

Abstract
In oxygenic photosynthesis, PSII carries out the oxidation of water and reduction of plastoquinone. The product of water oxidation is molecular oxygen. The water splitting complex is located on the lumenal side of the PSII reaction center and contains manganese, calcium, and chloride. Four sequential photooxidation reactions are required to generate oxygen from water; the five sequentially oxidized forms of the water splitting complex are known as the Sn states, where n refers to the number of oxidizing equivalents stored. Calcium plays a role in water oxidation; removal of calcium is associated with an inhibition of the S state cycle. Although calcium can be replaced by other cations in vitro, only strontium maintains activity, and the steady-state rate of oxygen evolution is decreased in strontium-reconstituted PSII. In this article, we study the role of calcium in PSII that is limited in water content. We report that strontium substitution or 18OH2 exchange causes conformational changes in the calcium ligation shell. The conformational change is detected because of a perturbation to calcium ligation during the S1 to S2 and S2 to S3 transition under water-limited conditions.
INTRODUCTION
In plants, algae, and cyanobacteria, oxygenic photosynthesis converts solar energy into chemical energy. PSII is one of the two photosynthetic reaction centers that carry out this process. PSII catalyzes the light-driven oxidation of water and reduction of plastoquinone. Reduced plastoquinone dissociates from PSII to act as a proton and electron carrier in the thylakoid membrane. The oxidation of water to oxygen occurs within the OEC of PSII. Four sequential oxidation reactions are required to generate oxygen from water. The sequentially oxidized forms of the OEC are called the S states. Oxygen release occurs during the S3 to S0 transition from an unstable intermediate, known as the S4 state. S4 is the most oxidized state of the OEC. The oxidation state of Mn in the S states has been studied by EPR and x-ray absorption near edge spectroscopy (reviewed in Britt, 1996 (1)). It is generally accepted that the S1 state contains a MnIII2MnIV2 cluster and that the S2 state contains a MnIII1MnIV3 cluster (for example, see Peloquin et al., 2000 (2) and Weng et al., 2004 (3) and references therein). The oxidation state of the Mn ions in the S3 state has been controversial, with some x-ray absorption studies suggesting that a Mn oxidation occurs (4,5) and some indicating that a ligand or μ-oxo bridge to the Mn is oxidized instead (6,7). Extended x-ray absorption fine structure and kinetic studies have shown that a structural change in the OEC occurs during the S2 to S3 transition (8–10). Recently, it has been suggested that this structural change includes the addition of a third di-μ-oxo bridge between two Mn atoms (11).
Calcium and chloride are essential cofactors in the OEC (12). Calcium is believed to bind close to the Mn cluster, and extended x-ray absorption fine structure measurements report Ca-Mn distances of 3.3–4.2 Å (13,14). Recent 87Sr+2 ESEEM experiments confirm that strontium is 3–5 Å from Mn (15). Four x-ray diffraction studies of PSII crystals from thermophilic cyanobacteria have been presented at 3.8 (16), 3.7 (17), 3.5 (18), and 3.2 Å (19), respectively. In the 3.5 Å structure, electron density has been assigned to calcium and manganese, which are 3.4 Å apart and μ-oxo bridged (18). Although ligating water molecules have been included in the calcium first ligation shell, all calcium ligands have not yet been assigned (18). In the other three structures, active site calcium has not yet been identified (16,17).
The role of calcium in PSII has been investigated by depletion and substitution techniques (20). Calcium removal blocks the S state cycle, generating an inhibited S3′ state (21,22). Only strontium can reconstitute oxygen evolution (23). Strontium-substituted preparations exhibit slower steady-state rates (23) and a slower turnover of the S state cycle (24–26). Substitution of strontium for calcium alters the EPR signals from the S2 state (for examples, see Kim et al., 2004 (15), Boussac et al., 1988 (27), and references therein). A 113Cd NMR study suggests that Ca+2 may have a symmetric array of ligands containing oxygen, nitrogen, and/or chlorine (28). It has also been suggested that calcium may play a role in water oxidation by binding a water or hydroxide ion, which then acts as a nucleophile in the reaction to form the oxygen-oxygen bond (29,30,). Mass spectrometry experiments showing that substitution of strontium for calcium alters the rate of substrate water exchange support this interpretation (31) as do cation substitution experiments (32).
In this report, we employ FTIR spectroscopy and strontium and 18OH2 exchange to investigate structural changes involving calcium in the OEC. FTIR spectroscopy has been used previously to study the S state cycle in cyanobacterial and plant PSII (for representative examples, see Hillier and Babcock (33) and Noguchi et al. (34)). Previous FTIR measurements have disagreed about the degree of coupling between Mn and Ca+2 during the S1 to S2 transition (see Noguchi et al. (35), Chu et al. (36), and Kimura et al. (37) and discussions therein), and calcium substitution experiments on the higher S states have not yet been reported.
In our experiments, we use PSII preparations under conditions in which the samples are limited for substrate water. Under these conditions, PSII does not undergo the S3 to S0 transition (38). These conditions have the benefit of decreasing the contribution from water in the 1800–1200 cm−1 region. Also, inhibiting conditions are commonly used to investigate aspects of enzyme reaction mechanisms. For example, in PSII, removal of calcium and chloride is known to generate new EPR signals, which have been used to probe mechanism and structure (reviewed in Yocum, 1992 (12)).
In this study, PSII samples were preflashed and dark adapted to generate the S1 state. Difference FTIR spectra assigned to each S state transition were constructed by manipulation of data obtained before and after each flash. These data support the interpretation that strontium substitution and 18OH2 exchange cause structural changes in peptide carbonyl ligands to calcium or in the protein environment of calcium. This conformational change is detected because of a perturbation to the calcium ligand environment, which occurs during the S2 to S3 transition. We also detect a perturbation to Mn-Ca+2 bridging ligands during the S1 to S2 transition. Therefore, our result provides support for the conclusion that a structural change occurs in the OEC during these S state transitions under water-limited conditions.
