

Abstract
Although gemcitabine is a potent therapeutic agent in the treatment of human non-small cell lung cancer (NSCLC), resistance to gemcitabine is common. In this study, we investigated the molecular mechanisms involved in acquired gemcitabine resistance against NSCLC cells. Gemcitabine-resistant NSCLC H1299 cells (H1299/GR) were selected by long-term exposure of parental H1299 cells to gemcitabine. The median inhibitory concentrations of gemcitabine in H1299 and H1299/GR cells were 19.4 nM and 233.1 nM, respectively. Gemcitabine induced activation of JNK in parental H1299 cells but not in H1299/GR cells after 48 h. Blocking JNK activation by pretreatment with SP600125, a specific JNK inhibitor, or by transfection with dominant-negative JNK vectors abrogated gemcitabine-induced apoptosis in parental H1299 cells as evidenced by interruption of caspase activation. Transient transfection with a JNKK2-JNK1 plasmid expressing constitutive JNK1 partially restored the effect of gemcitabine in H1299/GR cells. Our results indicate that gemcitabine-induced apoptosis in human NSCLC H1299 cells requires activation of the JNK signaling pathway. Attenuated JNK activation may contribute to development of acquired gemcitabine resistance in cancer cells.
Keywords: gemcitabine, JNK, apoptosis, lung cancer, drug resistance
Abbreviations: FACS, fluorescence-activated cell sorting; IC50, 50% inhibitory concentration; JNK, c-Jun NH2-Terminal Kinase; MOI, multiplicity of infection; NSCLC, non-small cell lung cancer; propidium iodide (PI); XTT, 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt
1. Introduction
Non-small cell lung cancer (NSCLC) is a major public health problem and one of the leading causes of cancer death worldwide [1]. Although various chemotherapeutic agents and treatment regimens have improved outcomes for patients with advanced NSCLC, the treatments ultimately fail in most patients because of resistance or intolerable toxicity. Chemoresistance, whether inherent or acquired, is known to be a major reason for the failure of anticancer therapies, and is due mainly to interruption of the apoptotic signaling pathway [2].
Gemcitabine (2′,2′-difluorodeoxycytidine) is a highly potent anticancer agent that is used as a single agent or in combination with other chemotherapeutic agents, such as cisplatin and docetaxel, against NSCLC [3–5]. The major effect of gemcitabine is arrest of DNA synthesis [6]. Gemcitabine also has been reported to induce apoptosis in multiple myeloma cells and pancreatic cancer cells through activation of caspase [7,8]. The exact mechanisms by which NSCLC cells acquire gemcitabine resistance are still unknown. Understanding this molecular mechanism may lead to better therapeutic strategies against NSCLC.
c-Jun NH2-terminal kinase (JNK), a subfamily of the mitogen-activated protein kinases (MAPKs), is an important mediator of apoptotic signaling [9,10]. JNK has at least 10 isoforms that are encoded by three genes, JNK1, JNK2, and JNK3.[11] JNK1 and JNK2 are expressed in various tissues and play crucial roles in many cellular events, including growth control, development, and apoptosis [12]. Importantly, JNK activation is required for induction of apoptosis by a number of stress stimuli, such as ultraviolet radiation, growth factor withdrawal, inflammatory cytokines, and chemotherapeutic agents [11,13,14]. Recent reports have shown that attenuation of JNK activation correlates with acquired resistance against several chemotherapeutic agents, such as cisplatin and vinblastine [15,16].
The purpose of our study was to characterize the mechanisms involved in acquired resistance to gemcitabine in NSCLC cell lines. We used gemcitabine-resistant lung cancer cells generated by continuous exposure of gemcitabine-susceptible H1299 cells to clinically relevant doses of gemcitabine. We also compared expressions of various antiapoptotic and proapoptotic molecules with JNK activation in parental H1299 and gemcitabine-resistant H1299/GR cells. Our data show that JNK activation is required for induction of apoptosis by gemcitabine in human lung cancer H1299 cells. In gemcitabine-resistant H1299/GR cells, JNK activation by gemcitabine treatment was markedly attenuated, which correlated with decreased induction of apoptosis. Transient transfection of a JNKK2-JNK1 plasmid expressing constitutive JNK1 partially restored the effect of gemcitabine in H1299/GR cells. Thus, JNK activation may contribute to the acquisition of gemcitabine resistance in NSCLC cells.
2. Materials and methods
2.1. Cells and culture conditions
Human NSCLC H1299 cells were routinely cultured in monolayers in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS), 25 mM HEPES, 100 units/ml of penicillin, and 100 mg/ml of streptomycin. Gemcitabine-resistant H1299 cells were established from these cells by initially adding 1 nM gemcitabine to the culture medium and thereafter escalating the dose to 100 nM. Cells were maintained in the presence of 5% CO2 at 37°C.
