

Abstract
A new strategy was established to determine the average orientation and dynamics of ergosterol in dimyristoylphosphatidylcholine model membranes. It is based on the analysis of chemical shift anisotropies (CSAs) averaged by the molecular dynamics. Static 13C CSA tensors were computed by quantum chemistry, using the gauge-including atomic-orbital approach within Hartree-Fock theory. Uniformly 13C-labeled ergosterol was purified from Pichia pastoris cells grown on labeled methanol. After reconstitution into dimyristoylphosphatidylcholine lipids, the complete 1H and 13C assignment of ergosterol's resonances was performed using a combination of magic-angle spinning two-dimensional experiments. Dynamically averaged CSAs were determined by standard side-band intensity analysis for isolated 13C resonances (C3 and ethylenic carbons) and by off-magic-angle spinning experiments for other carbons. A set of 18 constraints was thus obtained, from which the sterol's molecular order parameter and average orientation could be precisely defined. The validity of using computed CSAs in this strategy was verified on cholesterol model systems. This new method allowed us to quantify ergosterol's dynamics at three molar ratios: 16 mol % (Ld phase), 30 mol % (Lo phase), and 23 mol % (mixed phases). Contrary to cholesterol, ergosterol's molecular diffusion axis makes an important angle (14°) with the inertial axis of the rigid four-ring system.
INTRODUCTION
Cholesterol and ergosterol are well-known sterols, which play an essential role in the cell membrane of eukaryotic organisms. Cholesterol is the major sterol found in animal cells membrane and ergosterol is the predominant sterol in fungi and yeast; and they are absolutely required for viability and cell proliferation (1–4). One of the primary roles of sterols in eukaryotic cells is to modulate the physical properties of the plasma membrane phospholipid bilayer (2). For example, they reduce molecular surface area (5–9) and membrane permeability (2,10–12), alter lateral diffusion rates of proteins and lipids (13–17), broaden gel to liquid-crystalline phase transition (18–21) and modulate acyl-chain order in both the gel and liquid-crystalline phases (22–25). Many studies have been carried out to gain understanding of the molecular basis of cholesterol's biological properties (for excellent recent reviews on physical studies of cholesterol-phospholipid interactions, see Refs. 26 and 27). In contrast, there are very few detailed studies on effects of ergosterol on physical properties of phospholipid bilayer membranes. Early monolayer studies (28) noted that ergosterol was much less effective than cholesterol in condensing egg-phosphatidylcholine (PC) monolayers, whereas Demel and De Kruyff (10) found that ergosterol was less effective than cholesterol in reducing the permeability of egg-PC liposomes to glycerol, glucose, and rubidium ions. Semer and Gelerinter (24), using electron-paramagnetic-resonance of doxyl-fatty acid and cholestane spin-label probes in egg-PC membrane, reported the observation that ergosterol, in contrast with cholesterol, only ordered the acyl chain up to 15–20 mol %; above this percentage, ergosterol induced disorder of the acyl-chains. Ergosterol has a very low solubility (<3%) into soybean PC (containing 63% of di-unsaturated fatty acid) compared to cholesterol, which can be incorporated up to 23% (29). Urbina et al. (25) found that the ordering effects are slightly higher for ergosterol than for cholesterol, in sterol/dimyristoylphosphatidylcholine (DMPC) membrane at 30 mol % and at 25°C. More recently, Hsueh et al. (30) have determined the complete phase diagram of ergosterol/dipalmitoylphosphatidylcholine mixtures by deuterium nuclear-magnetic-resonance (NMR) and calorimetry.
Three main features of sterols have been linked to their characteristic effects on lipid bilayer membrane: a planar four-fused-ring motif, a 3β-OH group, and a hydrophobic side chain linked to C17. However, the mechanisms by which the slight differences in structures between cholesterol and ergosterol may be responsible for modification in the physical properties of the bilayer and in the sterol's dynamics are still largely unknown. Although the average orientation and dynamics of cholesterol have been well established by deuterium NMR (see Ref. 22 and references cited herein), very little is known concerning ergosterol on this aspect.
Solid-state NMR is uniquely suited to the determination of orientation, dynamics, and molecular structure of membrane compounds. Combination of solid NMR methods and stable isotope 15N labeling of the amide nitrogens (31–35) or 13C labeling of the amide carbonyls (36,37), are currently used to obtain orientational and structural constraints for membrane peptides and proteins. In the case of sterol, it has been shown that cholesterol order parameters can be extracted from the quadrupole splittings of specifically deuterated positions (22). This strategy has proven to be very powerful but requires critical steps such as chemical synthesis of specifically deuterated positions and assignment of the obtained quadrupolar splittings to be performed. 13C NMR could be an interesting alternative to study such compounds due to the large carbon chemical shift range and the sensitivity enhancement brought about by cross-polarization (CP) from neighboring protons. Moreover, modulation of biosynthetic labeling strategies can be used to increase both sensitivity and selectivity. In particular, methylotrophic yeast has been shown to be an excellent tool for the cost-efficient production of uniformly labeled biomass and membrane proteins (38–40). Another advantage for NMR is that this yeast can be easily grown on a minimal medium with methanol, i.e., a C1 compound as a sole carbon source. Randomly distributed 13C labeling to any level is therefore possible, and in particular low level 13C labeling (11%) is suitable to measure carbon chemical shifts or H–C dipolar couplings on isolated spin pairs with a reasonable sensitivity.
The 13C chemical shift anisotropy (CSA) is dependent on both molecular structure and dynamics and therefore represents a powerful probe of these two parameters. Furthermore, recent developments and implementation in quantum chemistry allow the accurate determination of theoretical chemical shift tensors (for reviews, see Refs. 41 and 42).
In this article, by combining quantum chemistry calculations with various magic-angle spinning (MAS) solid-state NMR experiments, carbon chemical shift anisotropies were used as restraints to probe orientation and dynamics of uniformly 13C-labeled ergosterol in DMPC membrane. The complete proton and carbon assignment of ergosterol in membrane has been achieved, based on a combination of one-dimensional and two-dimensional 13C−1H heteronuclear (J- and dipolar-heteronuclear correlation, i.e., HETCOR) and 13C−13C homonuclear (incredible-natural-abundance-double-quantum-transfer experiment; i.e., INADEQUATE) MAS experiments. Using this strategy we could determine ergosterol's order parameter and diffusion axis in the two major coexisting phases in ergosterol/DMPC mixtures, i.e., Ld and Lo phases. We analyzed at 313 K three different ergosterol/DMPC molar ratio; 16 mol % ergosterol (pure Ld phase), 30 mol % (pure Lo phase), and 23 mol % (two-phase coexistence) according to Hsueh et al. (30).
MATERIAL AND METHODS
Growth condition and culture media
Fermentation of Pichia pastoris methylotrophic yeast was used to produce ergosterol. The inoculum for fermentation was obtained by growing this microorganism in a liquid medium containing 2% (v/v) methanol, 3 g/l KH2PO4, 0.2 g/l MgSO4 7H2O, 1.5 g/l (NH4)2SO4, 2 ml/l of a trace element solution (50 g/l EDTA, 6 g/l ZnSO4 7H2O, 5.4 g/l CuCl2 2H2O, 0.39 g/l MnSO4 H2O, 4 g/l FeSO4 7H2O, 0.06 g/l (NH4)2 Mo2O7, 1.2 g/l CuSO4 5H2O, 0.8 g/l CoCl2 6H2O) and 1 ml/l vitamin solution (5 mg/l biotin and 300 mg/l thiamine chlorohydrate). The methanol and the vitamin solutions were sterilized by filtration. The pH was set to 6 with KOH 3N. Shake flask culture (200 ml) was performed at 37°C until OD640 reached 1. The production of large amount of 11% randomly labeled ergosterol was carried out in a 2.5-l fermentor. The initial fermentation culture media contained 10 g/l KH2PO4, 1 g/l MgSO4, 10 g/l (NH4)2SO4, 12.5 ml/l of trace element solution, 1 ml/l of vitamin solution, and 0.5% (v/v) of methanol solution (10% of 13C methanol and 90% of natural abundance methanol). The pH was regulated at 6.0 by addition of KOH 3N. The temperature was set to 37°C. During the fed batch phases, the flow rate of methanol was increased from 3 g/h to 30 g/h. At the end of fermentation, the methanol total consumption was 300 g. When the OD640 was 50, 50g of (NH4)2SO4 were added. The O2 pressure was regulated by increasing airflow and stirring speed. To maintain the dissolved O2 level above 25%, 40% oxygen-enriched air was used when the OD640 reached 70. At the end of fermentation, the cells were harvested by centrifugation and lyophilized.
Purification from Pichia pastoris yeast cells, and chemical characterization
Pichia pastoris yeast cells (10 g) were broken by freeze/thaw cycles and delipidated in methanol/chloroform/acetone solvent mixture (1:1:2 v/v/v) during 4 h under reflux and argon atmosphere. Polar and neutral lipids were separated by precipitation in acetone (2 days, 4°C). The mixture was then centrifuged at 4000g for 5 min and the supernatant saponified in a 1:1:4 potassium hydroxide/water/ethanol mixture. Sterols were extracted with hexane and further purified using silicic column chromatography (20 g silica-gel 60; Merck, Whitehouse Station, NJ).
