

Abstract
Glycine is a coagonist at the N-methyl-D-aspartate receptor. Changes in extracellular glycine concentration may modulate N-methyl-D-aspartate receptor function and excitatory synaptic transmission. The GLYT1 glycine transporter is present in glia surrounding excitatory synapses, and plays a key role in regulating extracellular glycine concentration. We investigated the kinetic and other biophysical properties of GLYT1b, stably expressed in CHO cells, using whole-cell patch-clamp techniques. Application of glycine produced an inward current, which decayed within a few seconds to a steady-state level. When glycine was removed, a transient outward current was observed, consistent with reverse transport of accumulated glycine. The outward current was enhanced by elevating intracellular or lowering extracellular [Na+], and was modulated by changes in extracellular [glycine] and time of glycine application. We developed a model of GLYT1b function, which accurately describes the time course of the transporter current under a range of experimental conditions. The model predicts that glial uptake of glycine will decay toward zero during a sustained period of elevated glycine concentration. This property of GLYT1b may permit spillover from glycinergic terminals to nearby excitatory terminals during a prolonged burst of inhibitory activity, and reverse transport may extend the period of elevated glycine concentration beyond the end of the inhibitory burst.
INTRODUCTION
Glycine has two distinct roles in neurotransmission. It acts as the main inhibitory neurotransmitter in the brain stem and spinal cord (1), and as an excitatory coagonist with glutamate at N-methyl-D-aspartate receptors (NMDARs) throughout the brain (2). After glycine is released at an inhibitory synapse, it is cleared by a combination of diffusion and reuptake into the synaptic terminal, neighboring neurons, and glial cells. Reuptake is performed by glycine transporters (GLYTs), of which several subtypes have been identified. GLYT2 is localized at inhibitory synaptic terminals, whereas GLYT1 is present in glia, including those that surround excitatory synapses (3,4). GLYT2 has a stoichiometry of 3 Na+/Cl−/glycine, which guarantees effective glycine accumulation under all physiological conditions, whereas GLYT1 has a stoichiometry of 2 Na+/Cl−/glycine, and glycine will be exported or imported depending on physiological conditions (5). No glycine-containing vesicles, or vesicular uptake proteins, are associated with excitatory presynaptic terminals (6–9). Instead, glycine may be released into the synaptic cleft from adjacent glia, via reverse transport through GLYT1 transporters. Evidence for nonvesicular, Ca2+-independent neurotransmitter release has been reported for γ-aminobutyric acid (GABA) (10) and glutamate transporters (11) and is reviewed in Attwell et al. (12). GLYT1-mediated glycine release has recently been demonstrated in a neuronal preparation (13), and GLYT1 can facilitate release of radiolabeled glycine from cell lines (14–16); in addition, reverse transport has been demonstrated in the oocyte expression system (5).
For GLYT1-mediated glycine release to modulate excitatory synaptic transmission, the resting glycine concentration in the synaptic cleft must not saturate the binding site on NMDARs. The minimum extracellular glycine concentration that GLYT1 can theoretically maintain (0.15 μM) is below the EC50 at the NMDAR (0.6–1.7 μM) (17). Inhibition of GLYT1 increases extracellular glycine concentration in the hippocampus of awake, freely moving rats (18). It also enhances NMDAR mediated synaptic currents in hippocampal pyramidal, prefrontal cortex, neonatal hypoglossal, and spinal cord (lamina X) neurons in brain slice (19–22). Thus, in some brain regions, GLYT1 maintains the glycine concentration in the synaptic cleft below the level needed to saturate NMDA receptors.
Glycine uptake by GLYT1 can downregulate excitatory synaptic transmission, but could reverse transport by GLYT1 enhance excitatory transmission under some circumstances? Reverse transport currents were observed in Xenopus laevis oocytes expressing rat GLYT1b after prolonged exposure to high levels of glycine (5). The current increased in amplitude with longer periods of glycine uptake, and increased exponentially with depolarization. Reapplying extracellular glycine inhibited the current. It remains unclear whether reverse transport through GLYT1b can influence extracellular glycine concentrations on the synaptic timescale. In oocyte recordings, the current lasts for several minutes, reflecting glycine accumulation in a very large intracellular volume. It is probable that glycine efflux from small mammalian cells will be more transient.
We investigated the kinetic and other biophysical properties of GLYT1b stably transfected in Chinese hamster ovary (CHO-GLYT1b) (23). Application of glycine produced an inward current that decayed to a steady-state level over several seconds. When glycine was removed, a transient overshoot current was observed, consistent with reverse transport of accumulated glycine. The reverse transport current was enhanced by elevating intracellular Na+, or by lowering the extracellular Na+ concentration and modulated by changes in extracellular glycine concentration as well as by changing the time exposed to glycine. We developed a model of GLYT1b, and the accumulation of glycine in a CHO cell. The model accurately predicts the time course of the transporter current under a range of experimental conditions, and provides insights into the role of GLYT1b transporters in modulating synaptic function.
MATERIALS AND METHODS
Cell culture media and solutions were supplied by GIBCO (Invitrogen, Grand Island, NY). All other chemicals were obtained from Sigma Chemicals (Sydney, Australia) unless otherwise stated. CHO-GLYT1b were a gift from John Morrow at Organon Laboratories (Newhouse, Scotland). N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine (NFPS) (also termed ALX-5407) was supplied by Allelix Neuroscience (NPS Pharmaceuticals, Salt Lake City, UT). NFPS was made into a stock solution of 1 mM in dimethylsulfoxide and diluted in to buffer as appropriate. The maximum dimethylsulfoxide concentration applied to cells was 0.1% (v/v), which did not generate a measurable current in untransfected CHO-K1 cells nor inhibit glycine transport currents when applied with glycine to CHO-GLYT1b cells.
Cell culture
CHO-GLYT1b cells were cultured in Dulbecco's modified Eagle's medium: Ham's F-12 nutrient mix (DMEM-F12) (1:1) media supplemented with 5% fetal bovine serum (CSL Biosciences, Parkville, Australia) and 0.6 mg/ml geneticin. Cells were incubated at 37°C in a humidified chamber with 5% CO2. Cells were grown to confluence and passaged with trypsin-EDTA. Experiments were performed 2–4 days after passage.
3H-Glycine uptake in CHO-GLYT1b cells
CHO-GLYT1b cells were passaged and plated out into 24 well plates (CulturPlate-24, Packard Instruments, Downers Grove, IL) at a density of 40 × 104 cells per well and left to settle for 3 h. Media were then removed and the cells were washed once with Hanks' buffer (37°C) to remove residual media. Uptake experiments were carried out at 37°C in the presence of 30 μM 3H-glycine for 20 min. 3H-glycine was applied alone or in the presence of NFPS (1 μM), a selective antagonist of GLYT1 transporters, or sarcosine (300 μM), a competitive substrate at GLYT1 transporters. NFPS acts slowly, so cells were preincubated for 5 min before the addition of 3H-glycine. Background 3H-glycine uptake was measured in untransfected CHO-K1 cells under the same conditions. Uptake was terminated by washing the cells twice with ice-cold Hanks' buffer, and cells were then lysed with 50 mM NaOH, and scintillation fluid (Ultima Gold, Packard Instruments) added. 3H-glycine uptake was quantified in counts per minute using a Unifilter-96 GF/C microplate scintillation counter (Packard Instruments).
Whole-cell patch-clamp recordings of CHO-GLYT1b cells
During whole-cell patch-clamp recordings, cell culture media were exchanged for an external salt solution containing in mM: NaCl 150; KCl 5; CaCl2 2; MgCl2 5; HEPES 10; glucose 10; adjusted to pH 7.4 (with NaOH), osmolality 320 mOsmol l−1. In experiments where extracellular Na+ was reduced, Na+ was substituted with equimolar choline chloride and pH was adjusted with KOH. Micropipette recording electrodes were pulled from borosilicate glass (1.5 mm outer diameter, 0.86 mm inner diameter, SDR Clinical Technology, Sydney, Australia) on a horizontal puller (Sutter Instruments, Navato, CA), fire-polished to a final resistance of 2–4 MOhm and coated with SigmaCoat before use. Electrodes were filled with an internal salt solution containing in mM: KCl 60; Kgluconate 80; CaCl2 0.2; EGTA 10; NaCl 5; HEPES 30; MgATP 5; adjusted to pH 7.4 (with KOH), osmolality 295 mOsmol l−1. In experiments where intracellular Na+ was increased, it was done at the expense of equimolar K+. The intracellular Na+ concentration ([Na+]i) is indicated in mM on each figure accompanying this article. In figures where the glycine gradient is altered, [glycine] is also indicated in mM.
