.75 units Herculase; 500 μm each dNTP; 1% dimethylsulfoxide; 0.5 μm of each phosphorylated primer; and 375 ng genomic DNA. PCR was performed in a PTC-100 thermocycler (MJ Research, Waltham, Massachusetts, USA) for 10 cycles: 10 s at 92°C; 30 s at 65°C; 13 min at 68°C, and was followed by 25 cycles with the extension time increased to 14 min 36 s. After electrophoretic fractionation, the PCR product was isolated from a 0.8% agarose, 1 × TAE gel (40mm Tris acetate, 2mm ethylenediaminetetraacetic acid, EDTA), purified with a Zymoclean gel DNA recovery kit (Zymo Research, Orange, California, USA) and quantitated by spectrofluorometery in the presence of picogreen (Invitrogen) before ligation with linearized pSMART-LC. DNA from the ligation reaction was used to transform electrocompetent Escherichia coli (Lucigen) using a BioRad Gene Pulser Xcell electroporator (Hercules, California, USA). Cells and DNA were placed in a 1.0mm cuvette and pulsed at 10 μF, 600ω and 1800 V; a time constant was not preset but typically ranged from 3.5 to 4.5 ms. Isolated DNA from the pSMART-LC clones was used as a template in genotyping reactions.
Primers for genotyping (Table 2) were designed to allow allelic discrimination by SSCP detection from a PCR product produced from common primers (g.10,951delG, p.V113fs), while allele-specific oligonucleotides (ASOs) were designed [23] for direct detection by real-time analysis in the presence of SYBR green [20] (g.7,700_7,702dupGAC, p.71Ddup and g.13,732C>T, p.S195L) or UV detection of ethidium bromide stained DNA after SDS-PAGE [19] (g.22,060T>G, p.N413 K).
PCR reactions were performed with either DyNAmo SYBR Green qPCR kit from Finnzymes Oy (Espoo, Finland) or with Platinum SYBR Green qPCR SuperMix UDG from Invitrogen. The volume was 20 μl, half of which was provided by the respective 2× master mix stocks containing everything except primers and template. The concentration of each primer in reactions performed with the DyNAmo enzyme was 0.3 μm, but was reduced to 0.2 μm in reactions performed with the Platinum enzyme mixture as recommended by the respective manufacturers. The template concentration was either 0.1 pg plasmid DNA, 20 ng genomic DNA or a combination of 0.1 pg plasmid DNA plus 20 ng genomic DNA. Controls without DNA were included to identify possible contaminants in the reaction mixture and primer–dimers. The heterozygous state was simulated for each mutation by combining equivalent quantities of either the cloned cDNA versions of the g.23,238C (p.Q472) allele and the synthesized mutant or reference genomic DNA (from the same individual from which the pENTR·hF2.1 cDNA clone was derived) and synthesized mutant DNA.
Real-Time PCR was performed and data analyzed with a DNA Engine Opticon 2 System from MJ Research. The g.7,700_7,702dupGAC (p.71Ddup) and g.10,951delG (p.V113fs) SNP detection was performed subsequent to DNA amplification with DyNAmo using an initial denaturation cycle at 95°C for 10 min. This was followed by 35 cycles denaturation at 94°C for 10 s, annealing at 54°C for 20 s and extension at 72°C for 10 s. The final cycle included extension at 72°C for 7 min, melting curve analysis from 60 to 95°C in 0.2°C increments and a final reannealing step at 72°C for 10 min. The same program was used to amplify the template containing the g.22,060T>G (p.N413 K) SNP with DyNAmo; however, the annealing temperature was reduced to 46°C and the cycle number was reduced from 35 to 30. The g.13,732C>T (p.S195L) SNP detection was performed by amplification using the Platinum enzyme mixture. The amplification and analysis cycles were identical to that for the g.7,700_7,702dupGAC and g.10,951delG variants, except that the initial cycle involved incubating at 50°C for 2 min to allow uracil-DNA glycosylase to function, and then at 95°C for 2 min to denature the template, inactivate the uracil-DNA glycosylase and activate the hot start polymerase. As part of methods development and confirmation of real-time results, electrophoretic resolution of PCR products from control reactions and select genomic samples was performed using 6% polyacrylamide gels (Invitrogen) in 1 × TBE buffer (89mm Tris base, 89mm boric acid, 2mm EDTA), with detection by UV-transillumination after staining with ethidium bromide.
SSCP analysis
SSCP was performed by electrophoresis of denatured PCR products (6 ng/well) amplified from common forward and reverse primers using GeneGel Excel 12.5/24 gels in a GenePhor electrophoresis unit. Electrophoresis was performed at 15°C with settings of 300 V, 15W and 25mA, until the dye front reached the anode (ca. 2 h). DNA was detected with the PlusOne DNA Silver staining kit. All SSCP steps were performed with products from Amersham Biosciences (Piscataway, New Jersey, USA) following the manufacturer’s recommendations.
Haplotype determination
Long PCR products, generated and cloned as described above from FMO2*1/*2 (g.23,238C>T) heterozygotes and one or more additional segregating SNPs, were utilized for haplotype determination. Plasmid DNA was isolated and utilized as template in genotyping reactions as described above; conditions were not altered except that the template concentration was 1.0 pg plasmid DNA in a 20 μl reaction.
Inferred haplotypes were estimated from genotype information determined from all of the Hispanic samples utilized in the study. A web-based (http://archimedes.well.ox.ac.uk/pise/) version of PHASE software (version 2.0.2) [24,25] was used to model the unphased haplotypes. The PHASE settings were MR (model allows recombination), 100 iterations, a thinning interval of 1, and a burn-in of 100.