Before the gas chromatography/mass spectrometry (GC/MS) experiment, ergosterol was acetylated in a 1:1 anhydrous pyridine/acetic anhydride mixture. GC/MS spectra were acquired on an HP 5989X glass column and interfaced to a Hewlett-Packard 5989A mass spectrometer (Palo Alto, CA). Acetylated ergosterol derivatives were analyzed by electron-impact/mass spectroscopy (ion source temperature 250°C, ionization voltage 70 eV). Labeling extent (10.7%) was calculated from GC/MS spectra on molecular ion (m/z = 441) and deacetylated fragment (m/z = 381). From GC/MS spectra, another minor sterol was identified (ergosta-5-7-22-24-tetraen-3β-ol, m/z = 379), which represented 4.8% of the total sterol content.
Sample preparation
Samples were prepared by drying down under nitrogen chloroform solution containing perdeuterated acyl chains dimyristoylphosphatidylcholine (DMPC-d54; Avanti Polar Lipids, Alabaster, AL) and purified ergosterol in the desired proportions (8.4:1.6, 7.7:2.3, and 7.0:3.0 mol/mol), then eliminating residual solvent under high vacuum overnight. The lipids were resuspended using deuterium-depleted water at 33 wt % solids, then subjected to five mixing and freeze (253 K) thaw (323 K) cycles, to ensure homogeneity of the multilamellar vesicles as described in Urbina et al. (25). Before NMR spectroscopy, sample hydration was adjusted at 50% weight by adding the appropriate amount of deuterium-depleted water. It was checked by deuterium NMR that the lipid chain's quadrupolar splittings had values in good accordance with those reported previously in the literature at equivalent sterol/lipid molar ratio (25). Spectra similar to those of Hsueh et al. (30) were obtained confirming the presence of Ld phase at 16 mol % ergosterol, Lo phase at 30 mol %, and Ld + Lo phase mixture at 23 mol % at 313 K (data not shown).
Solid-state NMR spectroscopy
NMR experiments were carried out on a Bruker Avance 500 spectrometer (Bruker Optics, Billerica, MA) equipped with a narrow-bore magnet operating at a resonance frequency of 125 MHz for 13C and 500.13 MHz for 1H. All solid-state NMR experiments were carried out at 313 K.
Static deuterium NMR spectra were recorded on a Bruker 7-mm double-resonance probe, with a solenoid coil oriented at 90° with respect to the magnetic field. The acyl-chain quadrupolar splittings of lipids were recorded by using a standard quadrupolar echo sequence. Deuterium π/2 pulses were equal to 4 μs; and the refocusing delay and the repetition time were set to 30 μs and 1 s, respectively.
MAS experiments were recorded with a double-tuned Doty XC5-MAS probe (Doty Scientific, Columbia, SC) equipped with a 5-mm spinning module. The 1H radio-frequency field-strength for heteronuclear two-pulse phase-modulation decoupling was 66 kHz for all experiments. All spectra were acquired at MAS spinning rate of 9 kHz with a repetition delay of 3.5 s to avoid sample heating.
Cross-polarization magic-angle spinning (CP-MAS) and cross-polarization off-magic-angle spinning (CP-OMAS) spectra were acquired using a 1H excitation pulse length of 4 μs and a CP spin-lock field-strength of ∼50 kHz. The CP contact time was 2 ms. The relaxation delay was set to 2 s for all experiments.
Two-dimensional non-refocused INADEQUATE (43) was obtained by recording 100 t1 increments with 320 scans each. The τ-delay was set equal to 0.86 ms. Contact time for the CP period and 1H radio-frequency field were identical to one-dimensional CP spectra.
Dipolar HETCOR spectra were obtained at a 9-kHz spinning rate. CP contact times were set to 2.5 ms or 250 μs. A total of 128 t1 increments with 50-μs dwell-time and 128 scans each, were recorded for each experiment.
Quantum chemistry calculation
The three β-hydroxyl rotameric ergosterol structures (gauche−, gauche+, and anti−; see Fig. 1) were optimized at the Hartree-Fock level using the STO-3G basis set. The carbon-shielding tensor was then computed by using the gauge-including-atomic-orbital approach within Hartree-Fock theory (6-31G(d,p) basis set). All calculations have been performed using the Gaussian 98 package (44). It has been previously shown that this computational strategy is sufficient to reproduce the experimental 1H and 13C isotropic NMR spectra of ergosterol in chloroform solution (45). The comparison of theoretical chemical shifts with those experimentally obtained has unambiguously revealed that one must take the theoretical average values among three isoenergetic β-hydroxyl rotamers to reproduce the 1H and 13C NMR spectra of ergosterol. Averaged carbon chemical shift tensors are thus calculated in a similar way. Diagonalization of the symmetric part of this tensor provides the principal values (δ11, δ22, and δ33) and their associated principal vectors  . The principal values are ordered as
. The principal values are ordered as  where the isotropic chemical shift is equal to
where the isotropic chemical shift is equal to  The static chemical shift anisotropy and the asymmetry parameter are defined as Δδstatic = δ33 − δiso and
The static chemical shift anisotropy and the asymmetry parameter are defined as Δδstatic = δ33 − δiso and  respectively. The principal vectors define the orientation of the chemical shift tensor principal axis system (denoted PASCS) in the molecular frame (denoted M and defined as
respectively. The principal vectors define the orientation of the chemical shift tensor principal axis system (denoted PASCS) in the molecular frame (denoted M and defined as 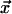 axis along the
axis along the  bond,
bond,  along
along 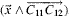 , and
, and 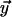 along
along  ). The molecular frame M is independent of the rotameric states and was thus preferred to the C3 molecular frame defined in Marsan et al. (22). The (
). The molecular frame M is independent of the rotameric states and was thus preferred to the C3 molecular frame defined in Marsan et al. (22). The (
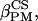 and
and  ) Euler angles characterize the rotation from PASCS to M (Fig. 2).
) Euler angles characterize the rotation from PASCS to M (Fig. 2).
FIGURE 1.

Newman representation of the three rotational isomers around the C3−O bond. These isomers are discriminated according to the C2−C3−O−H dihedral angle.
FIGURE 2.

Schematic representation of frame transformation and Euler angles definition. The values 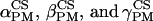 are the Euler angles associated to the rotation matrix (RPM) between the chemical shift tensor principal axis system PASCS and the molecular frame M (
are the Euler angles associated to the rotation matrix (RPM) between the chemical shift tensor principal axis system PASCS and the molecular frame M (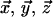 ). Columns of the RPM matrix correspond to chemical shift tensor principal axis defined in the molecular frame. Note that (α, β, and 0) are the Euler angles associated to the rotation matrix (RMN) between M and the reorientation frame (N). The values α and β correspond to polar coordinates of the ergosterol diffusion axis (
). Columns of the RPM matrix correspond to chemical shift tensor principal axis defined in the molecular frame. Note that (α, β, and 0) are the Euler angles associated to the rotation matrix (RMN) between M and the reorientation frame (N). The values α and β correspond to polar coordinates of the ergosterol diffusion axis ( ) in the molecular frame. The values (
) in the molecular frame. The values ( ) are the Euler angles corresponding to the rotation from PASCS to N. The values (
) are the Euler angles corresponding to the rotation from PASCS to N. The values (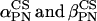 ) correspond to the polar coordinates of the ergosterol diffusion axis
) correspond to the polar coordinates of the ergosterol diffusion axis 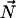 in PASCS. These angles are extracted from the rotation matrix RPN calculated as the product of RPM and RMN.
in PASCS. These angles are extracted from the rotation matrix RPN calculated as the product of RPM and RMN.
Static chemical shift tensor parameters are listed in Table 1 for ergosterol carbons 1–19 (see ergosterol structure in Fig. 3).
TABLE 1.
Theoretical static chemical shift tensor parameters for ergosterol ring carbons
| Carbon number | δ11, δ22, δ33 (ppm) | Δδstatic (ppm) | η | αPM, βPM, γPM (degrees) | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 55.9 | 38.5 | 18.5 | −19.1 | 0.91 | −183.4 | 129.4 | −53.3 |
| 2 | 44.7 | 34.8 | 17.1 | −15.1 | 0.66 | −113.4 | 138.3 | 75.8 |
| 3 | 90.2 | 81.5 | 32.3 | −35.7 | 0.24 | −95.6 | 94.8 | 75.5 |
| 4 | 59.4 | 41.9 | 20.3 | −20.2 | 0.87 | −68.1 | 63.8 | 89.4 |
| 5 | 35.4 | 128.2 | 247.4 | 110.4 | 0.84 | −100.9 | 118.4 | 82.1 |
| 6 | 38.5 | 96.9 | 224.3 | 104.4 | 0.56 | −104.3 | 129.4 | 80.6 |
| 7 | 29.0 | 96.9 | 221.8 | 105.9 | 0.64 | −79.9 | 54.0 | 78.9 |
| 8 | 33.4 | 136.8 | 247.8 | 108.4 | 0.95 | −75.9 | 60.9 | 73.9 |
| 9 | 30.1 | 41.3 | 62.9 | 18.1 | 0.61 | −19.0 | −70.4 | 19.5 |
| 10 | 31.0 | 34.4 | 45.9 | 8.8 | 0.38 | −198.6 | 131.1 | −29.7 |
| 11 | 14.4 | 20.6 | 35.2 | 11.8 | 0.53 | 69.1 | −100.4 | −12.5 |
| 12 | 48.8 | 40.8 | 22.1 | −15.1 | 0.53 | 14.8 | −99.1 | −32.1 |
| 13 | 28.7 | 36.9 | 57.6 | 16.5 | 0.50 | 76.9 | −74.8 | −15.9 |
| 14 | 69.3 | 50.4 | 31.3 | −19.1 | 0.99 | 68.7 | −121.0 | −85.1 |
| 15 | 10.3 | 20.8 | 44.0 | 19.0 | 0.56 | 85.6 | −74.0 | −1.5 |
| 16 | 9.4 | 22.1 | 55.9 | 26.8 | 0.48 | −84.7 | 147.1 | 11.0 |
| 17 | 69.6 | 59.2 | 28.5 | −23.9 | 0.43 | −111.6 | 93.9 | 77.0 |
| 18 | 22.7 | 18.2 | 5.9 | −9.7 | 0.46 | −83.4 | 48.4 | −192.2 |
| 19 | 8.4 | 16.6 | 38.1 | 17.0 | 0.48 | −77.9 | 80.7 | 81.8 |
The chemical shift principal values are given in parts per million relative to the absolute shielding of tetramethylsilane (TMS) (σTMS = 204.66 ppm) calculated at the same theoretical level.
