

Abstract
The essential HIV-1 regulatory protein Rev binds to the Rev responsive element (RRE) of the HIV-1 mRNA. A short α-helical peptide derived from Rev (Rev 34–50) and a truncated form of the RRE sequence (RRE IIB) provide a useful in vitro system to study the interactions between Rev and RRE. The current studies focus on evaluating the specificity of the binding interactions between Rev 34–50 and RRE IIB. The binding of L- and D-Rev peptides to natural and enantiomeric RRE IIB RNA was studied by fluorescence spectroscopy. D-Rev and L-Rev peptides bind to RRE IIB with similar affinities. CD measurements are consistent with a nonhelical, probably β-hairpin, conformation for D-Rev in the complex. The binding affinities of D/L Rev peptides to L-RRE IIB RNA are also similar to those with natural D-RRE IIB. Furthermore, the conformations of L- and D-peptides when bound to L-RRE are reciprocal to the conformations of these peptides in complex with D-RRE. RNA footprinting studies show that L- and D-Rev peptides bind to the same site on RRE IIB. Our results demonstrate lack of stereospecificity in RRE RNA–Rev peptide interactions. However, it is quite possible that the interactions between full-length Rev protein and RRE are highly specific.
Keywords: D and L Rev peptides, RRE IIB, HIV-1 RNA, fluorescence anisotropy, α-helix, β-hairpin, stereospecificity
INTRODUCTION
During the replication cycle of human immunodeficiency virus type 1 (HIV-1), alternative splicing of its primary transcript generates different mRNAs necessary for virus production (Karn 1991; Pollard and Malim 1998). Singly spliced and unspliced transcripts are transported to the cytoplasm, where they are either translated or packaged as genomic RNAs. HIV-1 Rev protein plays a central role both in the export of unspliced mRNAs and in the inhibition of splicing (Kjems et al. 1991; Pollard and Malim 1998). In the presence of Rev, the incompletely spliced mRNAs accumulate in the cytoplasm, whereas in its absence, these transcripts are retained in the cell nucleus (Frankel and Young 1998).
Rev is a 116–amino acid phosphoprotein, which accumulates primarily in the nucleoli of the host cell, and can shuttle between the nucleus and the cytoplasm (Frankel and Young 1998; Pollard and Malim 1998). An arginine-rich amino-terminal domain of Rev serves as a signal for nuclear localization (Henderson and Percipalle 1997; Pollard and Malim 1998). The same domain (positions 33–50) is responsible for the binding of Rev to its cis-acting RNA target sequence, the Rev response element (RRE) (Kjems et al. 1991, 1992; Tan and Frankel 1994; Frankel and Young 1998; Pollard and Malim 1998). RRE is encoded by sequences of the env gene, and is present in all unspliced and incompletely spliced viral mRNAs. Rev binding to the RRE targets the bound mRNA to the nuclear export machinery. Stem–loop IIB of RRE was identified as the high-affinity Rev binding site (Frankel and Young 1998), simplifying analysis of the binding interactions.
The molecular basis of Rev–RRE recognition requires the interaction of the 17-mer arginine-rich Rev 34–50 peptide with a bulge portion of RRE IIB RNA construct, as established by a variety of RNA footprinting techniques (Kjems et al. 1992; Tan and Frankel 1994). NMR structural analysis of the complex reveals that Rev peptide acquires an α-helical conformation while binding in the major groove of the RNA near the purine-rich bulge (Battiste et al. 1996). Several arginine side chains make base-specific contacts, and an asparagine residue contacts a non-Watson-Crick G•A base pair. The RNA major groove widens near another non-Watson-Crick G48•G71 base pair (Chen and Frankel 1995; Battiste et al. 1996). The structure formed by the two purine–purine base pairs of the RRE creates a distinctive binding pocket for specific Rev peptide recognition (Battiste et al. 1996).
Circular dichroism experiments indicate that the peptide α-helical structure is stabilized when bound specifically to RRE RNA, and that the RNA undergoes a conformational change upon binding. Mutation of any one of six amino acids (Thr 34, Arg 35, Arg 38, Arg 39, Asn 40, or Arg 44) strongly decreases specific RNA-binding affinity in vitro (Tan and Frankel 1994). Thus, the mode of Rev 34–50 peptide binding to RRE, and the conformation of the RNA in the complex have been generally established (Battiste et al. 1996). An issue of interest in the context of RNA-ligand interaction in general, and Rev–RRE interactions in particular, is just how specific the binding interactions are. To further elucidate the nature of the specificity of Rev peptide binding to RRE, a series of D- and L-Rev peptides were prepared, and their binding affinities to RRE IIB and its L-enantiomer were determined. Stereospecificity in binding processes is generally taken as the sine qua non for specific molecular recognition with proteins, and the lack of stereospecific interactions might suggest a lack of overall specificity. Surprisingly, the binding of Rev peptides to RRE IIB is nonstereospecific, as both L- and D-peptides bind with similar affinities. In addition, footprinting experiments demonstrate that the RNA-binding sites are similar, if not identical, for both peptides. However, this does not imply a lack of specificity in the binding process, but rather indicates how flexible peptides can achieve similar structural solutions in different ways. Whereas the L-Rev peptides bind to RRE IIB in an α-helical conformation, the D-peptide binds in what appears to be a β-turn conformation.
RESULTS
Binding of fluorescent Rev peptides to RRE IIB and RRE mutants
The initial experiments performed were aimed at exploring the specificity of interactions between Rev peptides and RRE RNA constructs. The RRE constructs chosen for study are shown in Scheme 1 ▶. RRE IIB is the wild-type RNA construct, corresponding to the high-affinity HIV Rev binding site on RRE RNA (Tan et al. 1993). It contains an A-form RNA duplex with a 5-nucleotide bulge (Scheme 1A; Tan et al. 1993). The M3 and M4 mutants (Scheme 1C,D) were included to study possible RNA sequence specificity of Rev–RRE interactions. The M3 mutant is known to bind to Rev 34–50 (Kjems et al. 1992; Hamasaki and Rando 1998) nearly as well as RRE IIB, whereas the affinity of Rev 34–50 for the M4 mutant is diminished significantly (Kjems et al. 1992; Hamasaki and Rando 1998).
SCHEME 1.