Whole-cell glycine transport currents were recorded from stably transfected CHO-GLYT1b cells. Cells presented were voltage clamped at 0 mV; control currents were also recorded at −30 mV for each experiment. Glycine transport currents were recorded using an Axopatch 200B amplifier (Axon Instruments, Molecular Devices, Union City, CA), with a CV203BU head stage (Axon Instruments) and AxoGraph 4.8 (Axon Instruments) acquisition software. Currents were low-pass filtered at 2 kHz. During recording, extracellular solution was delivered to the cell with a Warner Instruments (Hamden, CT) SF-77B perfusion fast step system, which produced a solution exchange time of <100 ms. Series resistance (<8 MOhm) was compensated by 80% and continuously monitored during experiments. Cell capacitance was compensated for manually by nulling capacitive transients evoked by applying a 5 mV pulse at 0 mV and ranged from 25–100 pF. Liquid junction potentials were calculated using JPCalc (written by P. Barry, University of New South Wales, 1994) and subtracted.
Data analysis
Current (I) as a function of glycine concentration ([gly]) was fitted by least-squares minimization to
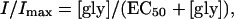 |
(1) |
where Imax is the maximal current and EC50 the concentration of glycine that generates a half-maximal current. To assess difference in means, analysis of variance (ANOVA), with the appropriate post hoc test or Student's t-test, was preformed using Microsoft Excel or GraphPad Prism software (24). Significant differences (p < 0.05) are indicated in the figures accompanying this article with an asterisk.
Modeling
A model was constructed of glycine accumulation in a CHO cell expressing GLYT1b. The two main goals were to provide a quantitative description of transporter function in our expression system, and to make predictions about GlyT1b performance in other systems with different geometry, transporter density, or solute gradients. It was necessary to set several model parameters to arbitrary but reasonable values. Thus, some parameters, such as substrate binding rate, are arbitrary, whereas others, such as substrate binding affinity, are constrained by the data and likely to be accurate. The transporters were represented using a Markov reaction scheme, which describes transitions between eight states. The model incorporated the buildup of glycine in the finite intracellular volume of the CHO cell, and the slow diffusion of glycine from the cellular compartment into the much larger volume of the patch electrode. The cell was treated as a single, well-stirred compartment, with a volume of Vcell liters, which can be determined from the cell radius, rcell,
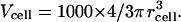 |
(2) |
The cell's membrane capacitance (determined from a test pulse) was used to estimate rcell. Specific membrane capacitance (Cm) is ∼1 μF cm−2, but the “apparent” value of this parameter can be distorted by membrane foldings, which are not visible under light microscopy (25). A high density of filopodia was observed on the surface of CHO cells under electron microscopy (results not shown), so apparent Cm was set to 3 μF cm−2, similar to the value obtained for HEK cells (25). For a GLYT1b transport rate of Tgly molecules per second into the cell, the change of intracellular glycine concentration Δ[gly] during a short time interval, Δt, is,
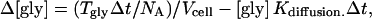 |
(3) |
where NA is Avogadro's number and Kdiffusion is the diffusion rate constant (s−1) from the cell to the patch electrode. The model was constructed using AxoGraph 4.9 (Axon Instruments), and the results were independently verified using the general-purpose modeling package, Berkeley Madonna. Information about these programs can be found at www.axon.com/cn_AxoGraph4.html and www.berkeleymadonna.com, respectively. Files implementing the AxoGraph or Berkeley Madonna models are available on request from John.Clements@anu.edu.au. Model parameters were systematically adjusted using a simplex algorithm to minimize the sum of squared errors between simulated and recorded current transients. The GLYT1b Markov model incorporated several simplifications and constraints to make it physically plausible, and to keep the number of free parameters manageable. Glycine and Na+ were assumed to bind in a narrow pore in the transporter, such that Na+ binding precedes glycine binding to the extracellular conformation, and Na+ unbinding precedes glycine unbinding from the intracellular conformation (26). The binding and cotransport of Cl− was not included in the model, because the concentration gradient was constant for all experiments, and Cl− binding/unbinding is not rate-limiting for inward glycine transport (27). However, cotransport of Cl− was considered when calculating the transporter current, Igly, which is generated by the transfer of one net positive charge per glycine molecule (one Cl− with two Na+):
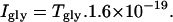 |
(4) |
There was no information in the data about the binding rate of glycine, so it was arbitrarily fixed at 5 μM−1 s−1, similar to its binding rate at ligand-gated receptors (28). Similarly, the Na+ binding and unbinding rates from the intracellular site were arbitrarily fixed at 1 μM−1 s−1 and 50,000 s−1, respectively, based on the observed sensitivity to intracellular Na+ for the related GABA transporter (10). The forward turnover rate of the fully loaded transporter (Na+ and glycine bound) was fixed at 5 s−1 based on data from the related GABA and serotonin transporters (29–31), and the reverse turnover rate was a free parameter. The forward and reverse turnover rates of the unloaded transporter were both free parameters. The quality of the fit was not sensitive to doubling or halving any one of the values chosen for the constrained reaction rate parameters (results not shown). Thus, if these parameters were not constrained, they could drift to physically unreasonable values during the fitting procedure. In total, the model had eight free parameters. Several observations confirm that it was not overparameterized. The optimum parameters showed relatively little cell-to-cell scatter, and the results for a given data set were independent of the starting guess. Also, the model was fit to a pair of transients that required 10 separate parameters to describe them, 5 parameters for each transient. These were inward current amplitude, decay time constant, steady-state inward current, outward current amplitude, and outward current decay time constant (see Fig. 8 C).
FIGURE 8.
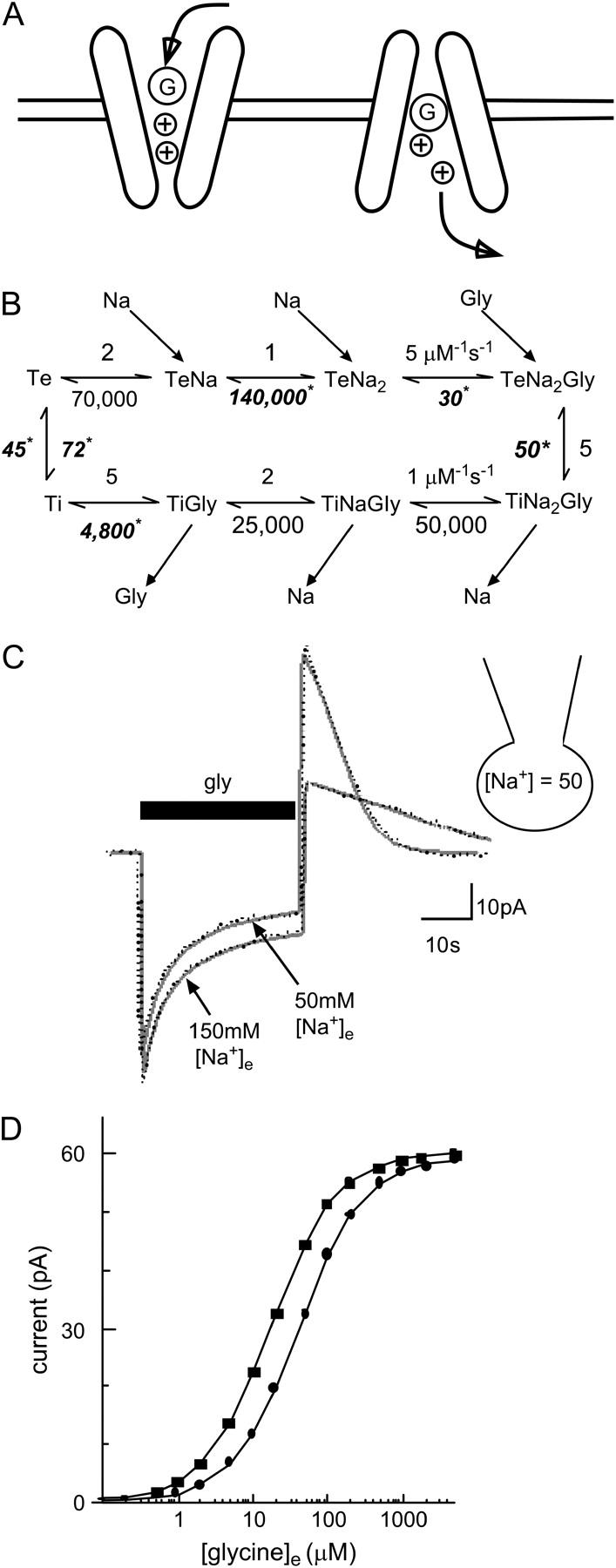
A model of glycine transport by GLYT1b. (A) Schematic showing glycine (G) and Na+ (+) binding sequentially in a narrow pore. Na+ binds before glycine to the extracellular conformation and is first to unbind from the intracellular conformation of the transporter. (B) Markov model of GLYT1b function; all reaction rates are given in s−1 unless indicated in the figure. Some reaction rates were constrained, but others were determined by fitting two separate responses to a pulse of glycine, one obtained in 50 mM [Na+]e, and the other in 150 mM [Na+]e. Unconstrained reaction rates are shown in bold italics with asterisks. (C) A typical fit obtained with the model (shaded line) of experimental data (dotted line) in two conditions, with 50 mM [Na+]i and either 150 mM or 50 mM [Na+]e. Fit parameters can be found in the Methods (fixed parameters) and Results sections (variable parameters). (D) Independent verification of the model was obtained by using the model to predict the glycine concentration-response curve with 50 mM [Na+]i and 50 mM [Na+]e (•), and with 5 mM [Na+]i and 150 mM [Na+]e (▪). The predicted curves are in good agreement with experimental results (presented in Figs. 1 C and 4 B).