MATERIALS AND METHODS
Sample preparation and strontium/18OH2 exchange
PSII membranes were prepared from market spinach, suspended in SMN buffer (0.40 M sucrose, 50 mM MES-NaOH, 15 mM NaCl, pH 6) at 3.0 mg chl per ml and stored at −70°C (39) PSII membranes were depleted of the 18 and 24 kDa subunits (salt-washed PSII samples) by dark incubation of PSII samples in high-salt (2.0 M NaCl) SM (0.40 M sucrose, 50 mM MES-NaOH, pH 6) buffer at 0.5 mg chl/ml for 30 min at 4°C (40). These membranes were washed with SM buffer and then resuspended in a SM buffer that contained either 20 mM CaCl2 or SrCl2. Aliquots of these samples were stored at −70°C until use. Just before use, samples were pelleted and exchanged twice into 50 μl aliquots of SM buffer, containing either 20 mM CaCl2 or SrCl2 (48,000 × G for 6 min at 4°C). For 18OH2 exchange, the same exchange procedure was followed through the use of 18OH2 SM buffer containing calcium chloride. In one case, PSII samples also contained 15 mM NaCl (see Fig. 2). In total, samples used for FTIR spectroscopy were incubated either in calcium or strontium buffer for more than 4 h before use (32). Note that the method employed here is not a calcium extraction procedure (24) but a strontium exchange method. This provides an advantage in the avoidance of chelators, which can bind to PSII and alter the structure of the OEC (see Kimura et al. (37) and references therein).
FIGURE 2.

Vibrational spectra of the OEC. Difference FTIR spectra are assigned to the S1 to S2 (A), S2 to S3 (B), S3 to S0 (C), and S0 to S1 (D) transitions in salt-washed PSII containing calcium. In E, a dark-minus-dark control is presented. The tick marks on the y axis are 2 × 10−4 absorbance units. Other conditions are given in the Materials and Methods section.
Oxygen evolution measurements
Oxygen evolution activity was monitored using a Clark electrode, as previously described (41). The steady-state rate of oxygen evolution under saturating light intensity was 680 μmol O2 (mg-chl hr)−1 for PSII samples employed in these studies. For salt-washed PSII, reconstituted either with CaCl2 or with SrCl2, the rates were measured to be 440 and 110 μmol O2 (mg-chl hr)−1, respectively. The decrease in oxygen evolution rate in the presence of calcium after salt washing (from 680 to 440 μmol O2 (mg-chl hr)−1) has been noted previously (40) and may be consistent with an increase in an acceptor-side rate limitation after salt washing (42). This rate limitation may be due to the removal of detergent in the additional wash steps.
The decrease in the steady-state oxygen evolution rate observed in the presence of strontium, compared to calcium, is consistent with substitution of strontium for calcium at the active site for water oxidation (20,32). In the absence of added calcium in the oxygen evolution assay, calcium-exchanged samples exhibited no significant oxygen evolution activity (∼60 μmol O2(mg-chl hr)−1).
Gel electrophoresis
Samples were subjected to electrophoresis using the modified SDS-PAGE Neville method (42). Gels were fixed, stained, and destained in an aqueous solution containing 45% methanol, 10% acetic acid, and ∼0.1% Coomassie Brilliant Blue R (Aldrich, Milwaukee, WI). This procedure was used to verify removal of the 18 and 24 kDa subunits (data not shown).
EPR experiments
EPR experiments were performed at 10 K to verify that strontium substitution. EPR experiments were conducted according to the method of Kim et al., 2004 (15). A Bruker (Billerica, MA) EMX spectrometer, an Oxford (Concord, MA) cryostat, and a standard Bruker TE cavity were employed. Samples were dark adapted at room temperature and an EPR background scan was performed. Samples were then illuminated at 200 K with red-filtered and heat-filtered light from a Dolan Jenner (Lawrence, MA) 150 W illuminator, and another EPR spectrum was recorded. Fig. 1 presents the result of the subtraction: after illumination-minus-before illumination. Samples contained 0.25 mM recrystallized 2,6-dichlorobenzoquinone (DCBQ) and 0.5 mM potassium ferricyanide. The spectral conditions were frequency, 9.44 GHz; microwave power, 3.2 mW; field modulation, 16 G; frequency modulation, 100 kHz; time constant, 2.6 s; scan time, 336 s; and sweep width, 6240 G. The data shown in Fig. 1 A were recorded on the calcium-exchanged PSII control (four scans per sample; average of three PSII samples), and the data shown in Fig. 1 B were recorded on the strontium-exchanged PSII samples (four scans per sample; average of two PSII samples). As expected (15), an increase in the g = 4.1 signal and a change in the hyperfine splittings of the multiline signal are observed in the strontium-exchanged sample (Fig. 1). This EPR result verifies that strontium has been exchanged at the calcium site.
FIGURE 1.

Light-minus-dark EPR spectra obtained on (A) calcium-exchanged PSII and (B) strontium-exchanged PSII. The frequency was 9.44 GHz. Data points in the g = 2.0 region, which contains a large signal from oxidized tyrosine residues, have been deleted for presentation purposes. Other conditions are given in the Materials and Methods section.
FTIR experiments
Samples were defrosted on ice immediately before use, and the electron acceptors, potassium ferricyanide, and recrystallized DCBQ were added to the suspension with final concentrations of 6.0 mM and 500 μM, respectively. Salt-washed PSII samples were then pelleted (48,000 × G for 6 min at 4°C). Compared to our earlier work (42,44,45), this set of conditions slightly raised the acceptor to reaction center ratio, which resulted in a lower contribution of 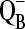 to the spectrum (46).
to the spectrum (46).
Approximately 2.0 mg of PSII was sandwiched between two CaF2 windows. The windows were slid apart, leaving a thin uniform film of sample, which was wiped from a 3 mm boundary around the window edge with a cotton swab. The sample was then dried under nitrogen flow (2500 cc/min) on ice. The windows were sandwiched again, and the space between them was sealed with silicone high-vacuum grease, applied in a continuous bead that did not contact the sample. A layer of parafilm was used to seal the circumference of the windows. Using this procedure, dehydration of the sample over the course of data acquisition was minimal, with <2.5% change in the ratio of the ∼3400 cm−1 band to the protein amide II (1545 cm−1) band. The average ratio of the 3400/1545 cm−1 bands in the FTIR spectrum was >3.0 in Fig. 2 and 2.4 in Figs. 3–5. In samples with the lower ratio, the final sucrose concentration was estimated to be 1.3 M from the weight change after dehydration. We have previously shown that this concentration of sucrose gives optimal steady-state oxygen rates in solution (42). Oxygen rates before and after the FTIR experiments were consistent with retention of activity, as previously reported (42,44).