2.2. Chemicals and antibodies
Gemcitabine was purchased from Eli Lilly (Indianapolis, IN), and dissolved in phosphate-buffered saline solution (PBS) to a concentration of 10 mM. JNK-specific inhibitor SP600125 was purchased from Calbiochem (La Jolla, CA), dissolved in dimethyl sulfoxide, stored at −20°C, and protected from light. An equal volume of solvent was used as a control. The following antibodies were used in western blot analysis: anti-Bcl-2, Bcl-xL, Mcl-1, Bax, and caspase-3 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-XIAP (BD PharMingen, San Diego, CA); anti-phospho-Akt, Akt, phospho-ERK, ERK, phospho-JNK, JNK, phospho-p38, p38, phospho-c-Jun, phospho-ATF2, and anti-caspase-9 (Cell Signaling Technology, Beverly, MA); anti-caspase-8 (MBL International, Woburn, MA); and anti-β-actin and hemagglutinin (HA) (Sigma, St. Louis, MO).
2.3. Cell proliferation assay
The antiproliferative effects of gemcitabine on NSCLC cell lines were determined by the 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt (XTT) assay. Cells (5 × 103) were seeded in 100 μl of culture medium/well in 96-well flat-bottomed plates and treated the next day with the drugs at various concentrations. After the indicated times, cells were washed once with PBS, and cell viability was determined by colorimetric assay with the tetrazolium dye XTT using the Cell Proliferation Kit II (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer’s protocol. The experiments were performed at least three times for each cell line. Cell viability was calculated as 100 × Atreatment/Acontrol, where A is the absorbance measured by using a microplate reader (Model MRX; Dynatech Laboratories, Chantilly, VA) at 450 nm, with a reference wavelength at 650 nm. The concentration of gemcitabine that inhibited absorbance by 50% (IC50) was determined by using the CurveExpert Version 1.3 program.
2.4. Apoptosis assay
For detection of apoptosis, fixed cells were resuspended in PBS containing 10 μg/ml propidium iodide (PI) (Roche Diagnostics, Indianapolis, IN) and 10 μg/ml RNase A (Sigma-Aldrich) at 37°C for 30 min. Cell-cycle analysis was performed by using an Epics Profile II flow cytometer (Beckman Coulter, Fullerton, CA) with MultiCycle software (Phoenix Flow Systems, San Diego, CA). Accumulation of sub-G1 cells, a known indicator of DNA fragmentation and apoptosis, was used to quantify apoptosis. All experiments were repeated at least twice.
2.5. Plasmid transfection and adenovirus vector transduction
H1299 cells were transfected with a dominant-negative gene cloned as either pLNCX-3X HA-p46JNK1alpha (dnJNK1, encoding an hemagglutinin (HA)-tagged dominant-negative JNK1 mutant) or pLNCX-3X HA-p54JNK2alpha (dnJNK2, encoding an HA-tagged dominant-negative JNK2 mutant) [17] were kindly provided by Dr. L.E. Heasley (University of Colorado Health Sciences Center, Denver, CO). Stable transfectants were selected for growth in the presence of 500 μg/ml G418. For transient transfection, H1299/GR cells were transfected with pSRα3 HA-JNKK2-JNK1, which expresses JNKK2-JNK1 fusion protein that has constitutive JNK1 activity [18]. Plasmid transfection was done using FuGENE6 reagent (Roche Diagnostics).
2.6. Western blot analysis
For preparation of whole-cell extracts, cells were washed twice in cold PBS, collected, and subjected to lysis in lysis buffer (62.5-mM Tris [pH 6.8], 2% sodium dodecyl sulfate, and 10% glycerol) containing 1 × proteinase-inhibitor cocktail (Roche). The lysates were spun at 14,000 × g in a microcentrifuge at 4°C for 10 min, and the supernatants were used as whole-cell extracts. Protein concentrations were determined by using the BCA Protein Assay Kit (Pierce, Rockford, IL). Equal amounts (50 μg) of proteins were used for immunoblotting, as described previously [19].
3. Results
3.1. Effect of gemcitabine on cell proliferation in H1299 and H1299/GR cells
To investigate the mechanisms of acquired resistance to gemcitabine in cancer cells, we generated a tissue culture model (H1299/GR cells) by repeated treatment of H1299 cells with gemcitabine. The cells were initially treated with 1 nM gemcitabine, and then surviving cells were treated with escalating doses of gemcitabine, up to 100 nM. We first checked the cytotoxic effect of gemcitabine in H1299 and H1299/GR cells. Cells were treated with various doses of gemcitabine for 72 h, and then cells viability was determined by XTT assay. As shown in Figure 1, 100 nM gemcitabine inhibited the growth of H1299 cells after 72 h. In contrast, H1299/GR cells, which were selected in a final gemcitabine concentration of 100 nM, were markedly resistant to 100 nM gemcitabine compared with parental H1299 cells. The IC50 value of gemcitabine in H1299/GR cells was consistently more than 10 times higher than that in H1299 parental cells. The IC50 values of H1299 and H1299/GR cells were 19.4 nM and 233.1 nM, respectively.
Fig. 1.