RESULTS
The presence of GLYT1b transporters in the stably transfected CHO-GLYT1b cell line was confirmed by examining substrate-selective uptake, and sensitivity to the GLYT1 specific inhibitor NFPS. After exposure to 3H-glycine as described in Methods, CHO-GLYT1b cells accumulated 11 times more (5.982 ± 0.48 fmol/min/104 cells, n = 6) 3H-glycine than CHO-K1 cells (0.524 ± 0.07 fmol/min/104 cells, n = 6). Sarcosine (300 μM), a substrate of GLYT1 transporters, reduced 3H-glycine uptake by 79 ± 2% (n = 6), and NFPS (1 μM) completely inhibited GLYT1b mediated 3H-glycine uptake (Fig. 1 A).
FIGURE 1.
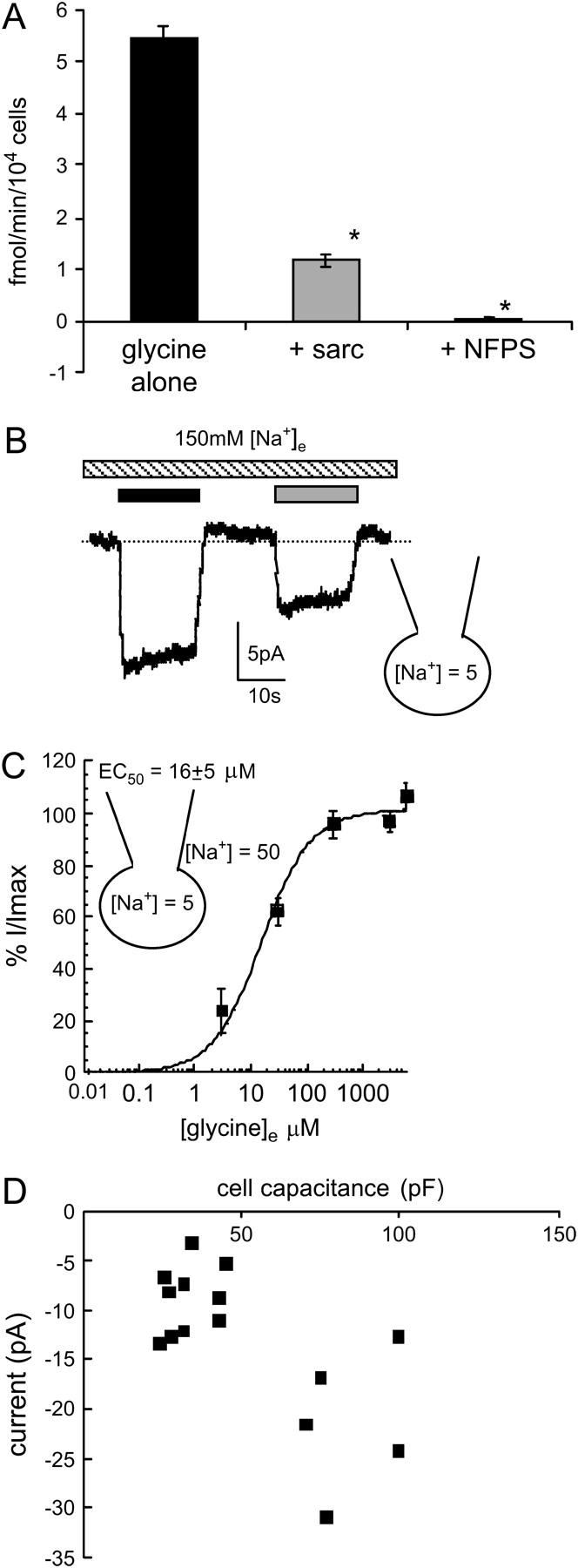
Glycine transport currents in CHO cells transfected with GLYT1b. (A) 3H-glycine uptake by CHO-GLYT1b cells (black bar); in the presence of the competitive GLYT1 substrate sarcosine (300 μM, shaded bar); and the GLYT1 specific inhibitor NFPS (1 μM, open bar) (n = 6, ANOVA). (B) A whole-cell patch-clamp recording at 0 mV of GLYT1b mediated currents. Sarcosine (300 μM, shaded bar) elicits a current that is smaller in amplitude than the current induced by glycine (300 μM, black bar). (C) Glycine concentration-response curve for transport currents mediated by CHO-GLYT1b cells voltage clamped at 0 mV. Glycine transport currents were normalized to the maximal current of each cell and fitted to the Michaelis-Menten equation (n = 10). (D) Correlation between cell capacitance and peak current amplitude (n = 15).
Application of maximal doses of glycine (300 μM) and sarcosine (300 μM) elicited inward currents in CHO-GLYT1b cells (Fig. 1 B). The peak amplitude of the current elicited by sarcosine is ∼80% of the amplitude of the current induced by the same dose of glycine, consistent with previous reports in cell lines (32) and in oocytes (K. R. Aubrey, P. Ju, and R. J. Vandenberg, unpublished data). The EC50 for glycine was 16 ± 5 μM (n = 10, Fig. 1 C), which is consistent with values obtained for GLYT1b expressed in oocytes (33,34). The inward current elicited by saturating glycine had an average amplitude of 18 ± 2 pA (n = 50, range from 4 to 60 pA), and the largest transporter currents were recorded 3 days after passage. Although we have recorded a range of glycine transporter current amplitudes, closer investigation of this data reveals that this range of amplitudes is more a reflection of a range of cell sizes, as opposed to changes in GLYT1b expression levels. Fig. 1 D illustrates the correlation between whole-cell capacitance and current amplitude recorded from n = 15 cells. Coapplication of NFPS with glycine resulted in a complete block of the transport current (Fig. 2 A).
FIGURE 2.

Characteristic features of glycine transport currents measured in the presence and absence of NFPS. (A) Application of glycine (300 μM, black bar) induces an inward current that decays to a sustained current. When glycine is removed, the current rapidly decays and transiently overshoots the baseline. When glycine is coapplied with NFPS (1 μM, open bar), both the inward and overshoot currents are blocked. The internal and external Na+ concentrations are indicated. Numbers mark the (1), baseline current; (2), peak current; (3), sustained current; (4), overshoot current; and (5), NFPS block of the overshoot current. (B) Glycine (black bar) induces an inward current, but NFPS (open bar), applied in the absence of glycine, has no effect on the baseline current.