FIGURE 3.

Effect of strontium/calcium replacement on the vibrational spectra of the OEC under substrate-limited conditions. Difference FTIR spectra are assigned to the S1 to S2 (A), S2 to S3 (B), S3 to S0 (C), and S0 to S1 (D) transitions. In the dashed lines, salt-washed PSII contained calcium in the OEC; in the solid line, salt-washed PSII contained strontium in the OEC. In E, a dark-minus-dark control is presented. The tick marks on the y axis are 1 × 10−4 absorbance units. Other conditions are given in the Materials and Methods section.
FIGURE 4.

Double difference spectra showing the effect of strontium/calcium (A and B) and 18OH2/16OH2 (C and D) replacement on the OEC. Data shown in (A and C) were constructed from the S2-S1 spectra, calcium-minus-strontium (A) and 16OH2-minus-18OH2 (C). Data shown in B and D were constructed from the S3-S2 spectra, calcium-minus-strontium (B) and 16OH2-minus-18OH2 (D). In E, a representative control double difference is shown, in which no vibrational bands are expected. This control double difference was constructed from the S3-minus-S2 data set. The tick marks on the y axis are 1 × 10−4 absorbance units.
FIGURE 5.

Effect of 18OH2/16OH2 exchange on the vibrational spectra of the OEC. Difference FTIR spectra are assigned to the S1 to S2 (A), S2 to S3 (B), S3 to S0 (C), and S0 to S1 (D) transitions. In the dashed lines, salt-washed PSII contained calcium and 16OH2; in the solid line, salt-washed PSII contained calcium and 18OH2. In E, a dark-minus-dark control is presented. The tick marks on the y axis are 1 × 10−4 absorbance units. Other conditions are given in the Materials and Methods section.
FTIR measurements were performed with a Bruker 66v spectrometer, as previously described (42,44,45). Samples were maintained at a constant temperature of 4°C through the use of a temperature-controlled Harrick (Ossining, NY) cell and a temperature-controlled recirculating bath. Samples were transferred to the cell at 14°C and immediately cooled. The sample compartment was purged with dry nitrogen for 60 min before data acquisition began and during data acquisition. Samples were irradiated with a depolarized Nd-YAG laser spot (Continuum, Santa Clara, CA; 7 ns, 532 nm), with an intensity of 10 mJ/cm2.
The measurement cycle began with dark adaptation of the samples for 60 min to give an equilibrium mixture of S0 and S1. This mixture was then given a single flash and allowed to equilibrate for 20 min to achieve a maximal concentration of the S1 state (see Halverson and Barry (42,44) and references therein). Single-channel spectra (100 scans, 15 s) were collected before S-state advancement flashes and after each of six advancement flashes. In all experiments, samples were used for three measurement cycles (19 flashes). The spectra recorded with each measurement cycle were found to be identical (data not shown). There was a 20 min equilibration between each set of six flashes. This dark adaptation time of 20 min was constant throughout the experiment to prevent spontaneous, dark-induced alterations in the S2-S1 spectrum, as previously described (44).
All single channel spectra were collected with a 4 mm aperture, 8 cm−1 resolution, a Happ-Genzel apodization function, four levels of zero-filling, and a Mertz phase correction. Difference spectra were calculated directly from the single channel spectra collected before and after each advancement flash. An FTIR absorption spectrum of each sample was also measured before and after data collection. In Fig. 2, spectra were normalized to an amide II absorbance of 0.5, correcting for pathlength and concentration. In Figs. 3–5, all data were scaled to the calcium S2-minus-S1 difference spectrum, using the intensity of a differential set of bands at 2112/2034 cm−1. These bands arise from the CN stretching vibration of ferricyanide and ferrocyanide, respectively (47). The data in Fig. 2 are an average of 24 individual difference spectra. The data in Figs. 3–5 are an average of 81 (calcium, 16OH2), 87 (strontium, 16OH2), or 93 (calcium, 18OH2) individual difference spectra.
RESULTS
Fig. 2 shows difference FTIR spectra acquired of calcium-containing samples with one (Fig. 2 A), two (Fig. 2 B), three (Fig. 2 C), and four (Fig. 2 D) saturating, 532 nm flashes. The data are assigned to S2-S1, S3-S2, S0-S3, and S1-S0 spectra, respectively. A dark-minus-dark control is shown for comparison (Fig. 2 E). In these experiments, the ratio of water to protein infrared bands was >3.0. These spectra resemble data previously reported under these conditions (see Hillier and Babcock (33), Noguchi et al. (34), and Yamanari et al. (48) and Discussion section).
Fig. 3 shows difference FTIR spectra acquired of calcium-containing samples under substrate-limiting conditions (water/protein ratio = 2.4). The data were recorded with one (Fig. 3 A, dashed line), two (Fig. 3 B, dashed line), three (Fig. 3 C, dashed line), and four (Fig. 3 D, dashed line) saturating, 532 nm flashes. The data are assigned to S2-S1, S3-S2, S0-S3, and S1-S0 spectra, respectively. In the solid lines, the same spectra were constructed for strontium-containing PSII samples. A dark-dark control spectrum in Fig. 3 E exhibits the level of noise. Only the S2-S1 and S3-S2 spectra show defined vibrational bands under these conditions (38), consistent with a limitation in the rate of PSII turnover caused by removal of water. This rate limitation does not alter the frequencies or relative amplitudes of bands in the S1 to S2 difference spectrum and does not have a dramatic effect on the S2 to S3 difference spectrum (compare Figs. 2 and 3). Note that this inhibitory effect is reversible, because oxygen evolution rates before and after the measurement were similar, as previously reported (42,44).