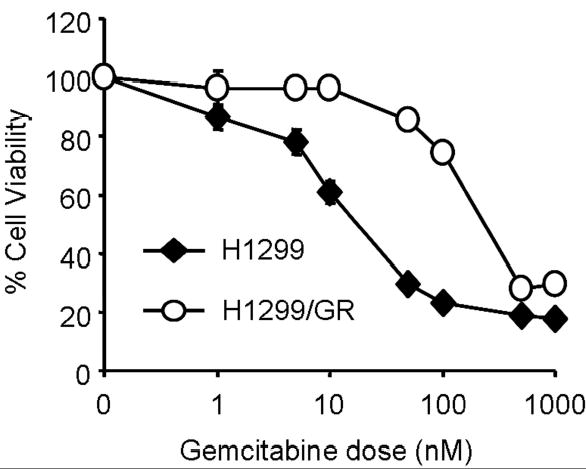
Cytotoxicity of gemcitabine in H1299 and H1299/GR cells. Cells were treated with various concentrations of gemcitabine for 72 h and cell viability was determined by XTT assay. Cells treated with PBS were used as a control, with viability set at 100%. Each data point represents the mean ± SD of three independent experiments.
3.2. Gemcitabine induced apoptosis via activation of caspases in H1299 cells
To elucidate the contribution of apoptosis induction to gemcitabine-mediated cell death, we determined the proportions of the sub-G1 populations in H1299 and H1299/GR cells treated with gemcitabine by flow cytometry assay or western blot analysis. Cells were treated with 100 nM gemcitabine for 24, 48, or 72 h. Cells were then harvested for quantitation of apoptotic cells by flow cytometry. In H1299 cells treated for 48 and 72 h, the portions of sub-G1 cells were 27.1% and 49.9%, respectively; however, no sub-G1 cells were observed in gemcitabine-treated H1299/GR cells (Fig. 2A). To further examine the ability of gemcitabine to induce apoptosis, we treated H1299 and H1299/GR cells with gemcitabine for 12, 24, or 48 h and evaluated cleavage of caspase by Western blotting. Cleavage of caspase-3, caspase-8, and caspase-9 was clearly detectable in H1299 cells after 48 h of treatment, but not in H1299/GR cells (Fig. 2B). These results suggest that gemcitabine induced apoptosis through caspase activation in H1299 cells.
Fig. 2.
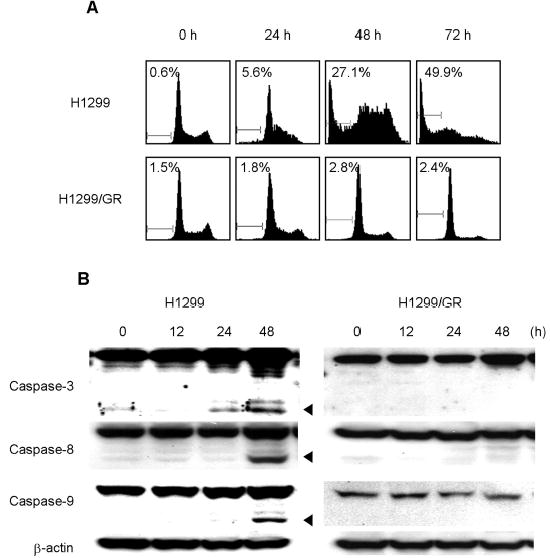
Gemcitabine induced apoptosis via caspase activation in H1299 cells. (A) Flow cytometry analysis of H1299 and H1299/GR cells treated with 100 nM gemcitabine for the indicated time periods. Cells were harvested, fixed, and stained with PI. Histograms represent DNA contents. The numbers represent the percentages of sub-G1 cells. (B) H1299 and H1299/GR cells were treated with 100 nM gemcitabine for the indicated time periods, and proteins in whole-cell lysates were analyzed by immunoblotting with anti-caspase-3, caspase-8, and caspase-9 antibodies. The arrowheads represent cleavage proteins. β-actin was used as a loading control.
3.3. Expression of antiapoptotic proteins in H1299 and H1299/GR cells
To examine the correlation between gemcitabine resistance and the levels of some antiapoptotic and proapoptoric proteins, we compared the expression levels of Bcl-2, Bcl-xL, Mcl-1, Bax, and XIAP in H1299 cells with those in H1299/GR cells by Western blot analysis. As shown in Figure 3, no differences were seen between parental H1299 cells and gemcitabine-resistant H1299/GR cells.
Fig. 3.
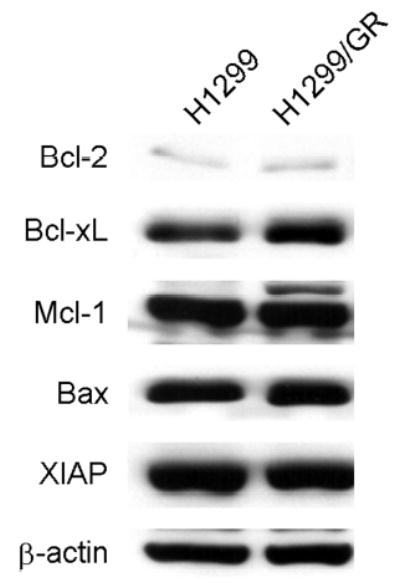
Expression of antiapoptotic and proapoptotic proteins in H1299 and H1299/GR cells. Whole-cell lysates were analyzed by immunoblotting with anti-Bcl-2, Bcl-xL, Mcl-1, Bax, and XIAP antibodies. β-actin was used as the loading control.