Ethnic differences in the occurrence of SNPs
A comparison of the allelic frequency of each SNP was made with the data from the Hispanic-American population from this study and the published results obtained for the same SNPs within an African-American population [17]. To enable direct comparisons of the ethnic groups, an assumption was made that all variant SNPs observed in each of the study populations occurred in association with the previously defined FMO2*2 variant. This reduced the number of Hispanic alleles from 306 to 276, and the number of African alleles from 100 to 89. Genotyping results for each SNP were expressed as frequency in the FMO2*2 population. SNP occurrences between groups were compared using the Pearson Chi-square test with exact (permutation) P-values generated by StatXact version 6.1 (Copyright 2003, Cytel Software Corp., Cambridge, Massachusetts, USA). Ninety-five percent confidence intervals (Clopper–Pearson binomial) were calculated based on the assumption that each sample approximates a simple random sample from the Hispanic or African-American population.
Protein expression and evaluation
The pENTR clones were used to produce baculovirus, which were in turn used to generate recombinant proteins. All procedures were as recommended by Invitrogen. Plasmid DNA was integrated with BaculoDirect linear DNA by LR-mediated clonase recombination. The resulting DNA was combined with Cellfectin reagent for lipid-mediated transfection of Sf9 cells and production of primary virus. Recombinant virus was amplified to yield tertiary or quaternary virus stocks. These stocks were used to infect Sf9 cells (2 × 106 cells/ml Sf900II SFM). FAD was added to the media (10 μg/ml) to ensure that cofactor levels would not be limiting during protein production. Microsomes were prepared from cells harvested 96 hr post-infection [12] and protein concentrations determined by the Bradford method [26]. The FAD content was determined by an HPLC based method already described [27]. The FMO content was estimated based on results from FAD analysis and was compared with estimates from detection by western analysis with rabbit anti-monkey FMO2 antibody [28] to confirm protein production and check for possible disruption of FAD binding by mutant proteins.
Preliminary assays were performed to determine the capacity of the expressed proteins for carrying out Soxidation reactions using a Cary 300 Bio UV-Visible double beam spectrophotometer as previously described [12]. Two batches of each protein were assayed. All samples were initially assessed by the methimazole assay [29] performed in tricine buffer (100mm tricine, pH 9.5; 1.0mm EDTA) with a final substrate concentration of 2.0mm. Microsomal protein concentrations ranged from 40 to 1000 μg/ml, depending on the level of activity detected. FMO2 methimazole-dependent S-oxidation specific activity was calculated using the FMO content determined with the FAD assay. When substantial methimazole-dependent activity was measurable, we also tested substrate-dependent NADPH oxidation of ethylenethiourea. Activity was determined in tricine buffer as noted, with an ethylenethiourea concentration of 75 μm using 80 to 100 μg of protein/ml. Additional testing was performed using four concentrations of ethylenethiourea ranging from 10 to 75 μm to permit calculation of kinetic parameters estimated from Lineweaver–Burk and Eadie–Hofstee plots. Enzyme activity of the mutant proteins was compared with p.Q472 (N=2 for each protein) using a simple t-test; however, because of the small sample size, assumptions of the t-test (e.g., normal data, equality of variance) were not assessed.
Results
Genotyping procedures
We developed genotyping procedures for the common polymorphisms using cloned FMO2*1 (reference sequence) cDNA and the appropriate synthetic mutant cDNA created from FMO2*1 cDNA. Thermocycling and detection conditions were tested using reference, mutant and pooled cDNAs to simulate the respective genomic homozygous and heterozygous genotypes. Because our only source of control DNA was the mutant cDNA constructs that we created, all primers were designed to anneal with DNA from the relevant exon. It was desirable to use ASOs coupled with real-time product detection in the presence of SYBR green for all of the polymorphisms, because, after methods validation, we could restrict our genotyping procedure to PCR with melting curve analysis and eliminate the need for subsequent electrophoretic product detection. We were able to develop procedures that could clearly differentiate the reference sequence from the respective cDNAs representing the g.7,700_7,702dupGAC (p.71Ddup), g.13,732C>T (p.S195L) and g.22,060T>G (p.N413 K) variants. When genomic DNA was used, real-time detection and electrophoresis worked equally well to distinguish products for the g.7,700_7,702dupGAC (p.71Ddup) (Fig. 1) and g.13,732C>T (p.S195L) loci (Fig. 2). However, the primers utilized for the g.22,060T>G (p.N413 K) variant also amplified additional DNA fragments that interfered with real-time detection and made electrophoretic assessment ambiguous (not shown). Because the two ASOs had good specificity with regard to distinguishing the underlying nucleotide change, we used sequential two-primer PCR amplification with either the allele-specific mutant primer or the allele-specific non-mutant primer and common forward primer (Table 2) followed by electrophoretic fractionation to screen for the underlying nucleotide (Fig. 3a and b).
Fig 1.
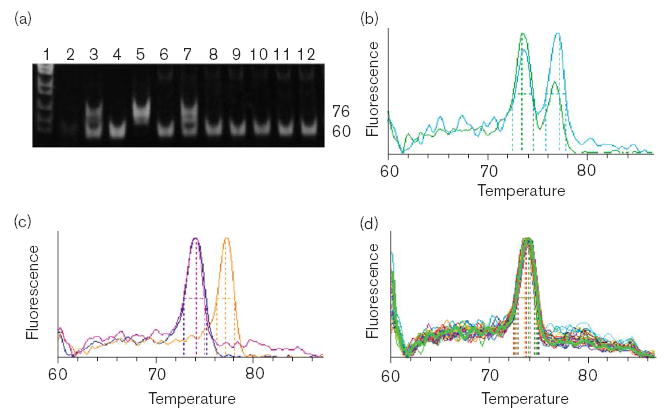
Identification of the g.7,700_7,702dupGAC SNP (encodes p.71Ddup) by (a) electrophoresis or (b–d) real-time detection of PCR products from 3-primer (hF2–44, hF2–45 and hF2–46r) allele-specific amplification. (a) Lane 1: MspI cut pBR322. Lanes 2–7 are control reactions: 2, minus template; 3, synthetic heterozygote composed of reference plasmid + g.7,700_7,702dupGAC plasmid DNA; 4, reference plasmid DNA; 5, g.7,700_7,702dupGAC plasmid DNA; 6, genomic DNA from a reference individual; 7, genomic DNA with added g.7,700_7,702dupGAC plasmid DNA. Lanes 8–12 are from genomic DNA from individual Hispanic-American samples. (b) Melting curve showing control reactions with synthetic heterozygotes; green, mixed plasmid DNA; teal, non-mutant genomic DNA with g.7,700_7,702dupGAC plasmid DNA. (c) Control reactions with simulated homozygotes and a reference homozygote: blue and pink 74°C peaks are from 60-bp products from reference plasmid DNA and genomic DNA, respectively; orange, 77°C peak is from 76-bp product from g.7,700_7,702dupGAC plasmid DNA. (d) Genomic DNA from 30 Hispanic- American individuals showing that the g.7,700_7,702dupGAC is absent.