FIGURE 3.

Structure of ergosterol. Carbons atoms are labeled with a number; hydrogen atoms, with the exception of the hydrogen of the hydroxyl group, are not shown.
Simulation of dynamically averaged chemical shift anisotropy
The carbon chemical shift value is given, in the case of fast axial diffusion along the normal bilayer 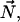 by (46)
by (46)
 |
(1) |
where βlayer is the angle between the bilayer normal and the B0 magnetic field axis, Smol the molecular order parameter (order parameter describing the amplitude of wobbling of the entire rigid moiety), and Sloc is a local order parameter to account for internal motions. It can be set equal to 1 for every ring atom as was shown previously (22). The values Δδstatic and η represent the static chemical shift anisotropy and the asymmetry parameters, respectively. The values (
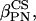 and
and 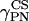 ) are the Euler angles associated to the rotation between the chemical shift tensor principal axis system (PASCS) and the diffusion frame (denoted N). The z′ axis of the N frame is along the ergosterol axis of diffusion (
) are the Euler angles associated to the rotation between the chemical shift tensor principal axis system (PASCS) and the diffusion frame (denoted N). The z′ axis of the N frame is along the ergosterol axis of diffusion (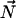 ). The values α, β are the polar coordinates of
). The values α, β are the polar coordinates of  in the molecular frame M. The angles (
in the molecular frame M. The angles (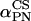 and
and  ) are then depending on both (
) are then depending on both (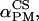
 and
and 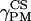 ), i.e., rotation from PASCS to M, and (α, β), i.e., orientation of N in M (see Fig. 2).
), i.e., rotation from PASCS to M, and (α, β), i.e., orientation of N in M (see Fig. 2).
In this article, theoretical carbon chemical shift anisotropy Δδcalc in the presence of fast axial diffusion is defined as
 |
(2) |
where δ90° and δ0° are the chemical shifts calculated for βlayer = 90° and βlayer = 0°, following Eq. 1.
Using static chemical shift tensor parameters obtained by quantum chemical computations, sets of Δδcalc values have been calculated for each ergosterol ring carbon (Sloc = 1) by varying α, β and Smol from 0° to 360°, 0° to 90°, and 0 to 1, respectively. The increments were 1° for angles and 0.1 for Smol. The obtained data were filtered according to the root mean-square difference (RMSD) calculated as
 |
(3) |
with Nσ the number of experimental CSA data (Nσ = 18), and 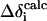 and
and  as the theoretical and experimental CSAs for the ith carbon, respectively. The values of
as the theoretical and experimental CSAs for the ith carbon, respectively. The values of  were determined as described in the next section.
were determined as described in the next section.
RESULTS AND DISCUSSION
Production of uniformly 10% and 100% 13C-labeled ergosterol
Methylotrophic yeast proved to be an excellent mean of producing randomly 13C-labeled ergosterol at any extent. Since it can be grown on methanol as a sole carbon source, the extent of labeling can be chosen simply by mixing adequate volumes of labeled and natural abundance methanol. Very high biomass (78 g) could be obtained in a few days, using 300 g of methanol. The purification of ergosterol from total lipids is straightforward, and it was found to be the major sterol, with a small contamination by a closely related sterol possessing one more double bond on its side chain (in position 24). Approximately 1 mg/g of cells (dry weight) could be purified. Two cultures were performed to obtain ergosterol with 10.7% and 99.5% 13C labeling extent.
Complete assignment of ergosterol in DMPC membrane
We have used a combination of one-dimensional and two-dimensional heteronuclear and homonuclear magic-angle spinning (MAS) solid-state NMR techniques—namely, scalar HETCOR, dipolar HETCOR, and INADEQUATE—to perform the complete assignment of ergosterol in membrane without any reference to the known liquid-state assignment (Table 2; structure of ergosterol in Fig. 3). The samples were 8.4:1.6 mol/mol DMPC-d54/ergosterol–11%13C multilamellar vesicles except for the INADEQUATE experiment for which ergosterol, 100% 13C-labeled, was used. Apart from the INADEQUATE experiment, the assignment strategy was identical to that recently described in the case of natural-abundance cholesterol in DMPC membrane (47). Two-dimensional pulse sequences used in this work are shown in Fig. 4. Carbon multiplicities have been determined by comparing one-dimensional carbon spectra recorded either via a CP scheme or via an insensitive-nuclei-enhanced-by-polarization-transfer (INEPT) scheme by varying the second delay of the refocusing period (Fig. 5). 1H−13C one-bond correlations were obtained via a liquidlike HETCOR experiment by using a refocused INEPT scheme. Skeletal information has been recovered by combining homonuclear 13C−13C correlations from the INADEQUATE experiment and long-range 1H-13C correlation from the dipolar HETCOR experiment. Fig. 6 shows the INADEQUATE spectrum of 8.4:1.6 perdeuterated acyl-chain DMPC/100% 13C-labeled ergosterol recorded in a total experimental time of 16 h. In the two-dimensional map, two directly bonded carbons share a common frequency in the double-quantum dimension. Starting from the easily identifiable resonance of the hydroxyl carbon C3 at 69.5 ppm, it is straightforward to sequentially assign the complete carbon spectrum. Two sets of correlation peaks are possible for the C3 connectivities (C3−C2, C3−C4), one at 31.5 ppm and another at 39.65 ppm. On the basis of chemical shift considerations, the upfield carbon signal at 39.65 ppm is identified as the methylene C4 carbon. Another way is to remark that each of them has a second clear correlation in the double-quantum frequency dimension with either a methylene for C2 (hence C1 at 38.32 ppm) or a quaternary C-signal (hence C5 at 141.18 ppm, data not shown) for the C4. Interestingly, our assignment differs in one point from what was obtained in the liquid state. In deuterated chloroform solution, carbon C8 was found to resonate at a higher frequency than carbon C5. This inversion was confirmed by analyzing the olefinic region of the dipolar HETCOR experiment (Fig. 7). This discrepancy underlines the importance of assigning compounds in their actual environment. These differences are currently under investigation by analyzing the environment effect (hydrogen bonding and rotameric state of the hydroxyl group) by combining solid-state NMR assignments with quantum chemistry calculations (47).
TABLE 2.
Assignment of carbon and proton chemical shifts for ergosterol inserted in a 8.4:1.6 (mol/mol) DMPC-d54/ergosterol lipid mixture
| Carbon number | 13C-chemical shift (ppm) | 1H-chemical shift (ppm) | Carbon number | 13C-chemical shift (ppm) | 1H-chemical shift (ppm) |
|---|---|---|---|---|---|
| 1 | 38.32 | 1.28a/1.8e | 15 | 23.19 | 1.68 |
| 2 | 31.15 | 1.52a/1.86e | 16 | 28.29 | 1.28/1.72 |
| 3 | 69.11 | 3.55 | 17 | 55.67 | 1.25 |
| 4 | 40.06 | 2.25/2.49 | 18 | 12.02 | 0.66 |
| 5 | 141.18 | — | 19 | 15.65 | 0.91 |
| 6 | 119.05 | 5.5 | 20 | 40.97 | 2.009 |
| 7 | 117.05 | 5.27 | 21 | 21.51 | 1.02 |
| 8 | 139.20 | — | 22 | 135.71 | 5.065 |
| 9 | 45.97 | 1.91 | 23 | 131.84 | 5.1 |
| 10 | 36.97 | — | 24 | 43.23 | 1.8 |
| 11 | 20.81 | 1.64 | 25 | 32.85 | 1.38 |
| 12 | 39.35 | 1.32a/2.018e | 26 | 17.76 | 0.89 |
| 13 | 42.48 | — | 27 | 19.44 | 0.79 |
| 14 | 54.41 | 1.9 | 28 | 21.51 | 0.79 |
Frequencies in the proton and carbon dimensions are given with respect to the methyl 18. C18 was set to12.08 ppm and H18 at 0.66 ppm with respect to tetramethylsilane (TMS) by analogy with a liquid-state spectrum recorded in deuterated chloroform at 295 K. Errors on the reported chemical shifts are estimated to be ∼±0.02 ppm. (The expressions a and e correspond to the stereospecific assignment of methylene protons, i.e., a, axial; e, equatorial.)