3.4. Activation of JNK signaling pathway by gemcitabine treatment in H1299 cells, but not in H1299/GR cells
To clarify the potential involvement of various protein kinase signaling pathways in gemcitabine-resistant cells, we next examined the effect of gemcitabine on MAPK and Akt activation in H1299 and H1299/GR cells. The cells were treated with 100 nM gemcitabine for 48 h, and then cell lysates were subjected to determination of phosphorylation status of JNK, ERK, p38, and Akt by Western blotting (Fig. 4A). The basal level of phosphorylated ERK expression in H1299 cells was markedly higher than that in H1299/GR cells. Gemcitabine had little effect on phosphorylated Akt, but markedly increased the level of phosphorylated JNK or p38 in H1299 cells compared with those treated with solvent alone. Phosphorylation of p38 was also observed in H1299/GR cells after gemcitabine treatment. However, gemcitabine-induced JNK phosphorylation was observed only in H1299 cells but not in H1299/GR cells. We further examined time-dependent changes in the molecules in JNK signaling pathway, including phosphorylated JNK, c-Jun, and ATF2 in gemcitabine-treated H1299 or H1299/GR cells (Fig. 4B). Although JNK was clearly detectable in both H1299 and H1299/GR cells, phosphorylated JNK was increased only in H1299 cells after 24 h or 48 h of treatment. Moreover, phosphorylated c-Jun and ATF2, which are downstream targets of the JNK signaling pathway, were also observed in H1299 cells after 24 h and 48 h of treatment, but not in H1299/GR cells. These results indicate that gemcitabine activates JNK and p38 signaling pathways, and that lack of JNK activation is likely to correlate with gemcitabine resistance in this cell system.
Fig. 4.
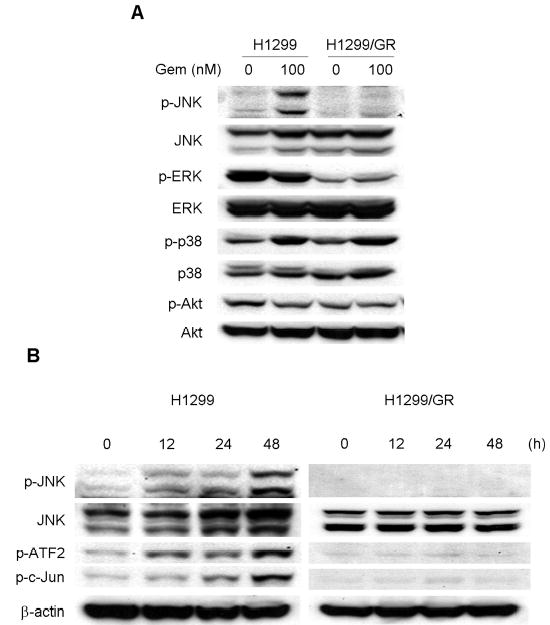
Gemcitabine treatment activated the JNK signaling pathway. (A) Effects of gemcitabine on MAPK and Akt activity in H1299 and H1299/GR cells. Cells were treated with 100 nM gemcitabine (Gem) for 48 h, and then whole-cell lysates were subjected to western blotting using phospho-specific and total protein antibodies to JNK, ERK, p38, and Akt. (B) Time-dependent activation of the JNK signaling pathway by gemcitabine treatment. H1299 and H1299/GR cells were treated with 100 nM gemcitabine for the indicated time periods. Cell lysates were then subjected to western blotting using total JNK and phospho-specific antibodies to JNK, c-Jun, and ATF2. Maximal phosphorylation of those molecules is observed in H1299 cells treated with gemcitabine for 48 h.
3.5. Inhibition of JNK activation blocks gemcitabine-induced apoptosis with inhibiting caspase activation
To clarify the role of JNK activation in gemcitabine resistance, we tested the effect of SP600125, a novel specific inhibitor of JNK, on JNK activation and apoptosis induction by gemcitabine treatment. Pretreatment with 20 μM of SP600125 markedly blocked phosphorylation of JNK, c-Jun, and ATF2 after 48 h, but not total JNK protein (Fig. 5A), suggesting that 20 μM of SP600125 was sufficient to block activation of JNK by gemcitabine. We then examined the effect of SP600125 on gemcitabine-induced apoptosis. H1299 cells were exposed to gemcitabine for 48 h in the presence or absence of 20 μM SP600125, and were quantified by fluorescence-activated cell sorting (FACS) analysis. SP600125 alone had little effect on H1299 cells. Pretreatment of H1299 cells with 20 μM SP600125 markedly diminished gemcitabine-induced apoptosis after 48 h (Fig. 5B). Moreover, cleavage of caspase-3 and caspase-9 was attenuated after 48 h in H1299 cells treated with gemcitabine in the presence of SP600125 (Fig. 5C). These results suggest that JNK activation is required for gemcitabine-mediated apoptosis through caspase activation in H1299 cells. To further confirm the effects of JNK inhibitor SP600125 on gemcitabine-induced apoptosis, we conducted similar studies with dominant-negative mutant vectors of JNK1 (dnJNK1) and JNK2 (dnJNK2). H1299 cells stably transfected with either dnJNK1 or dnJNK2 vector plasmid were treated with 1 μM gemcitabine for 72 h, fixed and stained with PI, and then analyzed by flow cytometry. The extent of gemcitabine-mediated apoptosis was reduced in both dnJNK1 and dnJNK2 transfectants than in an empty vector control (Fig. 5D).