When glycine was applied to CHO-GLYT1b cells voltage clamped at 0 mV, with no glycine added to the patch pipette, an inward current developed rapidly and then relaxed toward a steady-state level, referred to in the following as the sustained current (Fig. 2 A). The relaxation of the inward current is observed in other cell lines expressing GLYT1b (HEK cells (35) and quail fibroblast cells (K. R. Aubrey and R. J. Vandenberg, unpublished data), as well as oocytes expressing GLYT1b (K. R. Aubrey, P. Ju, and R. J. Vandenberg, unpublished data). When glycine was removed, the current transiently overshot the baseline before relaxing back to the baseline current level over several seconds (Fig. 2 A). The transient outward current is referred to in the following as the overshoot. It is observed to a variable extent in oocytes (unpublished data) and has not been reported in GLYT1 transfected cell lines. Coapplication of glycine with NFPS results in a characteristic slowly developing, irreversible block (36) of both the inward and overshoot currents, suggesting that the glycine transporter mediates the overshoot. Application of glycine or NFPS to untransfected CHO-K1 cells did not elicit a current (data not shown), confirming that GLYT1b expression is required for both inward and overshoot currents.
Other members of the Na+/Cl−-dependent neurotransmitter transporter family have constitutive, substrate-independent leak currents that can be blocked by transport inhibitors (for example, GABA (GAT) (37), dopamine (38), and serotonin (29) transporters); however, no leak currents or uncoupled currents have been observed for GLYT1 (33,34,39). NFPS was applied to CHO-GLYT1b cells under different experimental conditions, to rule out the possibility that the overshoot or any other component of the glycine activated currents were due to a leak current. These conditions were: 5 mM [Na+]i and 150 mM [Na+]e (n = 5), 50 mM [Na+]i and 150 mM [Na+]e (n = 5, Fig. 2 B), or 50 mM [Na+]i and 50 mM [Na+]e (n = 3). Under each of these conditions, the current measured during NFPS application was not significantly different from the baseline current (ANOVA, data not shown), confirming previous reports that GLYT1b does not have a constitutive leak current.
Glycine transport by GLYT1b is Na+ dependent (5), and the Na+ gradient across glial cell membrane in the central nervous system may vary, depending on local activity levels and other factors. We investigated the effects of altering [Na+]e and [Na+]i on GLYT1b function in CHO cells. With 5 mM [Na+]i and 150 mM [Na+]e, application of saturating glycine (300 μM) produced a sustained current amplitude that was 68 ± 5% of the peak current, and an overshoot current amplitude that was 19 ± 3% of the peak inward current (n = 12, Fig. 3 A). When [Na+]i was increased from 5 mM to 50 mM, the relative amplitude of the sustained current was unchanged at 63 ± 6% of peak, but the overshoot current was enhanced to 40 ± 6% of peak (n = 9, Fig. 3 B). Reducing [Na+]e from 150 mM to 50 mM further enhanced the relative amplitude of the overshoot current to 60 ± 6% of peak, but again did not alter the sustained current, which was 63 ± 4% of peak (n = 10, Fig. 3 B). A bar graph of the amplitude of the sustained and overshoot currents relative to the amplitude of the peak current is presented in Fig. 3 C. Note that the data presented in Fig. 3 A is from a different cell than that presented in Fig. 3 B; thus their current amplitudes are not directly comparable. In summary, the relative amplitude of the overshoot current was enhanced when [Na+]e was decreased and [Na+]i was increased, suggesting that the overshoot current results from reverse transport of accumulated glycine.
FIGURE 3.

Amplitude of overshoot current is enhanced by depletion of the Na+ gradient. Extracellular application of glycine stimulates an inward current (300 μM, black bar). (A) A glycine transport current recorded with 5 mM [Na+]i and 150 mM [Na+]e (striped bar) and (B) a different cell with 50 mM [Na+]i and either 150 mM or 50 mM (shaded bar) [Na+]e. (C) Bar graph of the sustained (black bars) and overshoot (open bars) current amplitudes as a percentage of peak current recorded in various Na+ gradients (n ≥ 10, ANOVA).
If this hypothesis is correct, then the amount of glycine accumulation in the cell should influence the amplitude of the sustained and overshoot currents. This was investigated in two ways: first by altering the applied glycine concentration and second by altering the time that glycine was applied.
Peak and sustained inward currents and peak overshoot current were measured at a range of applied glycine concentrations (Fig. 4 A). A standard pulse width of 15 s was used for all applications, and the interpulse recovery period was >60 s. Currents were recorded with 50 mM [Na+]i and 50 mM [Na+]e to enhance the relative amplitude of the overshoot current. Glycine concentration-response curves were constructed with current amplitudes normalized to the peak inward current at 300 μM glycine (Fig. 4 B). The peak current had an EC50 of 39 ± 3 μM, which was higher than the value obtained earlier (16 ± 5 μM, Fig. 1 C) due to the reduced [Na+]e (27,40). The sustained inward current had an EC50 of 50 ± 11 μM and saturated at 56 ± 9% of the peak current. The overshoot outward current had an EC50 of 48 ± 5 μM and saturated at 76 ± 3% of the peak current. The EC50 values for the peak, sustained, and overshoot currents do not differ significantly (n = 7, repeat measure ANOVA).
FIGURE 4.
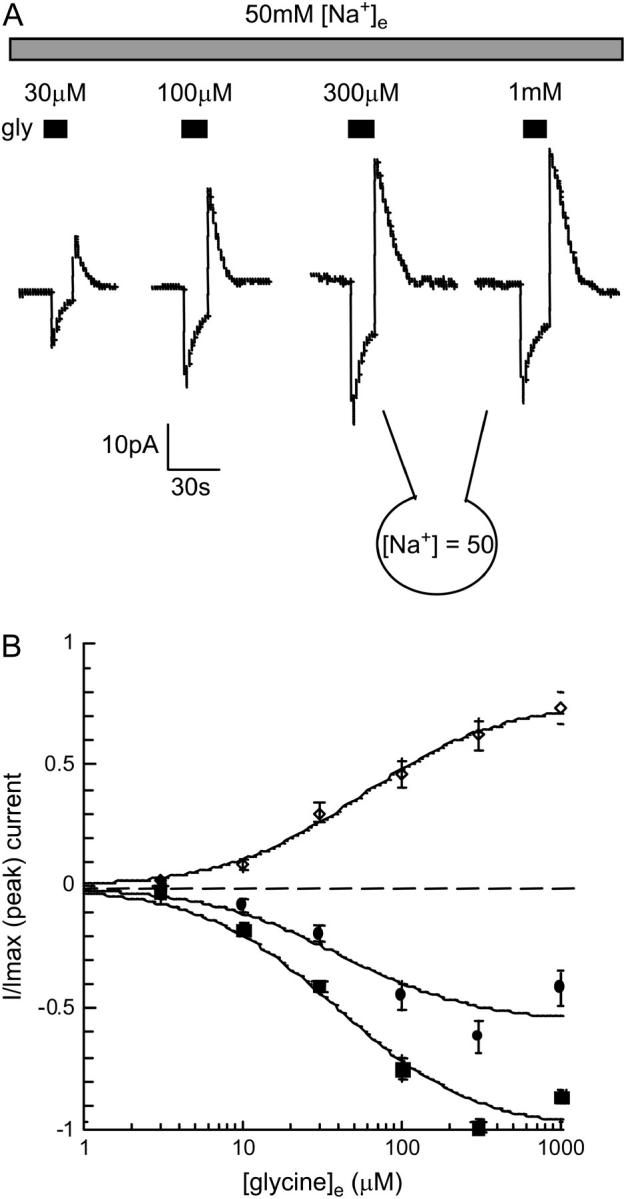
Amplitude of the peak, sustained, and overshoot currents is dependent on the extracellular glycine concentration. (A) Currents stimulated by increasing [glycine]e with 50 mM [Na+]i and 50 mM [Na+]e. (B) Concentration-response curves for the peak (▪), sustained (•), and overshoot (◊) currents normalized to the Imax of the peak current and fitted to the Michaelis-Menten equation (n = 7). The EC50 values for the peak, sustained, and overshoot currents were not significantly different (repeat measure ANOVA).