FIGURE 4.

Pulse sequences used in this work. (A) Dipolar HETCOR. During the evolution period, magnetization evolves under the 1H chemical shift and the residual proton dipolar couplings. 1H to 13C coherence transfer was then mediated via H–C dipolar couplings. Carbon chemical shift is probed during t2. (B) The CP transfer scheme is replaced by a refocused INEPT scheme and the transfer is mediated via 1J isotropic scalar couplings in the case of scalar HETCOR. (C) Two-dimensional INADEQUATE pulse sequence. Apart from the CP period, the experiment is completely analogous to liquid-state versions. After cross-polarization from 1H, the 13C magnetization evolves during the delay 2τ under the isotropic homonuclear J coupling Hamiltonian. The C–C homonuclear dipolar couplings are removed by fast MAS, and the chemical shift evolution is refocused by the 180° pulse. The double-quantum coherence created by the first carbon 90° pulse evolves during t1 at the sum frequency of the two spins, and is converted back into an antiphase transverse coherence by the last 90° pulse, which is detected during t2. (D) Lee-Golburg experiment was used to determine proton-carbon dipolar couplings and permit proton stereospecific assignments of methylene protons.
FIGURE 5.

Identification of carbon multiplicity. Comparison of one-dimensional carbon MAS spectra obtained with a CP transfer scheme (a) and a refocused INEPT scheme (b) allows us to identify missing resonances as quaternary carbon signals (denoted Q). Comparison of the refocused INEPT spectrum and the spectral edited refocused INEPT (c) shows the expected negative, but also the missing carbon resonances, which can be explained by fast transverse relaxation of the tightly coupled methylene protons of the ergosterol rings during the overall INEPT delay. Hence, negative and missing resonances can be assigned to methylene carbons. Lipids resonances are marked with an asterisk (*). These spectra were recorded on 8.4:1.6 DMPC-d54/ergosterol–11%13C multilamellar vesicles at 313 K.
FIGURE 6.

Assignment of the ring A of the ergosterol molecule. As in solution-state NMR, directly bonded carbon-13 resonances are identified by the fact that they generate a common double-quantum frequency. The connectivities between antiphase line shapes are indicated by solid lines. The spectrum was obtained in 18 h using 1.5 mg of 100% 13C-labeled ergosterol in DMPC-d54 membrane at 16% molar ratio at 313 K.
FIGURE 7.

Comparison of 1H−13C chemical shifts correlation extracted from scalar HETCOR (A) and dipolar HETCOR (B) centered onto the olefinic region of ergosterol. From the scalar HETCOR, the proton chemical shift directly bonded to carbon can be easily identified (C6−H6 119.05 ppm, 5.50 ppm, C7−H7 117.05 ppm, 5.27 ppm). The dipolar HETCOR carbon slice extracted at 141.18 ppm shows three cross-peaks: at 2.25, 2.49, and 5.50 ppm—with two correlations corresponding to long-range transfer from H4 protons and H6 proton, respectively. The resonance at 141.18 ppm can be thus identified as the C5 of ergosterol. Signal at 139.2 ppm corresponds then to C8 with a long-range proton transfer from H7 (5.27 ppm) and H9 (1.91 ppm). These spectra were recorded on 8.4:1.6 DMPC-d54/ergosterol–11%13C multilamellar vesicles at 313 K.
The ergosterol carbon assignments for the other two lipid sterol mixtures (7.7:2.3 and 7.0:3.0 mol/mol DMPC-d54/ergosterol–11%13C) show differences of <0.2 ppm (data not shown).
Orientation and dynamics of ergosterol in DMPC membrane: chemical shift anisotropy restraints
The determination of sterol's orientation and dynamics in lipid bilayers can be performed using several strategies. One can make use of the residual second rank tensor interactions, which are due to the incomplete motional averaging, by measuring 2H quadrupolar splittings (Ref. 22 and references cited herein), 1H−13C dipolar couplings (40), or chemical shift anisotropies on static liposomes (which give powder spectra) or mechanically oriented bilayer samples (48). One can also use MAS techniques to average out these tensor interactions in one dimension and selectively reintroduce them in another dimension (49–52). The present work was focused on the analysis of 13C CSA residual interactions on liposome samples, under MAS and OMAS conditions.
Critical chemical shift parameters
Molecular reorientations, on a timescale shorter than 10−5 s, will result in a reduction of the chemical shift anisotropy (CSA). The carbon chemical shift tensor can be characterized by six parameters: namely, three principal values (δ11, δ22, and δ33) and three Euler angles specifying the orientation of the principal axis system in the molecular frame. Fig. 8 shows simulated spectra for the cholesterol's carbon C4 resonance in the presence of fast axial diffusion around three axes. Fig. 8 a is for a static sample and Fig. 8, b–d, show the simulated line shape when the diffusion axis is parallel to  respectively. One can see that the chemical shift anisotropy is reduced in the presence of fast axial diffusion and depends on the orientation of the diffusion axis. Hence, given the principal values and orientation of at least three static chemical shift tensors within a rigid molecule, one can obtain the diffusion axis orientation and the molecular order parameter, by comparing the experimental motionally averaged anisotropy (Δδexp) to the theoretical one (Δδcalc).
respectively. One can see that the chemical shift anisotropy is reduced in the presence of fast axial diffusion and depends on the orientation of the diffusion axis. Hence, given the principal values and orientation of at least three static chemical shift tensors within a rigid molecule, one can obtain the diffusion axis orientation and the molecular order parameter, by comparing the experimental motionally averaged anisotropy (Δδexp) to the theoretical one (Δδcalc).
FIGURE 8.

Simulated 13C spectra of cholesterol–13C4, showing the effect of rapid axially symmetric reorientation on the powder pattern line shape: (a) the static case, with principal values and asymmetry parameter extracted from quantum chemistry calculation (δ11 = 58.3 ppm, δ22 = 40.8 ppm, δ33 = 20.5 ppm, Δδstatic = −19.4 ppm, and η = 0.9), and rapid axial reorientation about an axis (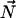 ) oriented (b) parallel to
) oriented (b) parallel to  (Δδcalc = −19.4 ppm), (c) parallel to
(Δδcalc = −19.4 ppm), (c) parallel to  (Δδcalc = 18.4 ppm), and (d) parallel to
(Δδcalc = 18.4 ppm), and (d) parallel to  (Δδcalc = 0.9 ppm).
(Δδcalc = 0.9 ppm).
Various experimental methods have been designed to estimate the principal values of the chemical shift tensor (see below). By contrast, the experimental determination of the chemical shift tensor's orientations is more difficult since it requires either single crystals (Ref. 53 and references therein) or chemical synthesis of specifically doubly labeled compounds, which allows us to orient the CSA tensor with respect to a dipolar tensor of known orientation (54–59). Fortunately, recent developments in quantum chemistry permit the accurate treatment of theoretical chemical shielding (for reviews see Refs. 41, 42, and 60–62). In this context, we have recently shown that isotropic carbon chemical shifts of cholesterol and ergosterol can be reproduced accurately, even though these calculations were performed for an isolated molecule, neglecting inner molecular vibrations and using fairly simple basis sets (45). However, isotropic chemical shift calculation only requires accurate principal values and does not depend on tensor orientations. To check the quality of eigen-values and eigen-vectors of the theoretical tensors we compared calculated and experimental motionally averaged chemical shift anisotropies of cholesterol's carbons C3 and C4, for which dynamical parameters in a DMPC membrane (7.0:3.0 mol/mol DMPC/cholesterol) have been previously determined (22). The carbon theoretical chemical shift tensors have been calculated following the same procedure as for ergosterol (Table 3). As shown in Table 3, there is a reasonably good agreement for each of the two carbon-averaged anisotropies showing that both the principal values and the orientation of the chemical tensors are efficiently provided by quantum chemistry calculations, and that the approximations associated with this approach lead to negligible effects in sterol's orientational analysis. It may be worth noting that the errors on the predicted Δδ (1.3 and 1.6 ppm for carbons C3 and C4, respectively) are comparable to the average error in isotropic chemical shift calculations—at ∼2 ppm, depending on the computation method and molecule, in Jolibois et al. (45). Therefore, orientation effects do not introduce a significantly larger error in chemical shift computation than principal values inaccuracies.
TABLE 3.
Theoretical (Δδcalc) and experimental (Δδexp) motionally averaged CSA of C3 and C4 carbons of cholesterol
| Carbon number | δ11, δ22, δ33 (ppm) | Δδstatic (ppm) | η | αPM | βPM | γPM | Δδcalc (ppm) | Δδexp (ppm) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 91.5 | 81.1 | 33.4 | −35.2 | 0.29 | −80.5 | 77.8 | −86.4 | −30 | −31.3 ± 1 |
| 4 | 58.3 | 40.8 | 20.5 | −19.4 | 0.90 | 78.9 | −115.3 | 82.0 | −14.9 | −13.3 ± 1 |
The Δδexp values were obtained by analyzing the spinning side-band pattern (at 2, 2.5, and 3-kHz spinning rates) of 7.0:3.0 (mol/mol) DMPC/cholesterol liposomes at 313 K. At this temperature, the molecular order parameter is equal to 0.94 and the polar coordinates of the diffusion axis in the molecular frame M are (α = 91°, β = 89°), corresponding to (α = 11°, β = 11°) in the molecular frame defined by Marsan (22).