Fig. 2.
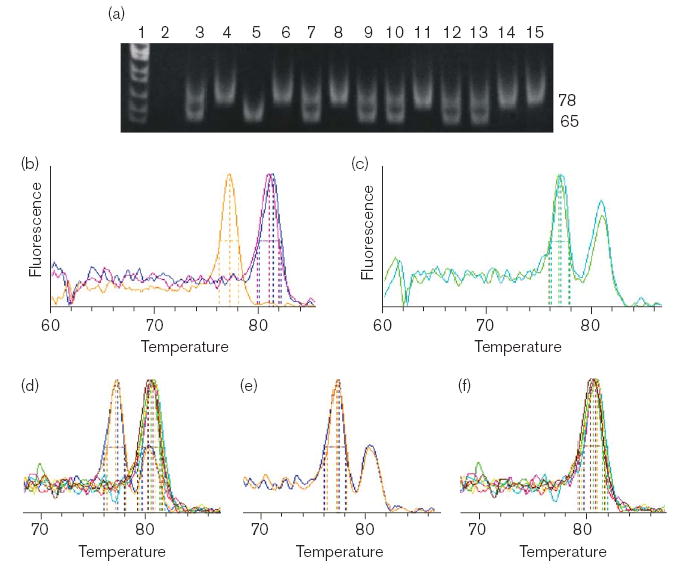
Identification of the g.13,732C>T SNP (encodes p.S195L) by (a) electrophoretic or (b–f) real-time detection of PCR products from 3-primer (hF2– 50, hF2–79 and hF2–52r) allele-specific amplification. (a) Lane 1: MspI cut pBR322. Lanes 2–7 are control reactions: 2, minus template; 3, synthetic heterozygote composed of reference plasmid + g.13,732C>T plasmid DNA; 4, reference plasmid DNA; 5, g.13,732C>T plasmid DNA; 6, genomic DNA from a reference individual; 7, genomic DNA with added g.13,732C>T plasmid DNA. Lanes 8–15 are products from genomic DNA from individual Hispanic-American samples. (b) Melting curve showing control reactions with simulated homozygotes and a non-mutant genomic sample: orange tracing of the 77°C peak is from the 65-bp g.13,732C>T plasmid DNA product; blue and pink 81°C peaks are from the 78-bp products from reference plasmid and genomic DNA, respectively. (c) Melting curve showing PCR products from control reactions with synthetic heterozygotes; green, mixed plasmid DNA; teal, non-mutant genomic DNA with g.13,732C>T plasmid DNA. (d) Superimposed melting curves of PCR products from the genomic DNA of eight Hispanic-American individuals. These melting curves are shown separated into their components including two heterozygotes (e), and six homozygotes encoding non-mutant p.S195 (f).
Fig. 3.
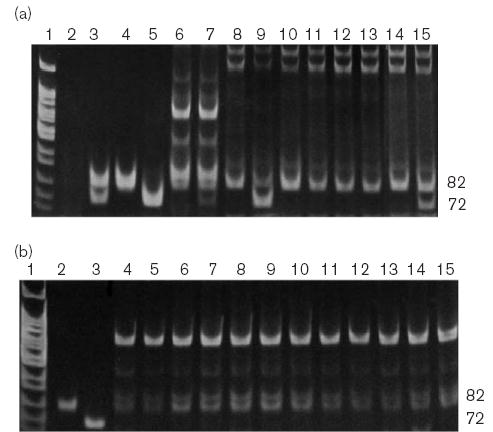
Identification of the g.22,060T>G SNP (encodes p.N413 K) by allelespecific 2-primer amplification with electrophoretic detection. (a) Control reactions and 2-primer screen for the 72-bp g.22,060T>G mutant PCR product. Lane 1: MspI cut pBR322. Lanes 2–7 are 3-primer (hF2–59, hF2–60r and hF2–61r) control reactions: 2, minus template; 3, synthetic heterozygote composed of reference plasmid + g.22,060T>G plasmid DNA; 4, reference plasmid DNA; 5, g.22,060T>G plasmid DNA; 6, genomic DNA from a reference individual; (7) genomic DNA with added g.22,060T>G plasmid DNA. Lanes 8–9 show 2-primer (hF2–59 and hF2–61r) control reactions: (8–9) template is the same as lanes 6–7, respectively. Lanes 9–15 are products from genomic DNA from individual Hispanic-American samples obtained by 2-primer (hF2–59 and hF2–61r) amplification. (b) Control reactions and 2-primer screen for the 82-bp g.22,060T nonmutant PCR product. Lane 1: MspI cut pBR322. Lanes 3–4 are 3- primer (hF2–59, hF2–60r and hF2–61r) control reactions: 2, reference plasmid DNA; 3, g.22,060T>G plasmid DNA. Lanes 4–15 are products from genomic DNA from individual Hispanic-American samples obtained by 2-primer (hF2–59 and hF2–60r) amplification.