To identify spectral changes induced by the substitution of strontium for calcium under inhibited conditions, the solid and dashed spectra in each panel were subtracted to generate double difference spectra. The results of this subtraction are shown in Fig. 4 for the S2-S1 (Fig. 4 A) and S3-S2 (Fig. 4 B) data. To construct the double difference spectra, the strontium difference spectrum was subtracted from the calcium difference spectrum. A representative control subtraction, in which no spectral bands are expected, is shown in Fig. 4 E. Spectral features, with signals above the noise in the measurements, are observed in the double difference spectra in Fig. 4, A and B. Vibrational bands are only present in these double difference spectra if substitution of strontium for calcium and photooxidation of the OEC perturb the vibrational spectrum. As shown in Fig. 4 A, photooxidation of Mn(III) during the S1 to S2 transition and substitution of strontium for calcium perturb the vibrational frequency of bands throughout the entire 1800–1200 cm−1 region of the infrared spectrum. This type of frequency shift is most likely to occur for vibrational bands of metal ligands in the OEC, which are sensitive to changes in Mn charge, Ca+2/Sr+2 ionic radius, and accompanying changes in bond distances.
As shown in Fig. 4 B, the S2 to S3 transition also perturbs the calcium/strontium site, but the double difference spectrum reflects only perturbations of amide I (C=O stretching) and II (CN stretching/NH bending) bands (48) and is distinct from the more complex spectral perturbation observed during the S1 to S2 transition (Fig. 4 A). These data are consistent with a μ-oxo/ligand oxidation reaction or with the oxidation of a different Mn ion during this S state transition (compared to the S1 to S2 transition). A possible assignment for the S2 to S3 double difference spectrum is that a positive band at 1675 cm−1 (Ca+2 sample, S3 state) shifts to give a negative band at 1644 cm−1 (Sr+2 sample, S3 state). The accompanying amide II changes exhibit a positive-negative-positive band pattern (1556 (+); 1538 (−); 1507 (+) cm−1).
Fig. 5 shows difference FTIR spectra acquired of 16OH2-containing samples with one (Fig. 5 A, dashed line), two (Fig. 5 B, dashed line), three (Fig. 5 C, dashed line), and four (Fig. 5 D, dashed line) saturating, 532 nm flashes. The data correspond to S2-S1, S3-S2, S0-S3, and S1-S0 spectra, respectively. In the solid lines, the same series of spectra were constructed for 18OH2-substituted salt-washed PSII samples. All samples contained calcium. A dark-dark control spectrum in Fig. 5 E shows the level of noise.
To identify spectral changes induced by the substitution of 18OH2 for 16OH2, the solid and dashed spectra in each panel were subtracted. The results of this subtraction are shown in Fig. 4 for the S2-S1 (Fig. 4 C) and S3-S2 (Fig. 4 D) spectra. A control subtraction, in which no spectral bands are expected, is shown in Fig. 4 E. Vibrational bands, with signals above the noise in the measurements, are observed only in the double difference spectrum corresponding to the S2 to S3 transition (Fig. 4 D). Therefore, we conclude that 18OH2 substitution and the S2 to S3 transition perturb the amide I and II vibrations (49) of the peptide backbone. The mechanism of this perturbation will be discussed below. Note that there are no significant effects of 18OH2 substitution on the S2-S1 spectrum (Fig. 4 C). Interestingly, the 18OH2 double difference spectrum is indistinguishable from the S3-S2 double difference spectrum generated by strontium substitution (Fig. 4, B and D), given the signal to noise ratio.
DISCUSSION
In this article, we report FTIR spectra of the S state transitions in salt-washed PSII. These spectra resemble data previously reported in plant and cyanobacterial PSII (33,34,42,44,45,48). Some of the bands in these S2-S1 and S3-S2 spectra have been assigned by global 15N and 13C labeling (48,50). For the S1 to S2 transition, specific isotopic labeling of the manganese stabilizing subunit have been reported; these experiments assign subsets of spectral features to particular amino acid residues in this subunit (45).
In some of our FTIR experiments, the water content of the PSII sample was limiting for the reaction mechanism. The fact that those data exhibit little spectral change after three flashes, which should induce the S3 to S0 transition, is consistent with an inhibition of this S state transition under these conditions. This phenomenon has been previously described in PSII samples maintained at low humidity (38). We attribute the inhibition to a rate limitation, which causes a reduction of the rate of the S3 to S0 transition relative to the 15 s data acquisition time. Under these conditions, PSII may be effectively trapped in the S3 state. The origin of the rate limitation at low water contents may be on the donor or acceptor side. Note that the frequencies and relative amplitudes of the FTIR spectra of the S1 to S2 and S2 to S3 transitions are not altered by removal of water. These conditions of low water content have the advantage that potential background water contributions in the 1640 cm−1 region are reduced in magnitude, and this reduction in the contribution of background water enables us to obtain information about the S1 to S2 and S2 to S3 transition under these inhibited conditions.
Previous FTIR experiments have disagreed about the interpretation of the S2-S1 spectrum and the degree of interaction between calcium and manganese in the OEC. It was originally suggested, on the basis of their frequency, that bands at 1400 (−) and 1360 (+) cm−1 could be assigned to a Mn/Ca+2 bridging carboxylate ligand (35). However, a study in which other divalent metal ions were apparently substituted for Ca+2 was reported to have no significant effect on the 1800–1200 cm−1 region of the spectrum (37). In Chu et al. (36), this discrepancy is also discussed. By contrast, in our strontium exchange experiments, we report strontium-induced frequency shifts throughout the S2-S1 spectrum. This difference between our results and those previously reported is not due to hydration level, because we observe the same strontium-substituted changes in spectra from fully hydrated PSII (A. De Riso, D. L. Jenson, and B. A. Barry, unpublished results). The origin of this difference is under investigation.