Motionally averaged chemical shift anisotropy measurements
The chemical shift anisotropy is a powerful probe of molecular structure and dynamics, and its measurement has lead to the design of various one-dimensional and two-dimensional static or MAS NMR methods. As shown above for assignment experiments, under fast rotation at the magic angle, all of the second rank interactions are effectively averaged to zero and high-resolution isotropic spectra can be obtained with high sensitivity and resolution in the case of magnetically dilute spins. In a slower spinning regime, when the spinning frequency νR is comparable to the spread in Larmor frequencies caused by the CSA, the NMR spectrum contains peaks at frequency coordinates viso and (viso+ k vr), where viso is the isotropic shift frequency of site, k an integer (called the side-band order), and vr the spinning frequency. The intensity distribution of the spinning side-bands is characteristic of the CSA tensor, and it may be analyzed to obtain the CSA parameters. Carbon 13 NMR spectra of DMPC-d54/ergosterol–11%13C multilamellar vesicles at three different ergosterol molar ratio (16, 23, and 30 mol %) were recorded under slow MAS conditions, at 2, 2.5, and 3 kHz. For each spectra, the intensities of the observed side-bands have been used in a Hertzfeld-Berger analysis to extract the components of the motionally averaged chemical shift tensor (Δδexp) associated to five carbon sites of the ergosterol molecule, namely C3 and C5−8, which are characterized by large anisotropies and well-resolved spinning side-bands (Table 4).
TABLE 4.
The motionally averaged chemical shift tensor (Δδexp) for ergosterol ring carbons of the three lipid mixtures at 313 K
| DMPC/Ergosterol (mol/mol)
|
|||
|---|---|---|---|
| Carbon number | 8.4:1.6 | 7.7:2.3 | 7.0:3.0 |
| 1 | 0.0 ± 3* | 0.0 ± 3* | 0.0 ± 3* |
| 2 | 0.0 ± 3* | 0.0 ± 3* | 0.0 ± 3* |
| 3 | −24.0 ± 1.6† | −26.0 ± 1.6† | −27.9 ± 1.6† |
| 4 | −13.3 ± 3* | −13.3 ± 3† | −13.3 ± 3* |
| 5 | 63 ± 1.6† | 80.7 ± 1.6† | 80.7 ± 1.6† |
| 6 | 37.7 ± 1.6† | 44.7 ± 1.6† | 44.5 ± 1.6† |
| 7 | 41.7 ± 1.6† | 50 ± 1.6† | 50.1 ± 1.6† |
| 8 | 66 ± 1.6† | 72.7 ± 1.6† | 69.6 ± 1.6† |
| 9 | 0.0 ± 3* | 0.0 ± 3* | 0.0 ± 3* |
| 10 | 0.0 ± 3* | 0.0 ± 3* | 0.0 ± 3* |
| 11 | −3.3 ± 3* | −3.3 ± 3* | −3.3 ± 3* |
| 12 | 5 ± 3* | 5 ± 3* | 6.7 ± 3* |
| 13 | −3.3 ± 3* | −3.3 ± 3* | −6.7 ± 3* |
| 14 | −6.6 ± 3* | −6.6 ± 3* | −6.7 ± 3* |
| 15 | −10 ± 3* | −10 ± 3* | −10 ± 3* |
| 16 | ND | ND | ND |
| 17 | −15 ± 3* | −15 ± 3* | −20 ± 3* |
| 18 | 3.3 ± 3* | 3.3 ± 3* | 6.7 ± 3* |
| 19 | 13.3 ± 3* | 13.3 ± 3* | 16.7 ± 3* |
Experimental values were extracted from OMAS experiments.
Experimental values were extracted from slow MAS experiments.
In the upfield region of the ergosterol carbon spectrum, spectral overlap, small CSAs, and perturbations from dipolar interactions prevented the extraction of reliable CSA parameters from side-band intensity analyses. To overcome this problem a series of two-dimensional experiments have been proposed, at low (Ref. 63 and references therein; and Refs. 64–66) and fast MAS rate (52,67,68), the common principle of these experiments being to use a second dimension to separate overlapping resonances according to their isotropic chemical shifts or side-band order. These experiments are widely used to determine CSAs for numerous nuclei, but they become technically difficult to implement for small CSAs (except for Ref. 68, where this point was addressed).
In this article, we have used a one-dimensional approach closely related to the idea of the switched-angle-sample-spinning (49,51) experiment: in a spectrum arising from fast-rotation OMAS, the CSA is scaled by a reduction factor (which is 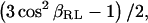 as long as βRL, the angle between the rotation axis and the static magnetic field, is kept close to magic angle; see Ref. 69). Two of the one-dimensional OMAS spectra at two different angles were then recorded to follow the deviations between δiso of the MAS spectrum and δ90° of the OMAS spectra (Fig. 9). Chemical shifts were extracted after calibration on the choline polar-head methyl's resonance, which is known to exhibit a small anisotropy (∼2 ppm) (48). OMAS scaling factors were calibrated by 31P NMR using the CSA of the DMPC phosphate. Table 4 summarizes the different values of carbon CSA with their uncertainty obtained for ergosterol signals extracted either from the slow MAS or from the OMAS experiments.
as long as βRL, the angle between the rotation axis and the static magnetic field, is kept close to magic angle; see Ref. 69). Two of the one-dimensional OMAS spectra at two different angles were then recorded to follow the deviations between δiso of the MAS spectrum and δ90° of the OMAS spectra (Fig. 9). Chemical shifts were extracted after calibration on the choline polar-head methyl's resonance, which is known to exhibit a small anisotropy (∼2 ppm) (48). OMAS scaling factors were calibrated by 31P NMR using the CSA of the DMPC phosphate. Table 4 summarizes the different values of carbon CSA with their uncertainty obtained for ergosterol signals extracted either from the slow MAS or from the OMAS experiments.
FIGURE 9.

Ergosterol–11%13C CP-MAS (a), and two CP-OMAS (b and c) spectra in DMPC-d54 multilamellar vesicles at 313 K (8.4:1.6 mol/mol DMPC/ergosterol). The OMAS angles were β90°,1 = 51.5° and β90°,2 = 49.5°. The motionally averaged CSAs (Δδexp) for ergosterol ring carbons are calculated using  (see Eq. 2 for Δδexp, δiso, and δ90° definitions). The values δiso, δ90°,1, and δ90°,2 are extracted for each carbon from the MAS (a), and the two OMAS (b and c) spectra, respectively. The spectra were calibrated on the choline polar-head methyl resonance, which has a very small CSA (<2 ppm; see Ref. 48). For each OMAS experiment, the scaling factor is determined from the 31P NMR spectra acquired on the same sample and same condition, and is equal to
(see Eq. 2 for Δδexp, δiso, and δ90° definitions). The values δiso, δ90°,1, and δ90°,2 are extracted for each carbon from the MAS (a), and the two OMAS (b and c) spectra, respectively. The spectra were calibrated on the choline polar-head methyl resonance, which has a very small CSA (<2 ppm; see Ref. 48). For each OMAS experiment, the scaling factor is determined from the 31P NMR spectra acquired on the same sample and same condition, and is equal to  The experimental error on Δδexp is estimated at ±3 ppm for each carbon and takes into account the accuracies of δ90°,1, δ90°,2 measurements in 31P and 13C spectra. Following this procedure we could calculate, for example, Δδexp (C4) = 7.5 ± 3 ppm and Δδexp (C9) = 0 ± 3 ppm.
The experimental error on Δδexp is estimated at ±3 ppm for each carbon and takes into account the accuracies of δ90°,1, δ90°,2 measurements in 31P and 13C spectra. Following this procedure we could calculate, for example, Δδexp (C4) = 7.5 ± 3 ppm and Δδexp (C9) = 0 ± 3 ppm.
Data analysis
The data analysis was performed in a similar way for three DMPC/ergosterol mixtures (8.4:1.6, 7.7:2.3, and 7.0:3.0 mol/mol). Using static chemical shift tensor parameters obtained by quantum chemical computations, sets of theoretical, motionally averaged, carbon chemical shift anisotropies (Δδcalc, Eq. 2) have been calculated by varying α, β, and Smol from 0° to 360°, 0° to 90°, and 0 to 1, respectively. For each (α, β, Smol) triplet, the 18 carbon's Δδcalc values were compared to the experimental ones and their RMSD was evaluated following Eq. 3. Over 32.4 106 possibilities, 386, 199, and 200 triplets correspond to an RMSD smaller than 3.3 ppm (upper limit of the experimental errors) for 8.4:1.6, 7.7:2.3, and 7.0:3.0 (mol/mol) DMPC/ergosterol samples, respectively. For each mixture, these solutions defined one single region in α and β, for which the average and the standard deviation values are given in Table 5. The linear regression between experimental anisotropies and theoretical ones obtained with these averaged values shows a good agreement, giving a Pearson coefficient equal to 0.99 and a slope of 1.00 for each of lipid mixture (data not shown).
TABLE 5.