Fig. 5.
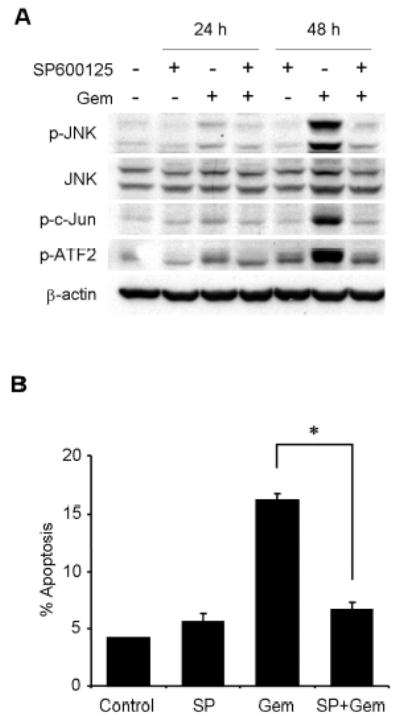
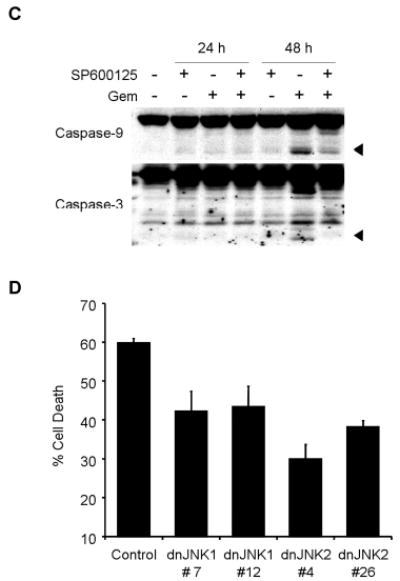
JNK activation has a key role in gemcitabine-induced apoptosis in H1299 cells. (A) H1299 cells were treated with 100 nM gemcitabine (Gem) in the presence or absence of 20 μM SP600125, and harvested at 24 h or 48 h. Whole-cell lysates were analyzed by immunoblotting with indicated antibodies as further evidence for blocking JNK activation and apoptosis. (B) H1299 cells were treated with 100 nM gemcitabine (Gem) in the presence or absence of SP600125 (SP) for 48 h, and sub-G1 population, which indicates apoptosis, was determined by FACS analysis. Data represent the means ± SD from three independent experiments. (C) H1299 cells were treated with 100 nM gemcitabine (Gem) in the presence or absence of 20 μM SP600125 for 24 h or 48 h. Whole-cell lysates were analyzed by immunoblotting with anti-caspase-9 and anti-caspase-3 antibodies. Arrowheads represent cleavage proteins. (D) H1299 cells were transfected with plasmid DNA encoding dominant-negative mutant JNK1 or JNK2 and then selected by G418 to obtain stable clones. Stably transfected H1299 clones were treated with 1 μM gemcitabine for 72 h, and then the apoptotic ratios were determined by flow cytometry. Results are the means ± SD from three independent experiments.
3.6. Transient transfection of a constitutive active JNK1 fusion construct partially reverses gemsitabine resistance in H1299/GR cells
To further investigate the role of JNK in gemcitabine resistance, we transiently transfected H1299/GR cells with the plasmid pSRα3HA-JNKK2-JNK1, which expresses JNKK2-JNK1, a fusion protein with constitutive JNK1 activitv [18]. The expression of JNKK2-JNK1 fusion protein was confirmed by Western blotting with anti-HA antibody (Fig. 6A). To further investigate whether JNKK2-JNK1 protein expression overcomes gemcitabine resistance, H1299/GR cells transfected with HA-JNKK2-JNK1 expressing plasmid were treated with 100 nM gemcitabine for 72 h, and then analyzed apoptotic ratio by flow cytometry (Fig. 6B). The expression of JNKK2-JNK1 fusion protein partially reversed gemcitabine resistance in H1299/GR cells. In contrast, JNKK2-JNK1 fusion protein alone had no detectable effect in apoptotic induction. Apoptotic cells after treatment with JNKK2-JNK1 plasmid alone, gemcitabine alone, and JNKK2-JNK1 plasmid plus gemcitabine were 2.4%, 2.3% and 17.3%, respectively. These results indicate that JNK1 activity is crucial for gemcitabine-mediated apoptosis and that lack of JNK1 activation will contribute to gemcitabine resistance.
Fig. 6.