Strontium-induced frequency shifts throughout the 1800–1200 cm−1 spectral region suggest that the majority of bands in the spectrum are affected by bond distance changes or changes in force constant when Mn is oxidized and strontium is substituted for calcium. This result is consistent with the idea that Mn and Ca+2 have bridging ligands, arising from amino acid side chains such as glutamate and aspartate, and that Mn oxidation on the S state transitions perturbs those bridging ligands. Note that although some Mn ligands have been assigned in PSII x-ray structures, the amino acid residues providing calcium ligation have not yet been assigned (16–19). Our result supports the idea that bridging Mn/Ca+2 ligands contribute to the 1800–1200 cm−1 region but also suggests that the spectral contributions caused by bond distance and force constant changes are complex. Local protein inhomogeneity may underlie this observation. This inhomogeneity may also underlie the width of spectral features, assigned to ligands to the Mn cluster and observed under illumination at 200 K. This inhomogeneity may originate in conformational degrees of freedom that are important for water oxidizing function (51,52,). Our previous work also showed that prolonged dark adaptation in the S1 state causes spontaneous, structural changes, again suggesting conformational flexibility in the OEC (44).
The FTIR data presented here provide comparative molecular information about the S1 to S2 and S2 to S3 transition. In the calcium-strontium data, the double difference spectrum constructed from the S1 to S2 spectra shows a complex set of derivative-shaped features. However, the double difference spectrum associated with the S2 to S3 transition is a relatively simple spectrum and reflects hydrogen bonding changes to one or more peptide bonds. Our 18OH2 data exhibit no 18O sensitive effects during the S1 to S2 transition but also generate a peptide bond perturbation spectrum during the S2 to S3 transition (see below). In each case, the derived double difference spectrum exhibits bands at 1675 (+), 1644/2 (−), 1556/49 (+), 1538/36 (−), and 1507/4 (+) cm−1, which are characteristic of perturbations to the C=O stretching (amide I) and C-N stretching/NH bending (amide II) bands of the peptide bond (49). A possible assignment for the S2 to S3 double difference spectrum is that a 1675 cm−1 band originating from the S3 state of the Ca+2-containing sample shifts to give a 1644/2 cm−1 band in the S3 state of the Sr+2-containing sample. The pattern of amide II shifts is consistent with a downshift of a derivative-shaped amide II vibration (+/−) when strontium is substituted for calcium.
Peptide carbonyl oxygens, as well as aspartate and glutamate side chains, are likely first shell ligands for calcium ions in proteins (for examples, see Bjornson et al. (53), Szebenyi and Moffat (54), Ilag et al. (55), and Dudev et al. (56). Previous FTIR studies of the Ca+2-ATPase have identified bands in the amide I region, which are associated with binding of calcium to the protein (57). In the available x-ray structures of the OEC, the ligand environment of calcium has not been completely assigned (16–19), but mutagenesis experiments suggest that aspartate and glutamate residues in the D1 subunit provide ligation to PSII calcium (58,59). In addition, the 3.5 Å structure of PSII shows that the carboxyl terminus of the D1 subunit is close to calcium, and the authors have suggested that the carboxyl terminus may be a ligand to calcium at some point in the S state cycle ((18), but see Chu et al. (60)).
The S3-minus-S2 FTIR spectrum presented here is consistent either with the addition of a C=O ligand to Ca+2 in the S3 state or with a conformational change at the calcium site, which is sensitive to the addition of Sr+2. Free C=O vibrations (in the S2 state) are not expected to contribute to the double difference spectrum, because the vibrational bands of the free C=O group would not be sensitive to strontium substitution. The downshift of the ligating C=O vibration in the presence of strontium could be caused by an inductive effect, in which strontium increases the basicity or the partial negative charge of the carbonyl oxygen, relative to the calcium-bound C=O (61). This inductive effect is observed, for example, in the pKa values of aqua ions bound to strontium (13.2), when compared to aqua ions bound to calcium (12.8; see Vrettos et al. (32) and references therein). This inductive effect decreases the frequency of the C=O stretching vibration, because ionic bonds are longer than covalent bonds. The larger ionic radius of strontium is also expected to decrease the M-O vibrational frequency, which is too low to be directly observed in our experiments (62).
The amide II bands exhibit a distinct positive-negative-positive pattern, compared to the derivative-shaped amide I bands. A positive-negative-positive band shape is consistent with the downshift of a derivative-shaped band in the S3-minus-S2 spectrum when strontium is substituted for calcium. The magnitude of the downshift is ∼10–20 cm−1. This spectral signature suggests that the amide II perturbation may be due to an additional interaction between the calcium ligation shell and one or more peptide bonds in the OEC or to a protein conformational change. This interaction may be due to a change in the hydrogen bonding of peptide NH groups in the calcium binding site. The fact that this amide II perturbation spectrum is observed only during the S2 to S3 transition suggests that hydrogen bonding is altered during this part of the S state cycle. Possible mechanisms which might give rise to such a perturbation have been discussed (3,5,7,10,29).
18OH2 water substitution results in an indistinguishable S3-minus-S2 double difference spectrum, when compared to calcium substitution. Hydrogen bond distances are sensitive to 18O substitution (63). Therefore, we hypothesize that changes in solvent-OEC hydrogen bonding slightly alter the ligand environment of the calcium site and lead to the double difference spectrum that we observe in 18OH2. However, this effect must be an indirect one, due to small changes in geometry at the calcium site, because the bond distances and force constants of water itself are not perturbed. For example, we observe no 18O sensitive bands in the 3500 cm−1 region under these conditions (64). An analogous situation is observed for enzymes in 2H2O buffers, which can induce small changes in the equilibrium structure of proteins due to changes in the hydrogen bonding distances (reviewed in Schowen and Schowen (65)).
In previous work, Brudvig and co-workers (32) concluded that the calcium site in PSII is similar to EF hand sites in other calcium-binding enzymes, with the exception that the PSII site is less selective for cation size. Although many EF hand sites are composed only of amino acid ligands, the PSII calcium ion may also bind water (29,30). The mobility of ligating water may be the cause of increased structural flexibility at the PSII calcium site, compared to EF hand proteins (32), and may be the underlying cause of the protein conformational changes observed in this work.
In summary, our data provide evidence that the ligand environment of calcium is perturbed during the S2 to S3 transition under conditions of low water content and that the S1 to S2 transition perturbs bridging ligands between Mn and Ca+2.
Acknowledgments
This work was supported by National Science Foundation grant MCB 03-55421.
Abbreviations used: PSII, photosystem II; chl, chlorophyll; EPR, electron paramagnetic resonance; ESEEM, electron spin echo envelope modulation; FTIR, Fourier-transform infrared spectroscopy; OEC, oxygen-evolving complex of photosystem II.