Diffusion parameters for ergosterol or cholesterol in DMPC bilayers at 313 K
| Diffusion axis parameters
|
||||
|---|---|---|---|---|
| α (°) | β (°) | Smol | Inertia-diffusion angle γI (°) | |
| DMPC/Ergosterol (mol/mol) | ||||
| 8.4:1.6 | 101 ± 5 | 89 ± 1 | 0.76 ± 0.02 | 15 ± 3 |
| 7.7:2.3 | 98 ± 5 | 88 ± 1 | 0.89 ± 0.02 | 13 ± 2 |
| 7.0:3.0 | 99 ± 5 | 88 ± 1 | 0.89 ± 0.02 | 14 ± 2 |
| DMPC/Cholesterol (mol/mol)* | ||||
| 7.0:3.0 | 91 ± 5 | 89 ± 1 | 0.94 ± 0.01 | 2 ± 2 |
The values α and β are the polar coordinates of the diffusion axis in the molecular frame M, Smol is the molecular order parameter. The value γI corresponds to the angle between the principal inertia axis  of the sterol ring system (from C1 to C19 with bonded protons and oxygen) and the diffusion axis.
of the sterol ring system (from C1 to C19 with bonded protons and oxygen) and the diffusion axis.
The polar coordinates in the molecular frame M correspond to (α = 11 ± 5°, β = 11 ± 1°) in the C3 molecular frame defined in Marsan et al. (22) for 7.0:3.0 (mol/mol) DMPC/cholesterol samples. Note that, in their publication, α is named γ.
The 18 experimental motionally averaged CSA restraints allow us to define orientational and dynamical ergosterol parameters with the same precision as one obtained on cholesterol using quadrupole splittings (the high number of CSA restraints compensating for their lower accuracy, as compared with quadrupole splittings). One important advantage of the CSA strategy is that it does not require chemical synthesis of specifically labeled sterols. One can note that the diffusion axis orientation is different for cholesterol and ergosterol mixtures but is independent of the DMPC/sterol molar ratio. The diffusion axis is indeed approximately parallel to the principal inertia axis  (defined by sterol tetra-cycle atoms) in the case of cholesterol (γI ≈ 2°), whereas it is tilted by ∼14° for the ergosterol mixture (Table 5). Various explanations can be proposed to understand this difference. The effective inertia axis of the whole molecule
(defined by sterol tetra-cycle atoms) in the case of cholesterol (γI ≈ 2°), whereas it is tilted by ∼14° for the ergosterol mixture (Table 5). Various explanations can be proposed to understand this difference. The effective inertia axis of the whole molecule 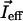 can be more tilted from
can be more tilted from  orientation for ergosterol than for cholesterol, due to the differences between the two sterols' side-chain structures and conformations. Alternatively, specific interactions between adjacent molecules (water or DMPC) and the sterol's hydroxyl group and/or the π system of the second ring could also be different for cholesterol and ergosterol and influence the relative orientation of
orientation for ergosterol than for cholesterol, due to the differences between the two sterols' side-chain structures and conformations. Alternatively, specific interactions between adjacent molecules (water or DMPC) and the sterol's hydroxyl group and/or the π system of the second ring could also be different for cholesterol and ergosterol and influence the relative orientation of  and
and  (45,47). Molecular dynamics simulations performed on a 8:1 DMPC/sterol (mol/mol) bilayer system have also suggested some difference in ergosterol and cholesterol orientation (70). In these simulations, the average tilt of the sterol molecules, defined as the angle between bilayer normal and the vector connecting carbon atoms C3 and C17, was found slightly higher for ergosterol than for cholesterol.
(45,47). Molecular dynamics simulations performed on a 8:1 DMPC/sterol (mol/mol) bilayer system have also suggested some difference in ergosterol and cholesterol orientation (70). In these simulations, the average tilt of the sterol molecules, defined as the angle between bilayer normal and the vector connecting carbon atoms C3 and C17, was found slightly higher for ergosterol than for cholesterol.
The two sterols also present significantly different molecular order parameters (Table 5), depending on DMPC/sterol ratio. At low concentration (16 mol %, Ld phase) ergosterol is highly mobile (Smol = 0.76 ± 0.02), and at high concentration (30 mol %, Lo phase) the dynamics of ergosterol and cholesterol are similar (Smol = 0.89 ± 0.02 compared to 0.94 ± 0.01). At intermediate concentration (23 mol %, coexistence of the Ld and Lo phases), ergosterol's molecular order parameter is identical to its value in the Lo phase.
Finally, our experiment gives an estimate for the upper limit of domain size at an intermediate molar ratio. The difference of isotopic chemical shift of ergosterol resonances in the Ld phase (16 mol % ergosterol) and the Lo phase (30 mol % ergosterol) is comprised between 15 and 20 Hz depending on carbon atom. Since for 23 mol % ergosterol sample (coexistence of the Ld and Lo phases) one single sharp resonance is observed for each carbon (fast averaging limit on the chemical-shift scale), the exchange rate of ergosterol between the Ld and Lo domains has to be higher than 102 Hz. Following the equation 〈Δx2〉1/2 = (4Dτ)1/2 (30) and using the same diffusion coefficient for phospholipid and sterol, 10−11 m2/s (15), we thus determine an upper limit for a domain size of 600 nm (taking τ = 10−2 s). This information is complementary to the lower limit of 20 nm obtained from deuterium NMR spectra for which slow exchange was observed on the deuterium-NMR timescale (30).
CONCLUSIONS
The chemical shift anisotropy is routinely used to determine membrane-peptide's orientation (71,72). It is also a powerful probe of molecular dynamics since molecular reorientations on a timescale shorter than 10−5 s reduce its amplitude. 31P CSA has been used to study phospholipid headgroup's dynamics (73) or to reveal specific protein-lipid interactions (74,75). Fast axial diffusion of transmembrane peptides has been demonstrated using 15N CSA (34) and 13C CSA (76) of peptide bonds. Nevertheless, to our knowledge the present work is the first really quantitative assessment of a membrane component's orientation and dynamics using theoretical static 13C CSA tensors and experimental motionally averaged values. For this study, we have first produced uniformly 100% and 11% 13C-labeled ergosterol from Pichia pastoris methylotrophically and performed complete proton and carbon assignment of ergosterol in membrane through a combination of one- and two-dimensional 13C−1H heteronuclear (J- and dipolar-HETCOR) and 13C−13C homonuclear (INADEQUATE) MAS experiments. Motionally averaged chemical shift anisotropies for all ring carbons have then been determined by spinning side-band intensity analyses and OMAS experiments. The experimental CSAs have finally been combined with theoretical static CSA tensors to give access to the orientation and molecular order parameters of the diffusion axis. These parameters have been obtained for ergosterol at three different DMPC/ergosterol molar ratios and compared to data obtained previously on cholesterol by deuterium NMR on oriented bilayers (22). It has been shown that, in DMPC bilayers, the inertial axis of ergosterol's four-ring system is tilted by ∼14° compared to the diffusion axis. Furthermore, ergosterol was shown to be highly mobile in the Ld phase (16 mol % ergosterol) and to present a dynamics similar to cholesterol in the Lo phase (30 mol % sterol).
Acknowledgments
We acknowledge Dr. N. Lindley, Institut National des Sciences Appliquées Toulouse, for his help with P. pastoris cultures.
The nuclear magnetic resonance spectrometers were financed by Centre National de la Recherche Scientifique (Ingénierie des MAcromolécules BIOlogiques program), the Région Midi-Pyrénées, and European Structural funds.
References
- 1.Dahl, C., and J. Dahl. 1988. Biology of Cholesterol. P. L. Yeagle, editor. Academic Publishers, Boca Raton, FL. 147–172.