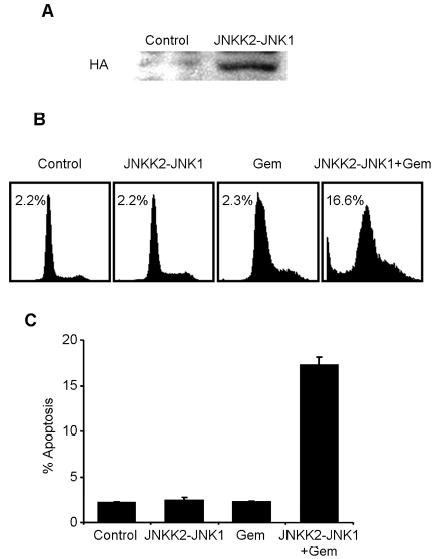
The effect of JNKK2-JNK1 fusion protein expression on H1299/GR cells. (A) H1299/GR cells were transiently transfected with JNKK2-JNK1 or plasmid for 24 h, and then whole-cell lysate were analyzed by immunoblotting with anti-hemagglutinin (HA) antibody. (B) H1299/GR cells transfected with JNKK2-JNK1 were treated with 100 nM gemcitabine for 72 h, and then fixed cells were analyzed by flow cytometry. Top panel, histograms show DNA content of cells. The numbers represent the percentages of sub-G1 cells. Bottom panel, apoptotic ratio was determined by flow cytometry. Data represent mean ± SD.
4. Discussion
Although gemcitabine is a promising treatment for NSCLC [20], the mechanisms of action for gemcitabine, especially the mechanism of acquired drug resistance, are still controversial, and there have been no reports about the correlation between attenuation of JNK activation and gemcitabine resistance. In the present study, we sought to clarify the mechanism of gemcitabine resistance by using NSCLC H1299 sublines (H1299/GR) in which gemcitabine resistance was generated by long-term exposure to gemcitabine.
A recent report showed that there is a strong inverse correlation between Bcl-xL expression and gemcitabine-induced apoptosis in some pancreatic cancer cell lines, indicating that the expression level of Bcl-xL was correlated with gemcitabine sensitivity [21]. It has also been reported that the level of Bcl-2 is a key factor in determining the sensitivity of cancer cells to gemcitabine treatment [22,23]. Our result reveals, however, that the expression levels of Bcl-xL and Bcl-2 were no different between H1299 parental cells and H1299/GR cells. This result suggests that the basal expression levels of those proteins were not involved in acquired gemcitabine resistance in H1299 cells.
Activation of p38 and Akt has been reported to correlate with the sensitivity to gemcitabine of pancreatic cancer cell lines [7,24,25], but our study revealed no differences in phosphorylated Akt and p38 levels at baseline and after gemcitabine treatment between H1299 parental cells and H1299/GR cells. It was also reported that inhibition of ERK activation by MEK inhibitor or MKP3 plasmid dramatically blocked gemcitabine–induced cell death in NSCLC cell lines [26]. Our data also showed that the level of phosphorylated ERK in H1299 cells was markedly higher than that in H1299/GR cells, indicating that reduced ERK activation may also be involved in gemcitabine resistance in H1299/GR cells. Nevertheless, gemcitabine-induced phosphorylations of JNK, c-Jun and ATF2 were completely absent in H1299/GR cells, but can be easily detected in parental H1299 cells, suggesting that JNK activation might be critical for resistance to gemcitabine. Although we cannot rule out the possibility that acquired gemcitabine resistance is associated with other substantial signaling pathways, this result led us to focus on clarifying the role of JNK activation in gemcitabine-induced cytotoxicity in our cell lines. To further investigate the role of JNK activation on the action of gemcitabine, we blocked the JNK signaling pathway in two ways, by a specific JNK inhibitor, SP600125, and by dominant-negative JNK vectors. Our data show that SP600125 blocked activation of JNK, c-Jun, and ATF2 phosphorylation by gemcitabine and strongly diminished gemcitabine-induced apoptosis by blocking caspase-9 and caspase-3 activation in H1299 cells. It also shows that dominant-negative vectors of JNK1 or JNK2 diminished gemcitabine-induced apoptosis. These results strongly indicate that JNK activation is required for induction of parental H1299 cell death by gemcitabine.
A previous report showed that JNK activation is involved in cisplatin-induced apoptosis and that its attenuation correlates with protection against cisplatin treatment [15]. It was also reported that JNK activation by cisplatin is lower in cisplatin-resistant sublines of human adenocarcinoma cells and mouse keratinocytes [27,28]. Another report suggested that inhibition of JNK by a specific inhibitor protects KB-3 epidermoid carcinoma cells from cytotoxicity of drugs such as vinblastine, doxorubicin, and etoposide [16]. Those findings indicate that lack of JNK activation may play a critical role in resistance of cancer cells to certain chemotherapeutic agents. Moreover, deactivation of JNK also has been mentioned in clinical studies in prostate [29] and colon [30] cancers. It was reported that JNK deactivation in advanced prostate tumors could be related to a lower apoptotic ratio [29]. An other report showed a consistent correlation between JNK activation and early stage NSCLC cancers [31]. The authors of this report suggested that JNK activation could increase the sensitivity of some NSCLC cells to chemotherapy and radiation therapy. Further investigation of the role of JNK activation in patients with NSCLC may give us crucial information about therapeutic strategies against these cancers.