Peak and sustained inward currents and peak overshoot currents were also measured in response to a fixed concentration of glycine (300 μM) over a range of application times (5, 15, and 60 s; Fig. 5 A). Once again currents were recorded with 50 mM [Na+]i and 50 mM [Na+]e to enhance the relative amplitude of the overshoot current, and the interpulse recovery period was >60 s. When glycine was applied for 5 s, the sustained current was 70 ± 4% and the overshoot 43 ± 16% of the peak current; when applied for 15 s, the sustained current was 52 ± 10% and the overshoot current 55 ± 11% of the peak current; these values are not significantly different from those reported in Fig. 3. When glycine was applied for 60 s, the amplitude of sustained current was 31 ± 16% and the overshoot 70 ± 19% of the peak current. The relative amplitude of the sustained and overshoot current compared to peak current increased significantly (5 vs. 60 s) as the time of glycine exposure increased. This data is presented in Fig. 5 B (n = 3, repeat measure ANOVA).
FIGURE 5.

Amplitude of the sustained and overshoot currents is dependent on the time of glycine application. (A) Currents stimulated by 300 mM [glycine]e with 50 mM [Na+]i and 50 mM [Na+]e applied for 5, 15, and 60 seconds (B) Bar graph of the sustained (black bars) and overshoot (open bars) current amplitudes as a percentage of peak current recorded for each of the time periods glycine was applied (n = 3; repeat measure ANOVA).
Based on the observation that GLYT1 does not mediate any leak conductances and given that sustained net reverse transport can be detected with NFPS, it should therefore be possible to apply standard transporter equilibrium equations to predict the behavior of the sustained outward current as a function of the Na+ concentration gradient.
The equilibrium membrane potential (Vr) of GLYT1b can be calculated from
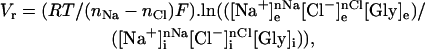 |
(5) |
where F = Faraday constant, R = gas constant, and T = 298 K. [Cl−]i = 65 mM; [Cl−]e = 170 mM. nNa and nCl are the number of Na+ and Cl− ions coupled to the transport of each glycine molecule (2 and 1, respectively). Setting [Gly]i to 2 mM, [Gly]e to 1 μM, [Na+]i to 5 mM, and [Na+]e to 150 mM, the predicted equilibrium potential Vr is +4 mV. If the Na+ gradient is reduced by lowering [Na+]e to 50 mM, the predicted Vr changes to −52 mV. Therefore, when the glycine gradient and [Na+]i are as described above, net reverse transport should be detected when CHO-GLYT1b cells clamped at 0 mV have 50 mM [Na+]e, but not at 150 mM [Na+]e. This prediction was tested experimentally. In 150 mM [Na+]e, stepping from 1 μM to 300 μM, glycine elicited an inward current (Fig. 6 A). Changing the extracellular solution to 50 mM [Na+]e produced a large outward shift in the baseline current, and extracellular glycine application again elicited an inward current. This glycine-elicited inward current appears larger in amplitude than the current stimulated by glycine in 150 mM [Na+]e, indicating that the inward glycine current in 50 mM [Na+]e results from a combination of glycine inhibition of the reverse transport current and glycine stimulation of forward glycine transport. Application of NFPS in 50 mM [Na+]e and 1 μM [gly]e induced a slowly developing inward current, consistent with progressive block of a substantial and sustained reverse transport current. However, when the [Na+]e was switched back to 150 mM the baseline current was not altered by the application of NFPS (Fig. 6 A). This illustrates that at 0 mV, reverse transport occurs in 50 mM [Na+]e, but not in 150 mM [Na+]e. This observation is consistent with the predictions of Eq. 1 and a summary of these results are presented in Fig. 6 B.
FIGURE 6.

Elevating [glycine]i stimulates an outward current that can be blocked with NFPS. (A) When the glycine and [Na+]i gradients are set as described on the figure, glycine (300 μM, black bar) stimulates an inward current in 150 mM [Na+]e (striped bar) and 50 mM [Na+]e (gray bar). NFPS (1 μM, open bar) blocks a current in 50 mM [Na+]e, but does not block a current in 150 mM [Na+]e (compare the baseline current at 150 mM [Na+]e before NFPS application and then after switching from 50 mM [Na+]e to 150 mM [Na+]e as NFPS inhibition of GLYT1b persists after washout of free drug (see Fig. 2 B)). (B) Bar graph comparing the amplitude of the NFPS blocked currents in 150 mM and 50 mM [Na+]e. (n = 4, paired Student's t-test). (C) When [glycine]i is 16 mM, NFPS (1 μM, open bar) blocks a sustained outward current. (D) The amplitude of the NFPS blocked current was enhanced by elevating [Na+]i and lowering [Na+]e (n ≥ 4).
Sustained reverse transport was seen in oocytes expressing GLYT1b when their intracellular glycine concentration was adjusted to 16 mM (5). This result was replicated in our study by patch clamping CHO-GLYT1b cells with 16 mM glycine in the pipette solution. When NFPS (1 μM) was applied, a slowly developing inward current was seen (Fig. 6 C). Reducing [Na+]e and increasing [Na+]i, enhanced the relative amplitude of the reverse transport current blocked by NFPS (Fig. 6 D). This result is similar to data presented earlier (Fig. 3 C), confirming that the overshoot arises from reverse transport through GLYT1b.
A concentration-response curve was constructed by altering [gly]i and measuring the amplitude of the outward current blocked by NFPS. The irreversible nature of NFPS meant that only one [gly]i concentration could be tested per cell. All recordings were made 2 or 3 days after passage, with 5 mM [Na+]i and 50 mM [Na+]e. Each data point represents data from n ≥ 5 cells. The intracellular glycine EC50 for reverse transport through GLYT1b was estimated to be 4.3 ± 0.6 mM (Fig. 7).
FIGURE 7.

Intracellular glycine concentration-response curve for reverse transport by GLYT1b. The amplitude of the NFPS blocked reverse transport current was measured as in Fig. 6 C, in the presence of 50 mM [Na+]e and 5 mM [Na+]i, across a range of internal glycine concentrations (0–48 mM). Raw GLYT1b reverse transport currents were fitted with the Michaelis-Menten equation (n ≥ 5).
We investigated the factors determining the kinetic properties of the inward, sustained, and overshoot currents in CHO-GLYT1b cells. Identifying these factors may provide insights into glycine dynamics in glia. A model of transporter function was constructed (see Methods), which accurately predicts the current transient elicited by glycine under a variety of experimental conditions. It assumed that Na+ and glycine bind sequentially inside a narrow pore in GLYT1b (Fig. 8 A). This assumption gives rise to an eight-state Markov reaction scheme describing GLYT1b function (Fig. 8 B), which was combined with a model of glycine dynamics in a CHO cell coupled to a patch pipette. Free parameters were systematically adjusted using a simplex algorithm, until an optimal fit to two different current transients was obtained. The two transients were recorded in the same cell with 50 mM [Na+]i and either 50 or 150 mM [Na+]e. They were elicited by a glycine pulse (300 μM, 30 s). The free parameters were the number of transporters, the diffusion rate from the cell into the patch electrode, and the six reaction rates indicated in Fig. 8 B. After parameter optimization, the model accurately predicted the complex, multiexponential inward and outward currents recorded under two different experimental conditions. As expected, there was considerable cell-to-cell variability in the number of transporters (from 1.2 × 107 to 8.5 × 107, n = 5), and the diffusion rate (0.007 to 0.047 s−1), most likely reflecting geometric differences between cells. Consistent with this, the estimated transporter density showed less variability (3200–8500 μm−2), and the amplitude of currents stimulated by glycine (300 μM) from cells recorded with 50 mM [Na+]i and 50 mM [Na+]e correlated strongly with cell capacitance (Fig. 1 D). The optimum reaction rates for the free parameters were: loaded transporter turnover rate fixed at 5 s−1, reverse rate 50 ± 7 s−1; unloaded turnover rate 46 ± 8 s−1, reverse rate 72 ± 15 s−1; glycine unbinding rate from the extracellular conformation 30 ± 5 s−1, and from the intracellular conformation 4800 ± 400 s−1; Na+ unbinding rate from the extracellular conformation 140,000 ± 20,000 s−1 (n = 5).