- 2.Yeagle, P. L. 1985. Cholesterol and the cell membrane. Biochim. Biophys. Acta. 822:267–287. [DOI] [PubMed] [Google Scholar]
- 3.Parks, L. W., S. J. Smith, and J. H. Crowley. 1995. Biochemical and physiological effects of sterol alterations in yeast—a review. Lipids. 30:227–230. [DOI] [PubMed] [Google Scholar]
- 4.Parks, L. W., J. H. Crowley, F. W. Leak, S. J. Smith, and M. E. Tomeo. 1999. Use of sterol mutants as probes for sterol functions in the yeast, Saccharomyces cerevisiae. Crit. Rev. Biochem. Mol. Biol. 34:399–404. [DOI] [PubMed] [Google Scholar]
- 5.Chapman, D., N. F. Owens, M. C. Phillips, and D. A. Walker. 1969. Mixed monolayers of phospholipids and cholesterol. Biochim. Biophys. Acta. 183:458–465. [DOI] [PubMed] [Google Scholar]
- 6.Joos, P., and R. A. Demel. 1969. The interaction energies of cholesterol and lecithin in spread mixed monolayers at the air-water interface. Biochim. Biophys. Acta. 183:447–457. [DOI] [PubMed] [Google Scholar]
- 7.Marsan, M. P., E. Bellet-Amalric, I. Muller, G. Zaccai, and A. Milon. 1998. Plant sterols: a neutron diffraction study of sitosterol and stigmasterol in soybean phosphatidylcholine membranes. Biophys. Chem. 75:45–55. [DOI] [PubMed] [Google Scholar]
- 8.Smaby, J. M., H. L. Brockman, and R. E. Brown. 1994. Cholesterol's interfacial interactions with sphingomyelins and phosphatidylcholines: hydrocarbon chain structure determines the magnitude of condensation. Biochemistry. 33:9135–9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockton, G. W., and I. C. Smith. 1976. A deuterium nuclear magnetic resonance study of the condensing effect of cholesterol on egg phosphatidylcholine bilayer membranes. I. Perdeuterated fatty acid probes. Chem. Phys. Lipids. 17:251–263. [DOI] [PubMed] [Google Scholar]
- 10.Demel, R. A., and B. De Kruyff. 1976. The function of sterols in membranes. Biochim. Biophys. Acta. 457:109–132. [DOI] [PubMed] [Google Scholar]
- 11.Clejan, S., R. Bittman, P. W. Deroo, Y. A. Isaacson, and A. F. Rosenthal. 1979. Permeability properties of sterol-containing liposomes from analogues of phosphatidylcholine-lacking acyl groups. Biochemistry. 18:2118–2125. [DOI] [PubMed] [Google Scholar]
- 12.Schuler, I., A. Milon, Y. Nakatani, G. Ourisson, A. M. Albrecht, P. Benveniste, and M. A. Hartmann. 1991. Differential effects of plant sterols on water permeability and on acyl chain ordering of soybean phosphatidylcholine bilayers. Proc. Natl. Acad. Sci. USA. 88:6926–6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bloom, M., and J. L. Thewalt. 1995. Time and distance scales of membrane domain organization. Mol. Membr. Biol. 12:9–13. [DOI] [PubMed] [Google Scholar]
- 14.Kuo, A. L., and C. G. Wade. 1979. Lipid lateral diffusion by pulsed nuclear magnetic resonance. Biochemistry. 18:2300–2308. [DOI] [PubMed] [Google Scholar]
- 15.Oradd, G., G. Lindblom, and P. W. Westerman. 2002. Lateral diffusion of cholesterol and dimyristoylphosphatidylcholine in a lipid bilayer measured by pulsed field gradient NMR spectroscopy. Biophys. J. 83:2702–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filippov, A., G. Oradd, and G. Lindblom. 2003. The effect of cholesterol on the lateral diffusion of phospholipids in oriented bilayers. Biophys. J. 84:3079–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubenstein, J. L., B. A. Smith, and H. M. McConnell. 1979. Lateral diffusion in binary mixtures of cholesterol and phosphatidylcholines. Proc. Natl. Acad. Sci. USA. 76:15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vist, M. R., and J. H. Davis. 1990. Phase equilibria of cholesterol/dipalmitoylphosphatidylcholine mixtures: 2H nuclear magnetic resonance and differential scanning calorimetry. Biochemistry. 29:451–464. [DOI] [PubMed] [Google Scholar]
- 19.Huang, T. H., C. W. Lee, S. K. Das Gupta, A. Blume, and R. G. Griffin. 1993. A 13C and 2H nuclear magnetic resonance study of phosphatidylcholine/cholesterol interactions: characterization of liquid-gel phases. Biochemistry. 32:13277–13287. [DOI] [PubMed] [Google Scholar]
- 20.McMullen, T. P., R. N. Lewis, and R. N. McElhaney. 1993. Differential scanning calorimetric study of the effect of cholesterol on the thermotropic phase behavior of a homologous series of linear saturated phosphatidylcholines. Biochemistry. 32:516–522. [DOI] [PubMed] [Google Scholar]
- 21.McMullen, T. P., and R. N. McElhaney. 1997. Differential scanning calorimetric studies of the interaction of cholesterol with distearoyl and dielaidoyl molecular species of phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine. Biochemistry. 36:4979–4986. [DOI] [PubMed] [Google Scholar]
- 22.Marsan, M. P., I. Muller, C. Ramos, F. Rodriguez, E. J. Dufourc, J. Czaplicki, and A. Milon. 1999. Cholesterol orientation and dynamics in dimyristoylphosphatidylcholine bilayers: a solid state deuterium NMR analysis. Biophys. J. 76:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sankaram, M. B., and T. E. Thompson. 1990. Modulation of phospholipid acyl chain order by cholesterol. A solid-state 2H nuclear magnetic resonance study. Biochemistry. 29:10676–10684. [DOI] [PubMed] [Google Scholar]
- 24.Semer, R., and E. Gelerinter. 1979. A spin label study of the effects of sterols on egg lecithin bilayers. Chem. Phys. Lipids. 23:201–211. [Google Scholar]
- 25.Urbina, J. A., S. Pekerar, H. B. Le, J. Patterson, B. Montez, and E. Oldfield. 1995. Molecular order and dynamics of phosphatidylcholine bilayer membranes in the presence of cholesterol, ergosterol and lanosterol: a comparative study using 2H-, 13C- and 31P-NMR spectroscopy. Biochim. Biophys. Acta. 1238:163–176. [DOI] [PubMed] [Google Scholar]
- 26.McMullen, T. P. W., and R. N. McElhaney. 1996. Physical studies of cholesterol-phospholipid interactions. Curr. Opin. Colloid Interface Sci. 1:83–90. [Google Scholar]
- 27.Ohvo-Rekila, H., B. Ramstedt, P. Leppimaki, and J. P. Slotte. 2002. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 41:66–97. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh, D., and J. Tinoco. 1972. Monolayer interactions of individual lecithins with natural sterols. Biochim. Biophys. Acta. 266:41–49. [DOI] [PubMed] [Google Scholar]
- 29.Schuler, I., G. Duportail, N. Glasser, P. Benveniste, and M. A. Hartmann. 1990. Soybean phosphatidylcholine vesicles containing plant sterols: a fluorescence anisotropy study. Biochim. Biophys. Acta. 1028:82–88. [DOI] [PubMed] [Google Scholar]
- 30.Hsueh, Y., K. Gilbert, C. Trandum, M. Zuckermann, and J. Thewalt. 2005. The effect of ergosterol on dipalmitoylphosphatidylcholine bilayers: a deuterium NMR and calorimetric study. Biophys. J. 88:1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovacs, F. A., J. K. Denny, Z. Song, J. R. Quine, and T. A. Cross. 2000. Helix tilt of the M2 transmembrane peptide from Influenza A virus: an intrinsic property. J. Mol. Biol. 295:117–125. [DOI] [PubMed] [Google Scholar]
- 32.Bak, M., R. P. Bywater, M. Hohwy, J. K. Thomsen, K. Adelhorst, H. J. Jakobsen, O. W. Sorensen, and N. C. Nielsen. 2001. Conformation of alamethicin in oriented phospholipid bilayers determined by (15)N solid-state nuclear magnetic resonance. Biophys. J. 81:1684–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marassi, F. M., C. Ma, J. J. Gesell, and S. J. Opella. 2000. Three-dimensional solid-state NMR spectroscopy is essential for resolution of resonances from in-plane residues in uniformly N-15-labeled helical membrane proteins in oriented lipid bilayers. J. Magn. Reson. 144:156–161. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi, S., D. Huster, A. Waring, R. I. Lehrer, W. Kearney, B. F. Tack, and M. Hong. 2001. Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys. J. 81:2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park, S. H., A. A. Morse, A. A. Nevzorov, M. F. Mesleh, M. Oblatt-Montal, M. Montal, and S. J. Opella. 2003. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein u (Vpu) from HIV-1. J. Mol. Biol. 333:409–424. [DOI] [PubMed] [Google Scholar]
- 36.Glaubitz, C., G. Grobner, and A. Watts. 2000. Structural and orientational information of the membrane-embedded M13 coat protein by C-13-MAS NMR spectroscopy. Biochim. Biophys. Acta. 1463:151–161. [DOI] [PubMed] [Google Scholar]
- 37.Creemers, A. F., S. Kiihne, P. H. Bovee-Geurts, W. J. DeGrip, J. Lugtenburg, and H. J. de Groot. 2002. (1)H and (13)C MAS NMR evidence for pronounced ligand-protein interactions involving the ionone ring of the retinylidene chromophore in rhodopsin. Proc. Natl. Acad. Sci. USA. 99:9101–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haon, S., S. Augé, M. Tropis, A. Milon, and N. Lindley. 1993. Low-cost production of perdeuterated biomass using methylotrophic yeasts. J. Label. Comp. Rad. 33:1053–1063. [Google Scholar]
- 39.Massou, S., S. Auge, M. Tropis, N. D. Lindley, and A. Milon. 1998. NMR analyses of deuterated phospholipids isolated from Pichia angusta. J. Chim. Phys. Phys. Chim. Biol. 95:406–411. [Google Scholar]
- 40.Massou, S., V. Puech, F. Talmont, P. Demange, N. D. Lindley, M. Tropis, and A. Milon. 1999. Heterologous expression of a deuterated membrane-integrated receptor and partial deuteration in methylotrophic yeasts. J. Biomol. NMR. 14:231–239. [DOI] [PubMed] [Google Scholar]
- 41.de Dios, A. C. 1996. Ab initio calculations of the NMR chemical shift. Prog. Nucl. Magn. Spectrosc. 29:229–278. [Google Scholar]
- 42.de Dios, A. C., and E. Oldfield. 1996. Recent progress in understanding chemical shifts. Solid State Nucl. Magn. Reson. 6:101–125. [DOI] [PubMed] [Google Scholar]
- 43.Lesage, A., M. Bardet, and L. Emsley. 1999. Through-bond carbon-carbon connectivities in disordered solids by NMR. J. Am. Chem. Soc. 121:10987–10993. [Google Scholar]
- 44.Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, Jr., J. C. Burant, S. Dapprich, J. M. Millam, et al. 1998. Gaussian 98, Rev. A9. Gaussian, Pittsburgh, PA.