One variant, g.10,951delG (p.V113fs) was not amenable to detection with ASOs as we were unable to develop primers with the required specificity (not shown). As an alternative we designed common forward and reverse primers to generate products that could be screened by SSCP. We amplified and tested a 120-bp product, and demonstrated ready separation of the g.10,951delG (p.X113) from g.10,951G (p.V113) variants by SSCP (Fig. 4).
Fig. 4.
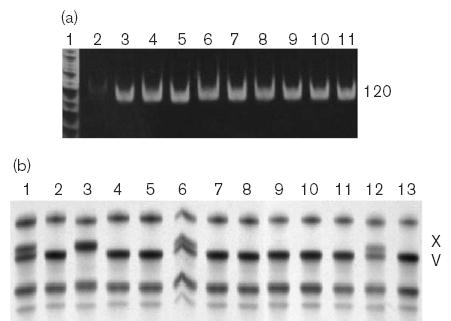
Identification of the g.10,951delG SNP (encodes p.V113fs) by SSCP detection (b) of products produced from standard 2-primer (hF2–83 and hF2–49r) PCR amplification (a). (a) Control reactions to confirm 120-bp PCR product amplification. Lane 1: MspI cut pBR322. Lanes 2–7 control reactions: 2, minus template; 3, synthetic heterozygote composed of reference plasmid + g.10,951delG plasmid DNA; 4, reference plasmid DNA; 5, g.10,951delG plasmid DNA; 6, genomic DNA from a reference individual; 7, genomic DNA with added g.10,951delG plasmid DNA. Lanes 8–11 are products from genomic DNA from individual Hispanic-American samples. (b) SSCP separation of control products from: 1, synthetic heterozygote composed of reference plasmid + g.10,951delG plasmid DNA; 2, reference plasmid DNA; 3, g.10,951delG plasmid DNA. Lanes 4–13 show SSCP separation of products from individual Hispanic-Americans.
Occurrence of polymorphisms
Previous genotyping studies of FMO2 from 632 Hispanic-Americans [16,18] identified 32 individuals with at least one FMO2*1 allele. We had sufficient DNA from 28 of the 31 FMO2*1/*2 individuals and one FMO2*1/*1 individual to genotype samples for the four most common polymorphisms identified in a population of 50 African-Americans [17]. Twenty-one of these individuals (72.4%) did not have any of the examined sequence variants (Table 3) and would be expected to produce active FMO2.1 protein. The remaining eight individuals (27.6%) were all heterozygotes for the g.13,732C>T (p.S195L) SNP. Three of the eight g.13,732C>T (p.S195L) heterozygotes (10.3%) were also heterozygous for two additional polymorphisms; however, the g.7,700_7,702dupGAC (p.71Ddup) polymorphism was not present in the tested Hispanic population.
Table 3.
Occurrence of FMO2 SNP combinations among Hispanic-American individuals and inferred haplotype pairs for observed genotypes
| Observed genotype (expressed as the AA encoded by the underlying SNP)
|
Genotype
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Exon 3 p.71Ddup | Exon 4 p.V113fs | Exon 5 p.S195L | Exon 8 p.N413K | Exon 9 p.Q472X | Exon 9p.C530fs | Occurrence | (%) | Haplotype pair (probability)a |
| FMO2*1 individuals (N = 29)b | ||||||||
| Q/Q | 1 | (3.4) | VSNQC VSNQC (1.000) | |||||
| Q/X | 20 | (69.0) | VSNQC VSNXC (1.000) | |||||
| S/L | Q/X | 3 | (10.3) | VSNQC VLNXC (0.990) | ||||
| S/L | Q/X | C/fs | 2 | (6.9) | VSNQC VLNXfs (0.990) | |||
| V/X | S/L | N/K | Q/X | 3 | (10.3) | VSNQC XLKXC (0.996)c | ||
| 0 | 10.3 | 27.6 | 10.3 | 96.6 | 6.9 | SNPs: % individuals | ||
| 0 | 5.2 | 13.8 | 5.2 | 48.3 | 3.4 | SNPs: % alleles | ||
| 0 | 10.7 | 28.6 | 10.7 | 100 | 7.1 | SNPs: % FMO2*2 allelesd | ||
| FMO2*2/*2 individuals (N = 124) | ||||||||
| X/X | NDe | 38 | (30.6) | VSNX VSNX (1.000) | ||||
| S/L | X/X | ND | 42 | (33.9) | VSNX VLNX (1.000) | |||
| L/L | X/X | ND | 13 | (10.5) | VLNX VLNX (1.000) | |||
| N/K | X/X | ND | 3 | (2.4) | VSNX VSKX (1.000) | |||
| V/X | S/L | N/K | X/X | ND | 19 | (15.3) | VSNX XLKX (0.999) | |
| V/X | L/L | N/K | X/X | ND | 9 | (7.3) | VLNX XLKX (1.000) | |
| 0 | 22.6 | 66.9 | 25.0 | 100 | ND | SNPs: % individuals | ||
| 0 | 11.3 | 42.3 | 12.5 | 100 | ND | SNPs: % alleles | ||
Haplotype pairs and the mean probability associated with the phase call were estimated using PHASE [24,25].
Undesignated alleles match the reference sequence (non-variant).
Cloning and repeat genotyping to confirm the indicated haplotype was performed for one of these individuals.
SNP occurrence observed (as percentage of FMO2*2 alleles) if all SNPs are limited to FMO2*2. This is also the expected allelic occurrence among the 124 FMO2*2/*2 individuals tested.
The presence or absence of this SNP was not determined.
We also genotyped 124 Puerto Rican-American individuals that were homozygous for the previously defined FMO2*2 allele (g.23,238C>T). Although none of these individuals would be capable of producing functional FMO2.1 protein, the unphased genotyping data is useful for phase estimation. Only 30.6% of the FMO2*2 homozygotes had none of the additional studied SNPs (Table 3). The SNP encoding p.S195L (g.13,732C>T) was present in 66.9% of the individuals and was present on both alleles of 26.5% of the individuals. Once again the SNP encoding p.V113fs (g.10,951delG) was only present in individuals that also had the g.13,732C>T (p.S195L) SNP, but was present in 22.6% of these individuals. These same individuals also had the SNP encoding p.N413 K (g.22,060T>G), but this SNP was also found in an additional 2.4% of the individuals as the sole additional variant. No individuals were identified that possessed the g.7,700_7,702dupGAC (p.71Ddup) variant.