References
- 1.Britt, R. D. 1996. Oxygen evolution. In Oxygenic Photosynthesis: The Light Reactions, Vol. 4. D. R. Ort and C. F. Yocum, editors. Kluwer Academic Publisher, Dordrecht, The Netherlands. 137–164.
- 2.Peloquin, J. M., K. A. Campbell, D. W. Randall, M. A. Evanchik, V. L. Pecoraro, W. H. Armstrong, and R. D. Britt. 2000. Mn-55 ENDOR of the S-2-state multiline EPR signal of photosystem II: implications on the structure of the tetranuclear Mn cluster. J. Am. Chem. Soc. 122:10926–10942. [Google Scholar]
- 3.Weng, T.-C., W.-Y. Hsieh, E. S. Uffelman, S. W. Gordon-Wylie, T. J. Collins, V. L. Pecoraro, and J. E. Penner-Hahn. 2004. XANES evidence against a manganyl species in the S3 state of the oxygen-evolving complex. J. Am. Chem. Soc. 126:8070–8071. [DOI] [PubMed] [Google Scholar]
- 4.Ono, T.-A., T. Noguchi, Y. Inoue, M. Kusunoki, T. Matsushita, and H. Oyanagi. 1992. X-ray detection of the period-four cycling of the manganese cluster in photosynthetic water oxidizing enzyme. Science. 258:1335–1337. [DOI] [PubMed] [Google Scholar]
- 5.Dau, H., L. Iuzzolino, and J. Dittmer. 2001. The tetra-manganese complex of photosystem II during its redox cycle—X-ray absorption results and mechanistic implications. Biochim. Biophys. Acta. 1503:24–39. [DOI] [PubMed] [Google Scholar]
- 6.Roelofs, T. A., W. Liang, M. L. Latimer, R. M. Cinco, A. Rompel, J. C. Andrews, K. Sauer, V. K. Yachandra, and M. P. Klein. 1996. Oxidation states of the manganese cluster during the flash-induced S-state cycle of the photosynthetic oxygen-evolving complex. Proc. Natl. Acad. Sci. USA. 93:3335–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Messinger, J., J. H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R. Cinco, K. L. Mc, B. E. Farlane, S. A. Pizarro, S. P. Cramer, K. Sauer, M. Klein, and V. K. Yachandra. 2001. Absence of Mn-center oxidation in the S2-S3 transition: implications for the mechanism of photosynthetic water oxidation. J. Am. Chem. Soc. 123:7804–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang, W., M. J. Latimer, H. Dau, T. A. Roelofs, V. K. Yachandra, K. Sauer, and M. P. Klein. 1994. Correlation between structure and magnetic spin state of the manganese cluster in the oxygen-evolving complex of photosystem II in the S2 state: determination by X-ray absorption spectroscopy. Biochemistry. 33:4923–4932. [DOI] [PubMed] [Google Scholar]
- 9.Karge, M., K.-D. Irrgang, and G. Renger. 1997. Analysis of the reaction coordinate of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O. Biochemistry. 36:8904–8913. [DOI] [PubMed] [Google Scholar]
- 10.Liang, W., T. A. Roelofs, R. M. Cinco, A. Rompel, M. J. Latimer, W. O. Yu, K. Sauer, M. P. Klein, and V. K. Yachandra. 2000. Structural change of the Mn cluster during the S2 to S3 state transition of the oxygen-evolving complex of photosystem II. Does it reflect the onset of water/substrate oxidation? Determination by Mn X-ray absorption spectroscopy. J. Am. Chem. Soc. 122:3399–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haumann, M., M. Grabolle, T. Neisius, and H. Dau. 2002. The first room-temperature X-ray absorption spectra of higher oxidation states of the tetra-manganese complex of photosystem II. FEBS Lett. 512:116–120. [DOI] [PubMed] [Google Scholar]
- 12.Yocum, C. F. 1992. The calcium and chloride requirements for photosynthetic water oxidation. In Manganese Redox Enzymes, V. L. Pecoraro, editor. VCH Publishers, New York. 71–83.
- 13.Penner-Hahn, J. E., R. M. Fronko, V. L. Pecoraro, C. F. Yocum, S. D. Betts, and N. R. Bowlby. 1990. Structural characterization of the manganese sites in the photosynthetic oxygen-evolving complex using X-ray absorption spectroscopy. J. Am. Chem. Soc. 112:2549–2557. [Google Scholar]
- 14.Latimer, M. J., V. J. DeRose, V. K. Yachandra, K. Sauer, and M. P. Klein. 1998. Structural effects of calcium depletion on the manganese cluster of photosystem II: determination by X-ray absorption spectroscopy. J. Phys. Chem. B. 102:8257–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, S. H., W. Gregor, J. M. Peloquin, M. Brynda, and R. D. Britt. 2004. Investigation of the calcium-binding site of the oxygen evolving complex of photosystem II using 87Sr ESEEM spectroscopy. J. Am. Chem. Soc. 126:7228–7237. [DOI] [PubMed] [Google Scholar]
- 16.Zouni, A., H.-T. Witt, J. Kern, P. Fromme, N. Krauß, W. Saenger, and P. Orth. 2001. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 409:739–743. [DOI] [PubMed] [Google Scholar]
- 17.Kamiya, N., and J.-R. Shen. 2003. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 Å resolution. Proc. Natl. Acad. Sci. USA. 100:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira, K. N., T. M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata. 2004. Architecture of the photosynthetic oxygen-evolving center. Science. 303:1831–1837. [DOI] [PubMed] [Google Scholar]
- 19.Biesiadka, J., B. Loll, J. Kern, K.-D. Irrgang, and A. Zouni. 2004. Crystal structure of cyanobacterial photosystem II at 3.2 Å resolution: a closer look at the Mn-cluster. Phys. Chem. Chem. Phys. 20:4733–4736. [Google Scholar]
- 20.Yocum, C. F. 1991. Calcium activation of photosynthetic water oxidation. Biochim. Biophys. Acta. 1059:1–15. [Google Scholar]