A recent report showed that selective JNK stimulation by transfection of JNK1 lead to apoptosis sensitization to cisplatin in cisplatin-resistant ovarian cancer cells [32]. Similarly, our data demonstrate that transient transfection of JNKK2-JNK1 fusion protein partially reverse gemcitabine resistance in H1299/GR cells. Previous study has revealed that JNKK2-JNK1 fusion protein has constitutive JNK activity [18]. Thus, susceptibility to gemcitabine-induced cytotoxicity can be restored by ectopic expression of JNK activity. Nevertheless, it is possible that the mechanisms of gemcitabine resistance could be different in different cells. Recent reports implicate expression of ribonucleotide reductase subunit 1 (RRM1) and deoxycytidine kinase (dCK), genes that are related to gemcitabine metabolism, in gemcitabine resistance [33–35]. It is certainly expected that metabolic inactivation of gemcitabine may lead to resistance to this agent.
In conclusion, our data demonstrate that JNK activation has a key role in gemcitabine-mediated cytotoxicity through induction of apoptosis in NSCLC cells. We also show that attenuation of JNK activation may contribute to the acquisition of gemcitabine resistance in lung cancer cells. Thus, the JNK signaling pathway may be studied further as a critical target for a novel approach of lung cancer treatment.
Acknowledgments
We thank for Kathryn Hale editorial review and Karen M. Ramirez for technical assistance with the flow cytometry analysis. This study was supported by grants from National Cancer Institute grants RO1 CA 092487-01A1 (to B. Fang), RO1 CA 098582-01A1 (to B. Fang), Lung SPORE development award, and NIH Core grant CA-16672.
References
- 1.Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 2.Guchelaar HJ, Vermes A, Vermes I, Haanen C. Apoptosis: molecular mechanisms and implications for cancer chemotherapy. Pharm World Sci. 1997;19:119–125. doi: 10.1023/a:1008654316572. [DOI] [PubMed] [Google Scholar]
- 3.Sandler A, Ettinger DS. Gemcitabine: single-agent and combination therapy in non-small cell lung cancer. Oncologist. 1999;4:241–251. [PubMed] [Google Scholar]
- 4.Bergman AM, Ruiz vH V, Veerman G, Kuiper CM, Peters GJ. Synergistic interaction between cisplatin and gemcitabine in vitro. Clin Cancer Res. 1996;2:521–530. [PubMed] [Google Scholar]
- 5.Georgoulias V, Kouroussis C, Androulakis N, Kakolyris S, Dimopoulos MA, Papadakis E, Bouros D, Apostolopoulou F, Papadimitriou C, Agelidou A, Hatzakis K, Kalbakis K, Kotsakis A, Vardakis N, Vlachonicolis J. Front-line treatment of advanced non-small-cell lung cancer with docetaxel and gemcitabine: a multicenter phase II trial. J Clin Oncol. 1999;17:914–920. doi: 10.1200/JCO.1999.17.3.914. [DOI] [PubMed] [Google Scholar]
- 6.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–6117. [PubMed] [Google Scholar]
- 7.Habiro A, Tanno S, Koizumi K, Izawa T, Nakano Y, Osanai M, Mizukami Y, Okumura T, Kohgo Y. Involvement of p38 mitogen-activated protein kinase in gemcitabine-induced apoptosis in human pancreatic cancer cells. Biochem Biophys Res Commun. 2004;316:71–77. doi: 10.1016/j.bbrc.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Nabhan C, Gajria D, Krett NL, Gandhi V, Ghias K, Rosen ST. Caspase activation is required for gemcitabine activity in multiple myeloma cell lines. Mol Cancer Ther. 2002;1:1221–1227. [PubMed] [Google Scholar]
- 9.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 11.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 12.Butterfield L, Zentrich E, Beekman A, Heasley LE. Stress- and cell type-dependent regulation of transfected c-Jun N-terminal kinase and mitogen-activated protein kinase kinase isoforms. Biochem J. 1999;338 ( Pt 3):681–686. [PMC free article] [PubMed] [Google Scholar]
- 13.Osborn MT, Chambers TC. Role of the stress-activated/c-Jun NH2-terminal protein kinase pathway in the cellular response to adriamycin and other chemotherapeutic drugs. J Biol Chem. 1996;271:30950–30955. doi: 10.1074/jbc.271.48.30950. [DOI] [PubMed] [Google Scholar]
- 14.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 15.Brozovic A, Fritz G, Christmann M, Zisowsky J, Jaehde U, Osmak M, Kaina B. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int J Cancer. 2004;112:974–985. doi: 10.1002/ijc.20522. [DOI] [PubMed] [Google Scholar]
- 16.Brantley-Finley C, Lyle CS, Du L, Goodwin ME, Hall T, Szwedo D, Kaushal GP, Chambers TC. The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem Pharmacol. 2003;66:459–469. doi: 10.1016/s0006-2952(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 17.Wojtaszek PA, Heasley LE, Siriwardana G, Berl T. Dominant-negative c-Jun NH2-terminal kinase 2 sensitizes renal inner medullary collecting duct cells to hypertonicity-induced lethality independent of organic osmolyte transport. J Biol Chem. 1998;273:800–804. doi: 10.1074/jbc.273.2.800. [DOI] [PubMed] [Google Scholar]