As an independent test of the model, the optimum parameters obtained from each cell, was used to simulate responses to a range of glycine concentrations, and concentration-response curves were constructed. Simulations were performed for two different Na+ gradients. Setting [Na+]e to 150 mM and [Na+]i to 5 mM gave a predicted EC50 of 14 ± 2 μM (n = 5), which is consistent with the experimentally observed EC50 of 16 ± 5 μM (Fig. 1 C). With 50 mM [Na+]e and 50 mM [Na+]I, an EC50 of 43 ± 6 μM was obtained, also consistent with the observed EC50 of 39 ± 3 μM (Fig. 4 B). These theoretical estimates of EC50 were generated purely from kinetic information, as the current transients used in the fitting procedure were obtained at a single saturating concentration of glycine.
DISCUSSION
The small electrogenic currents generated by transporters are typically studied in Xenopus oocytes, because their efficient protein expression and large membrane surface area make the currents relatively easy to detect and measure. However, the large size of the oocyte slows solution exchange and alters the kinetics of the response. Here we demonstrate that GLYT1b transport currents can be studied in a mammalian cell line, where results can be interpreted more confidently. Stably transfected CHO-GLYT1b cells express up to 108 functional GLYT1b per cell, permitting electrogenic transport currents to be measured with an excellent signal/noise ratio. Furthermore, rapid solution exchange (∼100 ms) can be achieved at the surface of these small cells, permitting detailed kinetic analysis of transporter currents.
When extracellular glycine was applied to whole-cell patch-clamped CHO-GLYT1b cells, a transporter-specific inward current was stimulated. The inward transport current rapidly relaxed to a sustained current and, when glycine was removed from the extracellular recording solution, a transient overshoot current was observed. We hypothesized that the relaxation of the GLYT1b current was due to intracellular glycine accumulation, and that the overshoot current was due to reverse transport of the accumulated glycine. Consistent with this interpretation, the amplitude of the sustained and overshoot currents was dependent on the Na+ gradients (Fig. 3) and were altered as predicted in conditions that changed the concentration of accumulated glycine (Figs. 4 and 5). Net reverse transport was stimulated by adding glycine to the patch pipette and shown to behave as predicted by standard transporter equilibrium equations (Fig. 6). A reverse transport concentration-response curve was then constructed (Fig. 7), which indicates that the glycine EC50 for reverse glycine transport by GLYT1b is ∼4.3 ± 0.6 mM, similar to previous reports for the GABA transporter (10). Based on these data, a model of GLYT1b function was constructed. The time course of the response of a CHO-GLYT1b cell to a long pulse of glycine can be explained in terms of this model of transporter function (Fig. 8, B and C).
Kinetics properties of GLYT1b
During continued application of glycine, the inward current decays exponentially as glycine accumulates in the CHO cell. The reduction in current is caused by rebinding of glycine to the transporter in the intracellular conformation, which slows recycling of the transporter to the extracellular conformation (via the Ti → Te pathway), and accelerates reverse glycine transport (via the TiNa2Gly → TeNa2Gly pathway). If the CHO cell were a sealed system, the current would decay to zero. However, intracellular glycine diffuses into the large sink provided by the recording electrode, and the current decays to a sustained level that is determined by the rate of diffusion. Thus, the sustained inward glycine transport is a recording artifact attributable to the patch electrode, and would not be present in an intact CHO-GLYT1b cell or glial cell. Glial cells are typically smaller and have many more small diameter processes than CHO cells, suggesting that GLYT1 mediated uptake may decay rapidly to zero in vivo during periods of strong or sustained glycinergic drive.
The diffusion time constant for the decay of the peak current to the sustained current recorded in CHO-GLYT1b cells ranged from 20 s to 150 s, which is in good agreement with other estimates (range 10–100 s) (41,42). The diffusion time constant (= 1/Kdiffusion) should be approximately proportional to series resistance, as both are limited by the inside diameter of the patch electrode, and should be proportional to cell volume. We plotted 1/Kdiffusion from each of the five analyzed cells versus series resistance multiplied by estimated volume of each cell (result not shown). The expected trend was apparent, but the correlation was not statistically significant.
When glycine is washed off a CHO-GLYT1b cell after a long pulse, a transient outward current is observed. We carefully characterized the overshoot current, and demonstrated that it is most likely to result from reverse transport of accumulated glycine through GLYT1b, consistent with previous observations using the oocyte expression system (5). The theoretical model accurately predicts the amplitude and time course of the overshoot current under a range of recording conditions. Decreasing [Na+]e increases the amplitude of the overshoot current and accelerates its decay. This observation can also be explained in terms of the model (Fig. 8 B). When [Na+]e is reduced, the transporter spends less time in the states with Na+ bound (TeNa, TeNa2), and more time in the unbound state (Te). This promotes the transition Te → Ti, which increases the availability of transporters in the intracellular conformation and enhances reverse transport (TiNa2Gly → TeNa2Gly). Faster reverse transport accelerates the depletion of the accumulated glycine, so the initially larger overshoot decays more rapidly. The model confirms that the overshoot current is consistent with reverse transport of accumulated glycine through GLYT1b.
The model predicts that reverse transport occurs faster (50 s−1) than inward transport (fixed at 5 s−1). If correct, then at equilibrium when [glycine]e is very low, GLYT1 will be oriented in the extracellular configuration most of the time. As a result, the number of transporters available to bind released glycine will be optimized for efficient removal of glycine from the synapse.
When glycine is applied, an inward current develops with a 20–80% rise time of 110 ± 8 ms (n = 8). In contrast, the model predicts that the response to an instantaneous step into glycine (300 μM) will have a 20–80% rise time of only 5 ms, suggesting that solution exchange at the cell membrane is rate limiting for transporter activation in our experiments. Our model supports the suggestion that GLYT1b in astrocyte processes proximal to inhibitory synapses will rapidly clear synaptically released glycine.
We carefully examined several alternative reaction schemes but rejected them, as they were not consistent with the data. A scheme based on the assumption that glycine binding must precede Na+ binding accurately fit the two transients recorded in 50 and 150 mM [Na+]e, but when the optimally fitted model was used to generate a concentration-response curve for glycine, the predicted EC50 was more than an order of magnitude too low (results not shown). If we assumed that the binding of Na+ and glycine occurs at two independent, noninteracting sites, the reaction scheme did not accurately fit the two transients. The preferred model (Fig. 8) is subject to all the usual caveats applied to Markov schemes. Several reaction rates could not be determined from our data, and were constrained to values determined in studies of closely related transporter proteins. This may introduce small systematic errors into our free parameter estimates. The model is a pragmatic simplification of the kinetic and pharmacological properties of GLYT1b. It effectively summarizes the main features of the transporter's dynamic behavior. The model is particularly useful for exploring the experimental results more deeply and for extrapolating beyond them to other scenarios.
Comparison with GABA transporters
Reverse transport currents mediated by GABA transporters (GAT) have been observed in expression systems and native preparations (10,37,43,44), and have similar properties to those reported here for GLYT1b. GAT-mediated reverse transport is dependent on intracellular GABA, Na+, and Cl−, and reverse transport is faster at positive potentials. The EC50 for intracellular GABA binding was 2 mM at 0 mV (10). Our experimental and theoretical results suggest a similar intracellular EC50 for glycine at GLYT1b. We also found that the intracellular Na+ binding site is not saturated at 5 mM, because increasing [Na+]i to 50 mM enhanced the overshoot current. GAT also has a very high intracellular EC50 for Na+ of 54.6 mM (10). The similar pharmacological and functional properties of GAT and GLYT1b suggest that they may play similar roles at inhibitory synapses. The kinetic properties of GLYT1b imply that, depending on the density of transporter in the membrane and the intracellular volume in which glycine accumulates; they may regulate excitatory synaptic function on the subsecond timescale.