Haplotype results
After determining individual genotypes of one FMO2*1 homozygote and 28 FMO2*1/*2 heterozygotes, we attempted to determine the haplotype of eight individuals in which the additional SNPs were detected. We successfully cloned the DNA from one FMO2*1/*2 individual that was also heterozygous for the SNPs in exons 4, 5 and 8 (g.10,951delG, g.13,732C>T, and g.22,060T>G, respectively). Repeat genotyping of the resulting clones indicated that all of the variant SNPs segregated with the g.23,238T variant previously used to define the FMO2*2 allele. Attempts to clone the DNA of the remaining seven FMO2*1/*2 individuals with additional SNP variants were not successful, due to instability of the 12.8 kb insert.
As an alternative approach, we randomly selected 124 Puerto Rican FMO2*2 homozygotes and determined their genotypes so that statistical methods could be used to infer individual haplotypes. In addition, the SNP frequency data from FMO2*2 homozygotes was compared to that from FMO2*1/*2 heterozygotes. The assumption was made that all SNPs observed among the heterozygotes segregated with the FMO2*2 allele as was observed directly for a single individual (see above). The frequency of individual SNPs was recalculated (Table 3); these frequencies were used to determine the expected SNP frequencies in the 248 chromosomes from FMO2*2 homozygotes previously genotyped. The observed frequency of SNPs (Table 3) that encode p.V113fs (g.10,951C>T) and p.N413 K (g.22,060T>G) was not significantly different than the expected frequency (P>0.5, both tests). The observed occurrence of the SNP that encodes p.S195L (g.13,732C>T) was higher than expected but not significantly so (P=0.22). These data confirm our assumption that these SNPs segregate with the previously defined FMO2*2 allele (g.23,238C>T). Inferred haplotypes were determined for all individuals and the predicted haplotype pairs and the associated phase call probabilities are provided in Table 3. Consistent with our assumption, the predicted phase for every individual reflects a chromosomal arrangement in which all variant SNPs segregate with the previously defined FMO2*2 allele.
Ethnic differences in the occurrence of SNPs
Assuming that inferred haplotyping results have correctly assigned the variant SNPs in the Hispanic-American population (Table 3) as segregating with the g.23,238C>T variant (FMO2*2), the actual allelic frequencies for the population can be reported exclusively as the occurrence within the subpopulation of FMO2*2 alleles (Table 4). We applied the same assumption to the data reported from a study of African- American individuals [17] to make a comparison of these ethnic groups possible without being confounded by the large difference in the occurrence of the g.23,238C>T variant (FMO2*2) between these ethnic groups. This assumption should be largely correct since posterior estimation of haplotypes for the African-American population completed since publication indicated that the common SNPs reported in their study segregated almost entirely with FMO2*2 g.23,238C>T variant (B. Furnes, personal communication). A single exception was an incidence of an FMO2*1 homozygote with a single copy of the SNP that encodes p.N413 K (g.22,060T>C). SNPs encoding p.71Ddup (g.7,700_7,702dupGAC), p.V113fs (g.10,951delG) and p.N413 K (g.22,060T>G) were all present among FMO2*2 (g.23,238C>T) alleles at a significantly lower frequency among Hispanic-Americans (Table 4) than was observed in the African-American group (P values of <0.0001, <0.0001 and 0.0001, respectively). This difference was most apparent for the SNP encoding p.71Ddup (g.7,700_7,702dupGAC), which was not detected in any of the Hispanic-American individuals. Only the SNP that encodes p.S195L (g.13,732C>T) occurred at a similar frequency in both populations (P=0.13).
Table 4.
Occurrence of SNPs among FMO2*2 alleles in a Hispanic- and African-American population
| Population | Exon 3g.7,700_7,702dupGAC | Exon 4 g.10,951delG | Exon 5 g.13,732C > T | Exon 8 g.22,060T > G |
|---|---|---|---|---|
| Africana, N = 89 | 27/89 | 27/89 | 51/89 | 27/89 |
| 30.3% | 30.3% | 57.3% | 30.3% | |
| (21.0–41.0%)b | (21.0–41.0%) | (46.4–67.7%) | (21.0–41.0%) | |
| Hispanic, N = 276 | 0/276 | 31/276 | 133/276 | 34/276 |
| 0% | 11.2% | 48.2% | 12.3% | |
| (0–1.3%) | (7.8–15.6%) | (42.2–54.3%) | (8.7–16.8%) |
Results from the African-American population were extracted from Furnes et al. [17].
Numbers in parentheses are 95% confidence intervals (Clopper–Pearson binomial) associated with the indicated SNP occurrence.
Expression and evaluation of mutant proteins
Plasmid constructs were created in pENTR for the variants of interest to serve as templates for protein production in Sf9 cells. We expected that expressed p.V113fs would lack enzyme activity and would fail to bind FAD because this is what is observed for the FMO2.2 (p.X472) protein [11,12] in which only background levels (not associated with FMO) of FAD are present [12], and this frame shifting mutation results in immediate replacement of Val with a stop codon. However, while the sequence alteration correctly encoded p.V113fs and virus successfully infected Sf9 cells, we were unable to detect the protein in either the soluble or membrane fractions using sodium dodecyl sulfate– polyacrylamide gel electrophoresis with Coomassie Blue staining or western blotting with anti-monkey FMO2 antibody (not shown). Additionally, FAD was not detectable (Table 5). The p.V113fs protein may be targeted for degradation in Sf9 cells. While detectable p.71Ddup protein was produced by Sf9 cells and targeted to the membrane fraction (not shown), it also failed to bind FAD and was devoid of activity toward methimazole (Table 5).