- 18.Zheng C, Xiang J, Hunter T, Lin A. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J Biol Chem. 1999;274:28966–28971. doi: 10.1074/jbc.274.41.28966. [DOI] [PubMed] [Google Scholar]
- 19.Teraishi F, Kadowaki Y, Tango Y, Kawashima T, Umeoka T, Kagawa S, Tanaka N, Fujiwara T. Ectopic p21sdi1 gene transfer induces retinoic acid receptor beta expression and sensitizes human cancer cells to retinoid treatment. Int J Cancer. 2003;103:833–839. doi: 10.1002/ijc.10892. [DOI] [PubMed] [Google Scholar]
- 20.Bunn PA, Jr, Kelly K. New chemotherapeutic agents prolong survival and improve quality of life in non-small cell lung cancer: a review of the literature and future directions. Clin Cancer Res. 1998;4:1087–1100. [PubMed] [Google Scholar]
- 21.Schniewind B, Christgen M, Kurdow R, Haye S, Kremer B, Kalthoff H, Ungefroren H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int J Cancer. 2004;109:182–188. doi: 10.1002/ijc.11679. [DOI] [PubMed] [Google Scholar]
- 22.Han JY, Hong EK, Choi BG, Park JN, Kim KW, Kang JH, Jin JY, Park SY, Hong YS, Lee KS. Death receptor 5 and Bcl-2 protein expression as predictors of tumor response to gemcitabine and cisplatin in patients with advanced non-small-cell lung cancer. Med Oncol. 2003;20:355–362. doi: 10.1385/MO:20:4:355. [DOI] [PubMed] [Google Scholar]
- 23.Rieger J, Durka S, Streffer J, Dichgans J, Weller M. Gemcitabine cytotoxicity of human malignant glioma cells: modulation by antioxidants, BCL-2 and dexamethasone. Eur J Pharmacol. 1999;365:301–308. doi: 10.1016/s0014-2999(98)00883-8. [DOI] [PubMed] [Google Scholar]
- 24.Ng SS, Tsao MS, Nicklee T, Hedley DW. Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin Cancer Res. 2001;7:3269–3275. [PubMed] [Google Scholar]
- 25.Ng SSW, Tsao MS, Chow S, Hedley DW. Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res. 2000;60:5451–5455. [PubMed] [Google Scholar]
- 26.Chang GC, Hsu SL, Tsai JR, Wu WJ, Chen CY, Sheu GT. Extracellular signal-regulated kinase activation and Bcl-2 downregulation mediate apoptosis after gemcitabine treatment partly via a p53-independent pathway. Eur J Pharmacol. 2004;502:169–183. doi: 10.1016/j.ejphar.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez-Perez I, Murguia JR, Perona R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene. 1998;16:533–540. doi: 10.1038/sj.onc.1201578. [DOI] [PubMed] [Google Scholar]
- 28.Cui W, Yazlovitskaya EM, Mayo MS, Pelling JC, Persons DL. Cisplatin-induced response of c-jun N-terminal kinase 1 and extracellular signal--regulated protein kinases 1 and 2 in a series of cisplatin-resistant ovarian carcinoma cell lines. Mol Carcinog. 2000;29:219–228. [PubMed] [Google Scholar]
- 29.Uzgare AR, Kaplan PJ, Greenberg NM. Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate. 2003;55:128–139. doi: 10.1002/pros.10212. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Ding Q, Dong Z, Ehlers RA, Evers BM. Downregulation of mitogen-activated protein kinases in human colon cancers. Anticancer Res. 2000;20:75–83. [PubMed] [Google Scholar]
- 31.Vicent S, Garayoa M, Lopez-Picazo JM, Lozano MD, Toledo G, Thunnissen FB, Manzano RG, Montuenga LM. Mitogen-activated protein kinase phosphatase-1 is overexpressed in non-small cell lung cancer and is an independent predictor of outcome in patients. Clin Cancer Res. 2004;10:3639–3649. doi: 10.1158/1078-0432.CCR-03-0771. [DOI] [PubMed] [Google Scholar]
- 32.Li F, Meng L, Zhou J, Xing H, Wang S, Xu G, Zhu H, Wang B, Chen G, Lu YP, Ma D. Reversing chemoresistance in cisplatin-resistant human ovarian cancer cells: A role of c-Jun NH(2)-terminal kinase 1. Biochem Biophys Res Commun. 2005;335:1070–1077. doi: 10.1016/j.bbrc.2005.07.169. [DOI] [PubMed] [Google Scholar]
- 33.Davidson JD, Ma L, Flagella M, Geeganage S, Gelbert LM, Slapak CA. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64:3761–3766. doi: 10.1158/0008-5472.CAN-03-3363. [DOI] [PubMed] [Google Scholar]
- 34.Bergman AM, Pinedo HM, Jongsma AP, Brouwer M, Ruiz vH V, Veerman G, Leyva A, Eriksson S, Peters GJ. Decreased resistance to gemcitabine (2′,2′-difluorodeoxycitidine) of cytosine arabinoside-resistant myeloblastic murine and rat leukemia cell lines: role of altered activity and substrate specificity of deoxycytidine kinase. Biochem Pharmacol. 1999;57:397–406. doi: 10.1016/s0006-2952(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 35.Heinemann V, Hertel LW, Grindey GB, Plunkett W. Comparison of the cellular pharmacokinetics and toxicity of 2′,2′-difluorodeoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1988;48:4024–4031. [PubMed] [Google Scholar]