- 21.Boussac, A., P. Setif, and A. W. Rutherford. 1992. Inhibition of S3 state in calcium-depleted and chloride-depleted photosystem II. Biochemistry. 31:1224–1233. [DOI] [PubMed] [Google Scholar]
- 22.Hallahan, B. J., J. H. A. Nugent, J. T. Warden, and M. C. W. Evans. 1992. Investigation of the origin of the “S3” EPR signal from the oxygen-evolving complex of photosystem 2: the role of tyrosine Z. Biochemistry. 31:4562–4573. [DOI] [PubMed] [Google Scholar]
- 23.Ghanotakis, D. F., G. T. Babcock, and C. F. Yocum. 1984. Calcium reconstitutes high rates of oxygen evolution in polypeptide depleted photosystem II preparations. FEBS Lett. 167:127–130. [Google Scholar]
- 24.Boussac, A., J.-L. Zimmermann, and A. W. Rutherford. 1989. EPR signals from modified charge accumulation states of the oxygen evolving enzyme in Ca+2-deficient photosystem II. Biochemistry. 28:8984–8989. [DOI] [PubMed] [Google Scholar]
- 25.Westphal, K. L., N. Lydakis-Simantris, R. I. Cukier, and G. T. Babcock. 2000. Effects of Sr+2 substitution on the reduction rates of Yz· in PSII membranes—evidence for concerted hydrogen-atom transfer in oxygen evolution. Biochemistry. 39:16220–16229. [DOI] [PubMed] [Google Scholar]
- 26.Boussac, A., F. Rappaport, P. Carrier, J.-M. Verbavatz, R. Gobin, D. Kirilovsky, A. W. Rutherford, and M. Sugiura. 2004. Biosynthetic Ca+2/Sr+2 exchange in photosystem II oxygen-evolving enzyme of Thermosynechococcus elongatus. J. Biol. Chem. 279:22809–22819. [DOI] [PubMed] [Google Scholar]
- 27.Boussac, A., and A. W. Rutherford. 1988. Nature of the inhibition of the oxygen-evolving enzyme of photosystem II induced by NaCl washing and reversed by the addition of Ca2+ or Sr2+. Biochemistry. 27:3476–3483. [Google Scholar]
- 28.Matysik, J., A. Alia, G. Nachtegaal, H. J. van Gorkom, A. J. Hoff, and H. J. M. der Groot. 2000. Exploring the calcium-binding site in photosystem II membranes by solid-state 113Cd NMR. Biochemistry. 39:6751–6755. [DOI] [PubMed] [Google Scholar]
- 29.Pecoraro, V. L., M. J. Baldwin, M. T. Caudle, W. Y. Hsieh, and N. A. Law. 1998. A proposal for water oxidation in photosystem II. Pure Appl. Chem. 70:925–929. [Google Scholar]
- 30.Vrettos, J. S., J. Limburg, and G. W. Brudvig. 2001. Mechanism of photosynthetic water oxidation: combining biophysical studies of photosystem II with inorganic model chemistry. Biochim. Biophys. Acta. 1503:229–245. [DOI] [PubMed] [Google Scholar]
- 31.Hendry, G., and T. Wydrzynski. 2003. 18O isotope exchange measurements reveal that calcium is involved in the binding of one substrate-water molecule to the oxygen-evolving complex in photosystem II. Biochemistry. 42:6209–6217. [DOI] [PubMed] [Google Scholar]
- 32.Vrettos, J. S., D. A. Stone, and G. W. Brudvig. 2001. Quantifying the ion selectivity of the calcium site in photosystem II: evidence for direct involvement of Ca+2 in O2 formation. Biochemistry. 40:7937–7945. [DOI] [PubMed] [Google Scholar]
- 33.Hillier, W., and G. Babcock. 2001. S-state dependent Fourier transform infrared difference spectra for the photosystem II oxygen evolving complex. Biochemistry. 40:1503–1509. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi, T., T. Tomo, and C. Kato. 2001. Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry. 40:1497–1502. [DOI] [PubMed] [Google Scholar]
- 35.Noguchi, T., T.-a. Ono, and Y. Inoue. 1995. Direct detection of a carboxylate bridge between Mn and Ca+2 in the photosynthetic oxygen-evolving center by means of Fourier transform infrared spectroscopy. Biochim. Biophys. Acta. 1228:189–200. [Google Scholar]
- 36.Chu, H.-A., W. Hillier, N. A. Law, and G. T. Babcock. 2001. Vibrational spectroscopy of the oxygen-evolving complex and of manganese model compounds. Biochim. Biophys. Acta. 1503:69–82. [DOI] [PubMed] [Google Scholar]
- 37.Kimura, Y., K. Hasegawa, and T.-a. Ono. 2002. Characteristic changes of the S2/S1 difference FTIR spectrum induced by Ca+2 depletion and metal cation substitution in the photosynthetic oxygen-evolving complex. Biochemistry. 41:5844–5853. [DOI] [PubMed] [Google Scholar]
- 38.Noguchi, T., and M. Sugiura. 2002. Flash-induced FTIR difference spectra of the water oxidizing complex in moderately hydrated photosystem II core films: effect of hydration extent on S-state transitions. Biochemistry. 41:2322–2330. [DOI] [PubMed] [Google Scholar]
- 39.Berthold, D. A., G. T. Babcock, and C. F. Yocum. 1981. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 134:231–234. [Google Scholar]
- 40.Ghanotakis, D. F., J. N. Topper, G. T. Babcock, and C. F. Yocum. 1984. Water-soluble 17 and 23 kDa polypeptides restore oxygen evolution activity by creating a high-affinity binding site for Ca+2 on the oxidizing side of photosystem II. FEBS Lett. 170:169–173. [Google Scholar]
- 41.Barry, B. A. 1995. Tyrosyl radicals in photosystem II. Methods Enzymol. 258:303–319. [DOI] [PubMed] [Google Scholar]
- 42.Halverson, K. M., and B. A. Barry. 2003a. Sucrose and glycerol effects on photosystem II. Biophys. J. 85:1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piccioni, R., G. Bellemare, and N. Chua. 1982. Methods of polyacrylamide gel electrophoresis in the analysis and preparation of plant polypeptides. In Methods in Chloroplast Molecular Biology. H. Edelman, R. B. Hallick, and N.-H. Chua, editors. Elsevier, Amsterdam. 985–1014.