Table 5.
Flavin content and activity of expressed proteins
| Enzyme activityb |
|||||||
|---|---|---|---|---|---|---|---|
| FAD contenta |
Methimazole
|
Ethylenethiourea
|
|||||
| Protein | Homogenate | Microsomes | SA | SA | Km | kcat | kcat/Km |
| p.71Ddup | 0.212 | 0.135 | NDc | NAd | |||
| 0.162 | 0.074 | ND | NA | ||||
| p.V113fs | 0.174 | 0.151 | ND | NA | |||
| 0.190 | 0.082 | ND | NA | ||||
| p.S195L | NA | 0.967 | 0.9 | NA | |||
| NA | 1.828 | 0.7 | NA | ||||
| p.N413K | NA | 1.142 | 46.2 | 70.2 | 32 | 118.7 | 3.7 |
| NA | 1.362 | 53.2 | 75.5 | 47 | 129.3 | 2.8 | |
| p.Q472 (FMO2.1) | NA | 1.336 | 38.1 | 32.4 | 14 | 38.7 | 2.8 |
| NA | 1.328 | 38.5 | 34.7 | 19 | 43.4 | 2.3 | |
| p.X472 (FMO2.2) | NA | 0.089 | ND | NA | |||
| NA | 0.217 | ND | NA | ||||
FAD content is nmol/mg total protein.
Enzyme assays were performed at pH 9.5 as indicated in Materials and methods. Kinetic constants were calculated using estimates of microsomal FAD content subsequent to subtraction of background levels of FAD (mean of four batches of p.X472, which does not bind FAD) from each batch. The specific activity (SA) (nmol/min/nmol FMO) was determined using 2mm methimazole and 75 mm ethylenethiourea. The Km (mm), and kcat (nmol/min/nmol FMO) were determined using 10 to 75 mm substrate.
ND indicates no activity was detected.
NA indicates the sample was not assayed.
The p.S195L and p.N413 K proteins were produced in abundance, were targeted to the membrane fraction, were associated with FAD levels comparable to that of FMO2.1 and exhibited measurable enzyme activity (Table 5). Under the conditions of our assay, p.S195L activity was severely compromised (P<0.0001), retaining only 2% of the activity exhibited by FMO2.1 (p.Q472). The p.N413 K activity exceeded that of FMO2.1 with both methimazole and ethylenethiourea, but the difference in activity was only significant for ethylenethiourea (P=0.0054) and not methimazole (P=0.0830). Although, the Km measured for p.N413 K was higher than that measured for FMO2.1 with ethylenethiourea as the substrate the difference was not significant (P=0.1006). However, the difference in kcat was highly significant (P=0.0048); thus, the catalytic efficiency (kcat/Km) was similar (P=0.3184) for both proteins.
Discussion
Research by Stephens et al. [30] reported haplotype variation in a sample of human genes and compared the distribution of SNPs and inferred haplotypes from individuals of African-American, Asian, Caucasian and Hispanic-Latino origin. They found that the population distribution of both haplotypes and SNPs was similar. The African-American population had the highest number of SNPs and the most SNPs that were unique to the population. By contrast, the Hispanic-Latino population had the lowest number of unique SNPs but the second highest number of total SNPs, as a result of having a large number of SNPs in common with either the African- American or Caucasian populations. The current study focused on the detection and characterization of four FMO2 SNPs in a Hispanic-American population that were previously found to be common in African-Americans [17]. Consistent with the global implications from the study by Stephens et al. [30], our finding that the variant encoding p.71Ddup is absent in the Hispanic-American study population may indicate that this SNP is unique to the African-American population.
The remaining three SNPs were detected in the Hispanic-American population so it is possible that they are cosmopolitan in nature and will be found in other ethnic groups. The SNP that encodes p.V113fs (g.10,951delG) always segregated with the g.13,732C>T SNP (p.S195L) and probably arose subsequent to it. The SNP encoding p.N413 K (g.22,060T>G) was less common than the SNP encoding p.S195L (g.13,732C>T) and usually, but not always, segregated with the SNPs encoding both p.V113fs and p.S195L (g.10,951delG and g.13,732C>T, respectively). These observations may indicate this SNP has occurred on a number of occasions or that recombination has occurred both between exons 5 and 8, as well as between exons 8 and 9. However, because the SNP encoding p.S195L was very common, if such recombination does occur, we would expect to find an occasional individual with the variant encoding p.S195L (g.13,732C>T) segregating with the g.23,238C (p.Q472) allele. Given the distance between the mutations on exons 5 and 9 (9.5 kb) and the difficulty we encountered acquiring stable clones of long PCR products (12.8 kb), it is likely that if recombinant individuals with the [g.13,732T; g.23,238C] haplotype ([p.S195L;Q472]) exist, they will be identified from a study population of African origin.
The current allelic designations for FMO2 polymorphisms were first proposed [15] after it was demonstrated that g.23,412dupT, when present was secondary to g.23,238C>T. We propose that the new haplotype information warrants introduction of additional designations to cover the known sequence combinations. Proposed designations (Table 6) adhere to international human gene nomenclature guidelines [31,32] as well as established criteria utilized for naming cytochrome P450 alleles [33], and bring the total of named FMO2 alleles to eight.
Table 6.
Proposed FMO2 allele nomenclature
| Allele | Protein | Genomic haplotype composition | cDNA haplotype composition |
|---|---|---|---|
| FMO2*1a | FMO2.1 | ||
| FMO2*2a | FMO2.2 | g.23,238C > T | c.1414C > T |
| FMO2*2Aa | FMO2.2A | [g.23,238C > T ; 23,412_23,413insT] | [c.1414C > T ; 1588_1589insT] |
| FMO2*2B | FMO2.2B | [g.13,732C > T ; 23,238C > T] | [c.584C > T ; 1414C > T] |
| FMO2*2C | FMO2.2C | [g.22,060T > G ; 23,238C > T] | [c.1239T > G ; 1414C > T] |
| FMO2*2D | FMO2.2D | [g.13,732C > T ; 23,238C > T ; 23,412_23,413insT] | [c.584C > T ; 1414C > T ; 1588_1589insT] |
| FMO2*2E | FMO2.2E | [g.10,951delG ; 13,732C > T ; 22,060T > G ; 23,238C > T] | [c.337delG ; 584C > T ; 1239T > G ; 1414C > T] |
| FMO2*3 | FMO2.3 | g.22,060T > G | c.1239T > G |
These alleles have been previously described [15].