Predicted secondary structures of RRE mutants used in the study. Arrows indicate nucleotide difference in constructs compared with RRE IIB (wild type). RRE IIB* depicts peptide-bound secondary structure of RRE IIB, based on the NMR (PDB access code 1ETF, 1ETG, (8)) model of RRE IIB/Rev33–55 complex.
In all of the binding experiments performed, a previously described fluorescence anisotropy methodology was utilized (Wang et al. 1997; Hamasaki and Rando 1998, Lacourciere 2000, Haddad et al. 2002). In this method, the increase in fluorescence anisotropy of a fluorescent ligand is measured as a function of ligand-RNA binding (Hamasaki and Rando 1998). Saturation is observed and competition experiments with unlabeled ligands can be carried out to determine affinity constants for binding (Hamasaki and Rando 1998). In the current studies, L-Rev peptide labeled with a rhodamine moiety at amino terminus, RevRh (rhodamine-TRQAR RNRRRRWRERQR) is used to probe binding.
In initial experiments, the binding of the fluorescent RevRh peptide to both RRE IIB enantiomers (the natural D- and its mirror-image L-enantiomer) and to the aforementioned M3 and M4 mutants (Scheme 1) was studied. Figure 1 ▶ shows typical binding isotherms for the interactions of RevRh to the D- and L-RRE IIB constructs. The dissociation constants for the binding of RevRh peptide to the RRE RNA constructs at 20°C are summarized in Table 1 ▶. Interestingly, the affinities of RevRh to the L- and D-enantiomers of RRE IIB are very close (Fig. 1B ▶; Table 1 ▶), demonstrating nonstereospecifity of fluorescent peptide binding.
FIGURE 1.