- 44.Halverson, K. M., and B. A. Barry. 2003b. Evidence for spontaneous structural changes in a dark-adapted state of photosystem II. Biophys. J. 85:2581–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sachs, R., K. M. Halverson, and B. A. Barry. 2003. Specific isotopic labeling and photooxidation-linked structural changes in the manganese-stabilizing subunit of photosystem II. J. Biol. Chem. 278:44222–44229. [DOI] [PubMed] [Google Scholar]
-
46.Zhang, H., G. Fischer, and T. Wydrzynski. 1998. Room-temperature vibrational difference spectrum for
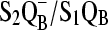 of photosystem II determined by time-resolved Fourier transform infrared spectroscopy. Biochemistry. 37:5511–5517. [DOI] [PubMed] [Google Scholar]
of photosystem II determined by time-resolved Fourier transform infrared spectroscopy. Biochemistry. 37:5511–5517. [DOI] [PubMed] [Google Scholar] - 47.Kim, S., and B. A. Barry. 1998. The protein environment surrounding tyrosyl radicals D· and Z· in photosystem II: a difference FTIR study. Biophys. J. 74:2588–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamanari, T., Y. Kimura, N. Mizusawa, A. Ishii, and T.-a. Ono. 2004. Mid- to low-frequency Fourier transform infrared spectra of S-state cycle for photosynthetic water oxidation in Synechocystis sp. PCC 6803. Biochemistry. 43:7479–7490. [DOI] [PubMed] [Google Scholar]
- 49.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. In Advances in Protein Chemistry, Vol. 38. C. B. Anfinsen, J. T. Edsall, and F. M. Richards, editors. Academic Press, New York. 181–364. [DOI] [PubMed]
- 50.Noguchi, T., and M. Sugiura. 2003. Analysis of flash-induced FTIR difference spectra of the S-state cycle in the photosynthetic water-oxidizing complex by uniform 15N and 13C isotope labeling. Biochemistry. 42:6035–6042. [DOI] [PubMed] [Google Scholar]
- 51.Steenhuis, J. J., and B. A. Barry. 1998. An FTIR study of photoassembly in the manganese catalytic site of photosystem II (Letter). J. Phys. Chem. 102:4–8. [Google Scholar]
- 52.Steenhuis, J. J., and B. A. Barry. 1997. The protein and ligand environment of the S2 state in photosynthetic oxygen evolution: a difference FTIR study. J. Phys. Chem. 101:6652–6660. [Google Scholar]
- 53.Bjornson, M. E., D. C. Corson, and B. D. Sykes. 1985. 13C and 113Cd NMR studies of the chelation of metal ions by the calcium binding protein parvalbumin. J. Inorg. Biochem. 25:141–149. [DOI] [PubMed] [Google Scholar]
- 54.Szebenyi, D. M., and K. Moffat. 1986. The refined structure of vitamin D-dependent calcium-binding protein from bovine intestine. Molecular details, ion binding, and implications for the structure of other calcium-binding proteins. J. Biol. Chem. 261:8761–8777. [PubMed] [Google Scholar]
- 55.Ilag, L. L., R. McKenna, M. P. Yadav, J. N. BeMiller, N. L. Incardona, and M. G. Rossmann. 1994. Calcium ion-induced structural changes in bacteriophage phi X174. J. Mol. Biol. 244:291–300. [DOI] [PubMed] [Google Scholar]
- 56.Dudev, T., Y. Lin, M. Dudev, and C. Lim. 2003. First-second shell interactions in metal binding sites in proteins: a PDB survey and DFT/CDM calculations. J. Am. Chem. Soc. 125:3168–3180. [DOI] [PubMed] [Google Scholar]
- 57.Troullier, A., K. Gerwert, and Y. Dupont. 1996. A time-resolved Fourier transform infrared difference spectroscopy study of the sarcoplasmic reticulum Ca+2-ATPase: kinetics of the high-affinity calcium binding at low temperature. Biophys. J. 71:2970–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chu, H. A., A. P. Nguyen, and R. J. Debus. 1995. Amino acid residues that influence the binding of manganese or calcium to photosystem II. 2. The carboxy-terminal domain of the D1 polypeptide. Biochemistry. 34:5859–5882. [DOI] [PubMed] [Google Scholar]
- 59.Chu, H. A., A. P. Nguyen, and R. J. Debus. 1995. Amino acid residues that influence the binding of manganese or calcium to photosystem II. 1. The lumenal interhelical domains of the D1 polypeptide. Biochemistry. 34:5839–5858. [DOI] [PubMed] [Google Scholar]
- 60.Chu, H.-A., W. Hillier, and R. J. Debus. 2004. Evidence that the C-terminus of the D1 polypeptide of photosystem II is ligated to the manganese ion that undergoes oxidation during the S1 to S2 transition: an isotope-edited FTIR study. Biochemistry. 43:3152–3166. [DOI] [PubMed] [Google Scholar]
- 61.Bellamy, L. J. 1980. The Infrared Spectra of Complex Molecules. Chapman and Hall, London.
- 62.Bopegedera, A. M. R. P., W. T. M. L. Fernando, and P. F. Bernath. 1990. Gas-phase inorganic chemistry: laser spectroscopy of calcium and strontium monoformamidates. J. Phys. Chem. 94:3547–3549. [Google Scholar]
- 63.Pinchas, S., and J. Shamir. 1969. Anomalous behavior of oxygen-18 labeled compounds. IV. Vibrational spectra of isotopic vanadate ions. Isr. J. Chem. 7:805–811. [Google Scholar]
- 64.Noguchi, T., and M. Sugiura. 2000. Structure of an active water molecule in the water-oxidizing complex of photosystem II as studied by FTIR spectroscopy. Biochemistry. 39:10943–10949. [DOI] [PubMed] [Google Scholar]
- 65.Schowen, K. B., and R. L. Schowen. 1982. Solvent isotope effects of enzyme systems. Methods Enzymol. 87:551–606. [PubMed] [Google Scholar]