- 45.Jolibois, F., O. Soubias, V. Réat, and A. Milon. 2004. Understanding sterol-membrane interactions. I. Hartree-Fock versus DFT calculations of 13C and 1H NMR isotropic chemical shifts of sterols in solution and hydrogen bonding effects analysis. Chem. Eur. J. 10:5996–6004. [DOI] [PubMed] [Google Scholar]
- 46.Davis, J. H., and M. Auger. 1999. Static and magic angle spinning NMR of membrane peptides and proteins. Progr. Nucl. Magn. Res. Spectrosc. 35:1–84. [Google Scholar]
- 47.Soubias, O., F. Jolibois, V. Réat, and A. Milon. 2004. Understanding sterol-membrane interactions. II. Complete 1H and 13C assignments by solid state NMR and determination of hydrogen bonding partners of cholesterol in a lipid bilayer. Chem. Eur. J. 10:6005–6014. [DOI] [PubMed] [Google Scholar]
- 48.Soubias, O., O. Saurel, V. Réat, and A. Milon. 2002. High resolution 13C NMR spectra on oriented lipid bilayers: from quantifying the various sources of line broadening to performing two-dimensional experiments with 0.2–0.3 ppm resolution in the carbon dimension. J. Biomol. NMR. 24:15–30. [DOI] [PubMed] [Google Scholar]
- 49.Bax, A., N. M. Szeverenyi, and G. E. Maciel. 1983. Chemical shift anisotropy in powdered solids studied by two-dimensional FT NMR with flipping of the spinning axis. J. Magn. Reson. 55:494–497. [Google Scholar]
- 50.Liu, S.-F., J.-D. Mao, and K. Schmidt-Rohr. 2002. A robust technique for two-dimensional separation of undistorted chemical-shift anisotropy powder patterns in magic-angle-spinning NMR. J. Magn. Reson. 155:15–18. [DOI] [PubMed] [Google Scholar]
- 51.Terao, T., T. Fujii, T. Onodera, and A. Saika. 1984. Switching-angle sample-spinning NMR spectroscopy for obtaining powder-pattern-resolved two-dimensional spectra: measurements of 13C chemical-shift anisotropies in powdered 3,4-dimethoxybenzaldehyde. Chem. Phys. Lett. 107:145–148. [Google Scholar]
- 52.Tycko, R., G. Dabbagh, and P. A. Mirau. 1989. Determination of chemical-shift-anisotropy lineshapes in a two-dimensional magic-angle-spinning NMR experiment. J. Magn. Reson. 85:265–274. [Google Scholar]
- 53.Shekar, S. C., A. Ramamoorthy, and R. J. Wittebort. 2002. Determination of the chemical shielding tensor orientation from two or one of the three conventional rotations of a single crystal. J. Magn. Reson. 155:257–262. [DOI] [PubMed] [Google Scholar]
- 54.Hong, M., J. D. Gross, C. M. Rienstra, R. G. Griffin, K. K. Kumashiro, and K. Schmidt-Rohr. 1997. Coupling amplification in two-dimensional MAS NMR and its application to torsion angle determination in peptides. J. Magn. Reson. 129:85–92. [DOI] [PubMed] [Google Scholar]
- 55.Leppert, J., B. Heise, and R. Ramachandran. 2000. 15N chemical shift tensor magnitude and orientation in the molecular frame of uracil determined via MAS NMR. J. Magn. Reson. 145:307–314. [DOI] [PubMed] [Google Scholar]
- 56.Wei, Y. F., D. K. Lee, and A. Ramamoorthy. 2000. One-dimensional dipolar-shift spectroscopy under magic angle spinning to determine the chemical-shift anisotropy tensors. Chem. Phys. Lett. 324:20–24. [Google Scholar]
- 57.Heise, B., J. Leppert, H. Wenschuh, O. Ohlenschlager, M. Gorlach, and R. Ramachandran. 2001. Two-dimensional relayed anisotropy correlation NMR: characterization of the C-13′ chemical shift tensor orientation in the peptide plane of the dipeptide AibAib. J. Biomol. NMR. 19:167–179. [DOI] [PubMed] [Google Scholar]
- 58.Yao, X., and M. Hong. 2002. Determination of Cα chemical shift tensor orientation in peptides by dipolar-modulated chemical shift recoupling NMR spectroscopy. J. Am. Chem. Soc. 124:2730–2738. [DOI] [PubMed] [Google Scholar]
- 59.Castellani, F., B.-J. van Rossum, A. Diehl, K. Rehbein, and H. Oschkinat. 2003. Determination of solid-state NMR structures of proteins by means of three-dimensional 15N–13C–13C dipolar correlation spectroscopy and chemical shift analysis. Biochemistry. 42:11476–11483. [DOI] [PubMed] [Google Scholar]
- 60.Havlin, R. H., D. D. Laws, H. M. L. Bitter, L. K. Sanders, H. H. Sun, J. S. Grimley, D. E. Wemmer, A. Pines, and E. Oldfield. 2001. An experimental and theoretical investigation of the chemical shielding tensors of C-13(α) of alanine, valine, and leucine residues in solid peptides and in proteins in solution. J. Am. Chem. Soc. 123:10362–10369. [DOI] [PubMed] [Google Scholar]
- 61.Oldfield, E. 2002. Chemical shifts in amino acids, peptides, and proteins: from quantum chemistry to drug design. Annu. Rev. Phys. Chem. 53:349–378. [DOI] [PubMed] [Google Scholar]
- 62.Walker, O., R. Varadan, and D. Fushman. 2004. Efficient and accurate determination of the overall rotational diffusion tensor of a molecule from N-15 relaxation data using computer program ROTDIF. J. Magn. Reson. 168:336–345. [DOI] [PubMed] [Google Scholar]
- 63.Antzukin, O. N. 1999. Side-band manipulation in magic angle spinning nuclear magnetic resonance. Prog. Nucl. Magn. Reson. Spectrosc. 35:203. [Google Scholar]
- 64.Alderman, D. W., G. McGeorge, J. Z. Hu, R. J. Pugmire, and D. M. Grant. 1998. A sensitive, high-resolution magic-angle turning experiment for measuring chemical shift tensor principal values. Mol. Phys. 95:1113–1126. [Google Scholar]
- 65.Bax, A., N. M. Szeverenyi, and G. E. Maciel. 1983. Correlation of isotropic shifts and chemical shift anisotropies by two-dimensional Fourier-transform magic-angle hopping NMR spectroscopy. J. Magn. Reson. 52:147–152. [Google Scholar]
- 66.Gan, Z. H. 1992. High-resolution chemical shift and chemical shift anisotropy correlation in solids using slow magic angle spinning. J. Am. Chem. Soc. 114:8307–8309. [Google Scholar]
- 67.Crockford, C., H. Geen, and J. J. Titman. 2001. Two-dimensional MAS-NMR spectra which correlate fast and slow magic angle spinning side-band patterns. Chem. Phys. Lett. 344:367–373. [Google Scholar]
- 68.Elena, B., S. Hediger, and L. Emsley. 2003. Correlation of fast and slow chemical shift spinning side-band patterns under fast magic-angle spinning. J. Magn. Reson. 160:40–46. [DOI] [PubMed] [Google Scholar]
- 69.Schmidt-Rohr, K., and H. W. Spiess. 1999. Multidimensional solid-state NMR and polymers. Academic Press, London.
- 70.Smondyrev, A. M., and M. L. Berkowitz. 2001. Molecular dynamics simulation of the structure of dimyristoylphosphatidylcholine bilayers with cholesterol, ergosterol, and lanosterol. Biophys. J. 80:1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Opella, S. J., A. Nevzorov, M. F. Mesleb, and F. M. Marassi. 2002. Structure determination of membrane proteins by NMR spectroscopy. Biochem. Cell Biol. 80:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bechinger, B. 2000. Biophysical investigations of membrane perturbations by polypeptides using solid-state NMR spectroscopy. Mol. Membr. Biol. 17:135–142. [DOI] [PubMed] [Google Scholar]
- 73.Dufourc, E. J., C. Mayer, J. Stohrer, G. Althoff, and G. Kothe. 1992. Dynamics of phosphate headgroups in biomembranes. Comprehensive analysis using phosphorus-31 nuclear magnetic resonance lineshape and relaxation time measurements. Biophys. J. 61:42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gale, P., and A. Watts. 1992. Effect of bacteriorhodopsin on the orientation of the headgroup of 1,2- dimyristoyl-sn-glycero-3-phosphocholine in bilayers: a 31P- and 2H-NMR study. Biochim. Biophys. Acta. 1106:317–324. [DOI] [PubMed] [Google Scholar]
- 75.Pinheiro, T. J., and A. Watts. 1994. Lipid specificity in the interaction of cytochrome-c with anionic phospholipid bilayers revealed by solid-state 31P NMR. Biochemistry. 33:2451–2458. [DOI] [PubMed] [Google Scholar]
- 76.Smith, R., F. Separovic, T. J. Milne, A. Whittaker, F. M. Bennett, B. A. Cornell, and A. Makriyannis. 1994. Structure and orientation of the pore-forming peptide, melittin, in lipid bilayers. J. Mol. Biol. 241:456–466. [DOI] [PubMed] [Google Scholar]