We hypothesized that p.S195L and p.N413 K proteins would retain enzyme activity. The p.S195L alteration lies within the conserved GXGXXG/A NADPH binding motif, which describes a beta-alpha-beta Rossman fold. Studies comparing amino acids that occur in proteins with this motif led to the prediction that any amino acid should be tolerated at the underlined position [34]. Yet among mammalian FMOs, amino acid position 195 is Ser in all isoforms examined. Thus, for FMO, substitution of a Ser with a Leu within this position of the NADPH binding motif seriously compromises enzyme activity under the assay conditions used. Alignment of human FMO2 position 413 with other mammalian FMO2s revealed 100% conservation of Asn at this position. Alignment and comparison with other FMOs demonstrated conservation of Lys at this position among mammalian FMO1 enzymes, Met or Arg in FMO3 enzymes, Glu in FMO4 enzymes and either Gln or Arg in FMO5 enzymes. The catalytic efficiency of expressed protein with the Lys substitution was as high as that of FMO2.1. Interestingly, this was achieved with an increased Km that was offset by a concomitant increase in velocity.
Expressed FMO2.1 is proficient at catalyzing the oxidation of chemicals such as thioureas [35] and thioethers [27]. Humans can be exposed to these chemicals in the form of drugs and insecticides, respectively. Individuals capable of producing FMO2.1 will potentially metabolize these and other FMO2 substrates. Approximately 26% of African- and 2–7% of Hispanic-American individuals can produce active FMO2.1; results from our current study indicate that these percentages are largely unaffected by the newly reported FMO2 SNPs [17]. Thus, approximately 9.9 million individuals in the United States alone (estimated from 2000 census figures) may uniquely metabolize FMO2 substrates.
Acknowledgments
We thank Genomics Collaborative, Inc. (GCI) for their sample donations and J. Van Dyke for laboratory assistance. The authors would like to thank the New York Cancer Project, which made biological samples and information available in connection with the publication of this study. The New York Cancer Project is administered and funded by Academic Medicine Development Corporation Foundation, Inc.
Footnotes
Disclaimers: Part of this work was presented at the Seventh International meeting of the International Society for the Study of Xenobiotics. Aug. 29–Sep. 2 2004, Vancouver, Canada.
This study was supported by PHS grant HL38650. We also acknowledge support from the Cell Culture Facility Core and the Statistical Core of the Oregon State University Environmental Health Sciences Center (ES 00210).
References
- 1.Cashman JR. The role of flavin-containing monooxygenases in drug metabolism and development. Curr Opin Drug Discov Devel. 2003;6:486–493. [PubMed] [Google Scholar]
- 2.Ziegler DM. An overview of the mechanism, substrate specificities, and structure of FMOs. Drug Metabol Rev. 2003;34:503–511. doi: 10.1081/dmr-120005650. [DOI] [PubMed] [Google Scholar]
- 3.Ziegler DM. Metabolic oxygenation of organic nitrogen and sulfur compounds. In: Mitchell JR, Horning MG, eds. Drug Metabolism and Toxicity New York: Raven Press; 1984. p. 33–53.
- 4.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Therap, accepted. [DOI] [PMC free article] [PubMed]
- 5.Hernandez D, Janmohamed A, Chandan P, Phillips IR, Shephard EA. Organization and evolution of the flavin-containing monooxygenase genes of human and mouse: identification of novel gene and pseudogene clusters. Pharmacogenetics. 2004;14:117–130. doi: 10.1097/00008571-200402000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Hines RN, Hopp KA, Franco J, Saeian K, Begun FP. Alternative processing of the human FMO6 gene renders transcripts incapable of encoding a functional flavin-containing monooxygenase. Mol Pharmacol. 2002;62:320–325. doi: 10.1124/mol.62.2.320. [DOI] [PubMed] [Google Scholar]
- 7.Lawton M, Cashman J, Cresteil T, Dolphin C, Elfarra A, Hines RN, et al. A nomenclature for the mammalian flavin-containing monooxygenase gene family based on amino acid sequence identities. Arch Biochem Biophys. 1994;308:254–257. doi: 10.1006/abbi.1994.1035. [DOI] [PubMed] [Google Scholar]
- 8.Lawton MP, Gasser R, Tynes RE, Hodgson E, Philpot RM. The flavincontaining monooxygenase enzymes expressed in rabbit liver and lung are products of related but distinctly different genes. J Biol Chem. 1990;265:5855–5861. [PubMed] [Google Scholar]
- 9.Shehin-Johnson SE, Williams DE, Larsen-Su S, Stresser DM, Hines RN. Tissue-specific expression of flavin-containing monooxygenase (FMO) forms 1 and 2 in the rabbit. J Pharmacol Exp Ther. 1995;272:1293–1299. [PubMed] [Google Scholar]
- 10.Tynes RE, Philpot RM. Tissue- and species-dependent expression of multiple forms of mammalian microsomal flavin-containing monooxygenase. Mol Pharmacol. 1987;31:569–574. [PubMed] [Google Scholar]
- 11.Dolphin CT, Beckett DJ, Janmohamed A, Cullingford TE, Smith RL, Shephard EA, et al. The flavin-containing monooxygenase 2 gene (FMO2) of humans, but not of other primates, encodes a truncated, nonfunctional protein. J Biol Chem. 1998;273:30599–30607. doi: 10.1074/jbc.273.46.30599. [DOI] [PubMed] [Google Scholar]