Physiological roles of GLYT1 in vivo
In this report, we have presented a plausible model for GLYT1 function based on biophysical studies of GLYT1b expressed in CHO cells. In the following section, we will discuss the significance of this work in terms of understanding the GLYT1 function in vivo. Transporter function can sometimes be sensitive to expression levels in recombinant experimental systems (45). The model estimates that the density of GLYT1 expression in CHO-GLYT1b cells is between 3200 μm−2 and 8500 μm−2. GLYT1b transporter density has not been measured in vivo, but the density of GABA transporters has been estimated at 800–1300 μm−2 in mouse cerebellum and hippocampal presynaptic boutons (46), and the density of glutamate transporters has been estimated at 2300 μm−2 and 8500 μm−2 in astrocytes of the hippocampus and cerebellum, respectively (47). These in vivo values are similar to our estimates of the GLYT1b density in CHO-GLYT1b cells (3200–8500 μm−2). GLYT1 transport currents recorded from mouse Bergmann glial cells are of similar amplitude to those measured here, suggesting a high density of GLYT1 expression in this in vivo preparation (13).
We can confidently extrapolate from the biophysical properties of GLYT1b observed in this study to predict GLYT function in vivo. The cell volume of astrocytes and other glia is smaller than CHO-GLYT1b cells (diameter 20–40 μm), resulting in faster glycine accumulation for a given GLYT density. Furthermore, the kinetics of glycine accumulation and efflux will be even faster in the microenvironment of the abundant low-diameter processes that project from the astrocyte cell body, due to their high surface area/volume ratio (48). Finally, GLYT kinetics will be faster in vivo at 37°C compared to our room-temperature experiments (49). Thus, the general characteristics of the transport currents observed in this study, including rapid saturation and transient reverse transport, are almost certain to occur in vivo. In microscopic domains of the brain and spinal cord, the kinetics of glycine accumulation and efflux are likely to be significantly faster than observed in this study, and may occur on a subsecond timescale.
Our findings demonstrate that GLYT1b is capable of mediating glycine release as well as glycine uptake in mammalian cells. The release of accumulated glycine has the potential to modulate synaptic function. For example, a sustained period of inhibitory synaptic activity may saturate GLYT1 uptake due to accumulation of glycine in the low-diameter processes that project from glial cells and surround the synapse. This would lead to local extracellular build up of glycine, which would then spill over to nearby excitatory synapses and enhance NMDAR sensitivity. When inhibitory activity decreases, the accumulated glycine may be released from glia via reverse transport, thereby prolonging the enhancement of NMDAR sensitivity.
The reversal potential for GLYT1b in astrocytes under resting physiological conditions is estimated at between −65 and −95 mV (50,51), and astrocyte resting membrane potential is approximately −80 mV (52). So GLYT1b operates close to its reversal potential. Activation of any of the receptor-gated ion channels known to be expressed in astrocytes, may change their resting membrane potential and alter GLYT1b function. Similarly, changes in intracellular Na+ or Cl− concentration may shift the reversal potential of GLYT1b, causing functional changes. For example, pathological states may elevate [Na+]i (53,54), which would stimulate glycine release and thereby increase neurotoxic NMDAR activation.
Reverse transport of glycine may also play a role at inhibitory synapses. It is hypothesized that at times of low synaptic activity, GLYT1 releases glycine from glial cells surrounding the synapse, which is taken up by GLYT2 on postsynaptic neurons for packaging into vesicles (6,55).
Acknowledgments
Thanks to Trevor Lewis, Stefan Broer, and Mark Connor for helpful discussion, and to Billy Chieng, Ernie Jennings, and Louise Roberts for technical advice. John Morrow at Organon Laboratories (Newhouse, Scotland) supplied the CHO cells stably transfected with human GLYT1b, and Allelix Neuroscience (NPS Pharmaceuticals, Salt Lake City, UT) provided the NFPS used in this study.
This work was supported by a project grant from the National Health and Medical Research Council of Australia (R.J.V.). K.R.A. is supported by the University of Sydney Medical Foundation and the Neuroscience Institute of Schizophrenia and Allied Disorders. J.D.C. is supported by an Australian Research Council Senior Research fellowship.
References
- 1.Legendre, P. 2001. The glycinergic inhibitory synapse. Cell. Mol. Life Sci. 58:760–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomson, A. M. 1990. Glycine is a coagonist at the NMDA receptor/channel complex. Prog. Neurobiol. 35:53–74. [DOI] [PubMed] [Google Scholar]
- 3.Poyatos, I., J. Ponce, C. Aragon, C. Gimenez, and F. Zafra. 1997. The glycine transporter GLYT2 is a reliable marker for glycine-immunoreactive neurons. Brain Res. Mol. Brain Res. 49:63–70. [DOI] [PubMed] [Google Scholar]
- 4.Zafra, F., C. Aragon, L. Olivares, N. C. Danbolt, C. Gimenez, and J. Storm-Mathisen. 1995. Glycine transporters are differentially expressed among CNS cells. J. Neurosci. 15:3952–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roux, M. J., and S. Supplisson. 2000. Neuronal and glial glycine transporters have different stoichiometries. Neuron. 25:373–403. [DOI] [PubMed] [Google Scholar]
- 6.Supplisson, S., and M. J. Roux. 2002. Why glycine transporters have different stoichiometries. FEBS Lett. 529:93–101. [DOI] [PubMed] [Google Scholar]
- 7.Sagne, C., S. El Mestikawy, M. F. Isambert, M. Hamon, J. P. Henry, B. Giros, and B. Gasnier. 1997. Cloning of a functional vesicular GABA and glycine transporter by screening of genome databases. FEBS Lett. 417:177–183. [DOI] [PubMed] [Google Scholar]
- 8.McIntire, S. L., R. J. Reimer, K. Schuske, R. H. Edwards, and E. M. Jorgensen. 1997. Identification and characterization of the vesicular GABA transporter. Nature. 409:870–876. [DOI] [PubMed] [Google Scholar]
- 9.Dumoulin, A., P. Rostaing, C. Bedet, S. Levi, M. F. Isambert, J. P. Henry, A. Triller, and B. Gasnier. 1999. Presence of the vesicular inhibitory amino acid transporter in GABAergic and glycinergic synaptic terminal boutons. J. Cell Sci. 112:811–823. [DOI] [PubMed] [Google Scholar]
- 10.Lu, C. C., and D. W. Hilgemann. 1999. GAT1 (GABA:Na+:Cl−) cotransport function. Steady state studies in giant Xenopus oocyte membrane patches. J. Gen. Physiol. 114:429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi, D. J., T. Oshima, and D. Attwell. 2000. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 403:316–321. [DOI] [PubMed] [Google Scholar]
- 12.Attwell, D., B. Barbour, and M. Szatkowski. 1993. Nonvesicular release of neurotransmitter. Neuron. 11:401–407. [DOI] [PubMed] [Google Scholar]
- 13.Huang, H., L. Barakat, D. Wang, and A. Bordey. 2004. Bergmann glial GlyT1 mediates glycine uptake and release in mouse cerebellar slices. J. Physiol. 560:721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayor, F., Jr., J. G. Marvizon, M. C. Aragon, C. Gimenez, and F. Valdivieso. 1981. Glycine transport into plasma-membrane vesicles derived from rat brain synaptosomes. Biochem. J. 198:535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aragon, M. C., and C. Gimenez. 1986. Efflux and exchange of glycine by synaptic plasma membrane vesicles derived from rat brain. Biochim. Biophys. Acta. 855:257–264. [DOI] [PubMed] [Google Scholar]
- 16.Sakata, K., K. Sato, P. Schloss, H. Betz, S. Shimada, and M. Tohyama. 1997. Characterization of glycine release mediated by glycine transporter 1 stably expressed in HEK-293 cells. Brain Res. Mol. Brain Res. 49:89–94. [DOI] [PubMed] [Google Scholar]
- 17.Hollmann, M., and S. Heinemann. 1994. Cloned glutamate receptors. Annu. Rev. Neurosci. 17:31–108. [DOI] [PubMed] [Google Scholar]
- 18.Martina, M., Y. Gorfinkel, S. Halman, J. A. Lowe, P. Periyalwar, C. J. Schmidt, and R. Bergeron. 2004. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J. Physiol. 557:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergeron, R., T. M. Meyer, J. T. Coyle, and R. W. Greene. 1998. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc. Natl. Acad. Sci. USA. 95:15730–15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradaia, A., R. Schlichter, and J. Trouslard. 2004. Role of glial and neuronal glycine transporters in the control of glycinergic and glutamatergic synaptic transmission in lamina X of the rat spinal cord. J. Physiol. 559:169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen, L., M. Muhlhauser, and C. R. Yang. 2003. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J. Neurophysiol. 89:691–703. [DOI] [PubMed] [Google Scholar]
- 22.Lim, R., P. D. Hoang, and A. J. Berger. 2004. Blockade of glycine transporter-1 (GLYT-1) potentiates NMDA-receptor mediated synaptic transmission in hypoglossal motorneurons. J. Neurophysiol. 92:2530–2537. [DOI] [PubMed] [Google Scholar]
- 23.Morrow, J. A., I. T. Collie, D. R. Dunbar, G. B. Walker, M. Shahid, and D. R. Hill. 1998. Molecular cloning and functional expression of the human glycine transporter GlyT2 and chromosomal localisation of the gene in the human genome. FEBS Lett. 439:334–340. [DOI] [PubMed] [Google Scholar]
- 24.Motulsky, H. J. 1999. Analyzing with GraphPad Prism. GraphPad Software, San Diego, CA.