- 12.Krueger SK, Martin SR, Yueh M-F, Pereira CB, Williams DE. Identification of active flavin-containing monooxygenase isoform 2 in human lung and characterization of expressed protein. Drug Metab Dispos. 2002;30:34–41. doi: 10.1124/dmd.30.1.34. [DOI] [PubMed] [Google Scholar]
- 13.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: encyclopedia for genes, proteins and diseases. Rehovot, Israel: Weizmann Institute of Science, Bioinformatics Unit and Genome Center; 1997; GeneCard for FMO2; update Feb 25, 2004; world wide web URL: http://bioinformatics.weizmann.ac.il/cards-bin/carddisp?FMO2
- 14.Cashman JR. The implications of polymorphisms in mammalian flavincontaining monooxygenases in drug discovery and development. Drug Discov Today. 2004;9:574–581. doi: 10.1016/S1359-6446(04)03136-8. [DOI] [PubMed] [Google Scholar]
- 15.Whetstine JR, Yueh M-F, McCarver DG, Williams DE, Park C-S, Kang JH, et al. Ethnic differences in human flavin-containing monooxygenase 2 (FMO2) polymorphisms: detection of expressed protein in African-Americans. Toxicol Appl Pharmacol. 2000;168:216–224. doi: 10.1006/taap.2000.9050. [DOI] [PubMed] [Google Scholar]
- 16.Krueger SK, Williams DE, Yueh M-F, Martin SR, Hines RN, Raucy JL, et al. Genetic polymorphisms of flavin-containing monooxygenase (FMO) Drug Metab Rev. 2002;34:523–532. doi: 10.1081/dmr-120005653. [DOI] [PubMed] [Google Scholar]
- 17.Furnes B, Feng J, Sommer SS, Schlenk D. Identification of novel variants of the flavin-containing monooxygenase gene family in African Americans. Drug Metab Dispos. 2003;31:187–193. doi: 10.1124/dmd.31.2.187. [DOI] [PubMed] [Google Scholar]
- 18.Krueger SK, Siddens LK, Martin SR, Yu Z, Pereira CB, Cabacungan ET, et al. Differences in FMO2*1 allelic frequency between Hispanics of Puerto Rican and Mexican Descent. Drug Metab Dispos. 2004;32:1337–1340. doi: 10.1124/dmd.104.001099. [DOI] [PubMed] [Google Scholar]
- 19.Sommer SS, Groszbach AR, Bottema CD. PCR amplification of specific alleles (PASA) is a general method for rapidly detecting known single-base changes. Biotechniques. 1992;12:82–87. [PubMed] [Google Scholar]
- 20.Germer S, Higuchi R. Single-tube genotyping without oligonucleotide probes. Genome Res. 1999;9:72–78. [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y-M, Henne KR, Charmley P, Kim RB, McCarver DG, Cabacungan ET, et al. Genotyping and site-directed mutagenesis of a cytochrome P450 meander Pro-X-Arg motif critical to CYP4B1 catalysis. Toxicol Appl Pharmacol. 2003;186:119–126. doi: 10.1016/s0041-008x(02)00028-5. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell MK, Gregersen PK, Johnson S, Parsons R, Vlahov D. The New York Cancer Project: rationale, organization, design, and baseline characteristics. J Urban Health. 2004;81:301–310. doi: 10.1093/jurban/jth116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rust S, Funke H, Assmann G. Mutagenically separated PCR (MS-PCR): a highly specific one step procedure for easy mutation detection. Nucleic Acids Res. 1993;21:3623–3629. doi: 10.1093/nar/21.16.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens M, Smith N, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 27.Henderson MC, Krueger SK, Siddens LK, Stevens JF, Williams DE. S-oxygenation of the thioether organophosphate insecticides phorate and disulfoton by human lung flavin-containing monooxygenase 2. Biochem Pharmacol. 2004;68:959–967. doi: 10.1016/j.bcp.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 28.Krueger SK, Yueh M-F, Martin SR, Pereira CB, Williams DE. Characterization of expressed full-length and truncated FMO2 from rhesus monkey. Drug Metab Dispos. 2001;29:693–700. [PubMed] [Google Scholar]
- 29.Dixit A, Roche TE. Spectrophotometric assay of the flavin-containing monooxygenase and changes in its activity in female mouse liver with nutritional and diurnal conditions. Arch Biochem Biophys. 1984;233:50–63. doi: 10.1016/0003-9861(84)90600-3. [DOI] [PubMed] [Google Scholar]
- 30.Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, et al. Haplotype variation and linkage disequilibrium in 313 human genes. Science. 2001;293:489–493. doi: 10.1126/science.1059431. [DOI] [PubMed] [Google Scholar]
- 31.Shows TB, McAlpine PJ, Boucheix C, Collins FS, Coneally PM, Frezal J, et al. Guidelines for human gene nomenclature. Cytogenet Cell Genet. 1987;46:11–28. doi: 10.1159/000132471. [DOI] [PubMed] [Google Scholar]
- 32.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 33.Daly AK, Brockmöller J, Broly F, Eichelbaum M, Evans WE, Gonzalez FJ, et al. Nomenclature for human CYP2D6 alleles. Pharmacogenetics. 1996;6:193–201. doi: 10.1097/00008571-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Wierenga RK, Terpstra P, Hol WG. Prediction of the occurrence of the ADPbinding beta alpha beta-fold in proteins, using an amino acid sequence fingerprint. J Mol Biol. 1986;187:101–107. doi: 10.1016/0022-2836(86)90409-2. [DOI] [PubMed] [Google Scholar]
- 35.Henderson MC, Krueger SK, Stevens JF, Williams DE. Human flavincontaining monooxygenase form 2 S-oxygenation. Sulfenic acid formation from thioureas and oxidation of glutathione. Chem Res Toxicol. 2004;17:633–640. doi: 10.1021/tx034253s. [DOI] [PubMed] [Google Scholar]