- 25.Gentet, L. J., G. J. Stuart, and J. D. Clements. 2000. Direct measurement of specific membrane capacitance in neurons. Biophys. J. 79:314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jardetzky, O. 1966. Simple allosteric model for membrane pumps. Nature. 211:969–970. [DOI] [PubMed] [Google Scholar]
- 27.Loo, D. D., S. Eskandari, K. J. Boorer, H. K. Sarkar, and E. M. Wright. 2000. Role of Cl− in electrogenic Na+-coupled cotransporters GAT1 and SGLT1. J. Biol. Chem. 275:37414–37422. [DOI] [PubMed] [Google Scholar]
- 28.Gentet, L. J., and J. D. Clements. 2002. Binding site stoichiometry and the effects of phosphorylation on human alpha1 homomeric glycine receptors. J. Physiol. 544:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mager, S., C. Min, D. J. Henry, C. Chavkin, B. J. Hoffman, N. Davidson, and H. A. Lester. 1994. Conducting states of a mammalian serotonin transporter. Neuron. 12:845–859. [DOI] [PubMed] [Google Scholar]
- 30.Mager, S., J. Naeve, M. Quick, C. Labarca, N. Davidson, and H. A. Lester. 1993. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 10:177–188. [DOI] [PubMed] [Google Scholar]
- 31.Wadiche, J. I., J. L. Arriza, S. G. Amara, and M. P. Kavanaugh. 1995. Kinetics of a human glutamate transporter. Neuron. 14:1019–1027. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Corcuera, B., R. Martinez-Maza, E. Nunez, M. Roux, S. Supplisson, and C. Aragon. 1998. Differential properties of two stably expressed brain-specific glycine transporters. J. Neurochem. 71:2211–2219. [DOI] [PubMed] [Google Scholar]
- 33.Aubrey, K. R., A. D. Mitrovic, and R. J. Vandenberg. 2000. Molecular basis for proton regulation of glycine transport by glycine transporter subtype 1b. Mol. Pharmacol. 58:129–135. [DOI] [PubMed] [Google Scholar]
- 34.Ju, P., K. R. Aubrey, and R. J. Vandenberg. 2004. Zn2+ inhibits glycine transport by glycine transporter subtype 1b. J. Biol. Chem. 279:22983–22991. [DOI] [PubMed] [Google Scholar]
- 35.Laube, B. 2002. Potentiation of inhibitory glycinergic neurotransmission by Zn2+: a synergistic interplay between presynaptic P2X2 and postsynaptic glycine receptors. Eur. J. Neurosci. 16:1025–1036. [DOI] [PubMed] [Google Scholar]
- 36.Aubrey, K. R., and R. J. Vandenberg. 2001. N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine (NFPS) is a selective persistent inhibitor of glycine transport. Br. J. Pharmacol. 134:1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cammack, J. N., S. V. Rakhilin, and E. A. Schwartz. 1994. A GABA transporter operates asymmetrically and with variable stoichiometry. Neuron. 13:949–960. [DOI] [PubMed] [Google Scholar]
- 38.Sonders, M. S., S. J. Zhu, N. R. Zahniser, M. P. Kavanaugh, and S. G. Amara. 1997. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J. Neurosci. 17:960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roux, M. J., R. Martinez-Maza, A. Le Goff, B. Lopez-Corcuera, C. Aragon, and S. Supplisson. 2001. The glial and the neuronal glycine transporters differ in their reactivity to sulfhydryl reagents. J. Biol. Chem. 276:17699–17705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stein, W. D. 1990. Channels, charriers, and pumps. An introduction to membrane transport. Academic Press, San Diego, CA.
- 41.Mathias, R. T., I. S. Cohen, and C. Oliva. 1990. Limitations of the whole cell patch clamp technique in the control of intracellular concentrations. Biophys. J. 58:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pusch, M., and E. Neher. 1988. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflugers Arch. 411:204–211. [DOI] [PubMed] [Google Scholar]
- 43.Risso, S., L. J. DeFelice, and R. D. Blakely. 1996. Sodium-dependent GABA-induced currents in GAT1-transfected HeLa cells. J. Physiol. 490:691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barakat, L., and A. Bordey. 2002. GAT-1 and reversible GABA transport in Bergmann glia in slices. J. Neurophysiol. 88:1407–1419. [DOI] [PubMed] [Google Scholar]
- 45.Ramsey, I. S., and L. J. DeFelice. 2002. Serotonin transporter function and pharmacology are sensitive to expression level: evidence for an endogenous regulatory factor. J. Biol. Chem. 277:14475–14482. [DOI] [PubMed] [Google Scholar]
- 46.Chiu, C. S., K. Jensen, I. Sokolova, D. Wang, M. Li, P. Deshpande, N. Davidson, I. Mody, M. W. Quick, S. R. Quake, and H. A. Lester. 2002. Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions. J. Neurosci. 22:10251–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehre, K. P., and N. C. Danbolt. 1998. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J. Neurosci. 18:8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bushong, E. A., M. E. Martone, Y. Z. Jones, and M. H. Ellisman. 2002. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tong, G., and C. E. Jahr. 1994. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 13:1195–1203. [DOI] [PubMed] [Google Scholar]
- 50.Verkhratsky, A., and C. Steinhauser. 2000. Ion channels in glial cells. Brain Res. Brain Res. Rev. 32:400–412. [DOI] [PubMed] [Google Scholar]
- 51.Berger, S., J. Carter, and O. Lowry. 1977. The distribution of glycine, GABA, glutamate and aspartate in rabbit spinal cord, cerebellum and hippocampus. J. Neurochem. 28:149–158. [DOI] [PubMed] [Google Scholar]
- 52.Sontheimer, H., J. A. Black, and S. G. Waxman. 1996. Voltage-gated Na+ channels in glia: properties and possible functions. Trends Neurosci. 19:325–331. [DOI] [PubMed] [Google Scholar]
- 53.Bordey, A., and H. Sontheimer. 1998. Electrophysiological properties of human astrocytic tumor cells in situ: enigma of spiking glial cells. J. Neurophysiol. 79:2782–2793. [DOI] [PubMed] [Google Scholar]
- 54.O'Connor, E. R., H. Sontheimer, D. D. Spencer, and N. C. de Lanerolle. 1998. Astrocytes from human hippocampal epileptogenic foci exhibit action potential-like responses. Epilepsia. 39:347–354. [DOI] [PubMed] [Google Scholar]
- 55.Gomeza, J., K. Ohno, S. Hulsmann, W. Armsen, V. Eulenburg, D. W. Richter, B. Laube, and H. Betz. 2003. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron. 40:797–806. [DOI] [PubMed] [Google Scholar]