Fluorescence anisotropy isotherms of RevRh binding to D-RRE IIB (A) and L- RRE IIB (B) RNA. The tracer concentration is 10 nM.
TABLE 1.
Kd, nM, of RNA-peptide complexes at 20°C
| Peptide | D-RRE IIB | L-RRE IIB | M3 | M4 |
| LRevRh | 14.4 ± 0.6 | 11 ± 3 | 59 ± 7 | 47 ± 6 |
| LRev | 31 ± 1 | 21.8 ± 0.6 | 179 ± 5 | 231 ± 4 |
| DRev | 14.6 ± 0.3 | 17.0 ± 0.4 | 29 ± 2 | 377 ± 23 |
| LRevErse | 42 ± 2 | 14.8 ± 0.1 | 38 ± 2 | 1160 ± 69 |
| DRevErse | 28 ± 1 | 22.6 ± 0.8 | 14.1 ± 0.4 | 121 ± 5 |
| DrevMut1 | 1027 ± 52 | — | — | — |
Competition between RevRh and Rev peptides
Given the stereochemical results described above, the unmodified Rev peptides were tested as competitive inhibitors for RevRh binding to the RRE constructs (Table 1 ▶). By measuring the decrease in the fluorescent anisotropy of the RevRh–RNA complex in the presence of Rev peptides, the affinities of these peptides to the RNA were determined (see Materials and Methods; Hamasaki and Rando 1998). Natural L-Rev peptide and its D-enantiomer were tested for binding to both D- and L-RRE IIB. Figure 2 ▶ shows examples of the fluorescence anisotropy isotherms of D- and L-Rev in competition with RevRh for D- and L-RRE IIB binding. As seen here, straightforward competition for L- and D-RRE IIB binding occurs with similar measured KDs (Table 1 ▶). Further experiments with the M3 and M4 mutants indicate that D-Rev binds with a greater affinity than L-Rev to M3, whereas their affinities to M4 are similar and ~10–20 times lower than to RRE IIB. The results with the L-peptides are quantitatively similar to the data obtained previously with fluorescein-labeled L-Rev used as a tracer (Hamasaki and Rando 1998). Finally, the D-Rev mutant, N40|ZyA, was prepared, and its binding to RRE IIB was studied to access elements of binding specificity. In the L-series, this mutant only binds weakly to RRE IIB (Tan et al. 1993). The measurements indicate that this D-Rev (N|ZyA) mutant behaves similarly to L-Rev (N|ZyA) mutant, and binds with much lower affinity to RRE IIB.
FIGURE 2.

Competition of LRev (A) and DRev (B) with RevRh for D-RRE IIB binding. Competition of LRev (C) and DRev (D) with RevRh for L-RRE IIB binding.
To further probe the specificity of the binding reaction, the D- and L-Rev peptides with reverse sequences (L- and D-RevErse) were prepared and tested. The results of the competition experiments performed with L- and D-RevErse peptides are summarized in Table 1 ▶. Both the L- and D-peptides with the reverse amino acid sequences bind with similar affinities to D- and L-RRE IIB and M3 constructs, whereas their affinities to M4 proved to be significantly lower (Table 1 ▶). Moreover, they also bind to the constructs with similar affinities, as do the L- and D-Rev peptides.
Binding sites of Rev peptides on RRE IIB
Given the results described above, it was important to determine whether the enantiomeric peptides recognize similar features of the RRE IIB RNA-binding site. In these studies, three RNA footprinting techniques (lead acetate, RNase A, and RNase T1 footprinting) were used to reveal the binding sites for the peptides on RRE IIB (Fig. 3A,B,C ▶). Lead acetate induces hydrolytic cleavage at phosphate moieties, whereas RNase A cleaves at purines, and RNase T1 cleaves at the 3′ position of guanosines and adjacent nucleotides.
FIGURE 3.



RRE IIB footprinting studies of the Rev peptides binding sites on the RNA construct. (A) Lead acetate footprinting. (B) RNase T1 footprinting. (C) RNase A footprinting. (0) Untreated RRE IIB, 32P-labeled at 5′; (Ctrl) control experiment in the absence of binders; (L-Rev, D-Rev, L-RevErse, D-RevErse) RNA footprinting in the presence of peptide binders; (NeoB) RNA footprinting in the presence of neomycin B. Concentrations of the peptides, 2.5 and 12.5 μM. Concentration of neomycin B 25 and 125 μM.
Figure 4 ▶ shows a quantitative analysis of the lead acetate and RNase A footprinting results. Lanes 2 (control), 3 (L-Rev), 5 (D-Rev), 7 (L-RevErse), 9 (D-RevErse), and 11 (neomycin B) were used in the analysis. Band intensities of each lane were normalized to G61 band intensity in the lead acetate footprinting gel (Fig. 3A ▶), and were normalized to G42 in the RNase A footprinting gel (Fig. 3B ▶). The intensities of each band were then compared with the intensities of the corresponding band in the control lane (lane 2). Lower intensities indicate protection at the particular nucleotide, whereas increased intensities indicate an increase in cleavage at the nucleotide. Increases in cleavage may be due to a conformational change in the RNA molecule, induced by ligand binding. Differences in band intensities exceeding control by 10% or more are plotted (Fig. 4 ▶). The sites on RRE IIB, which appear protected by lead acetate, RNase A, and RNase T1 footprinting, are assumed to correlate with the binding sites for the ligands.
FIGURE 4.


Quantitation of RRE IIB footprinting studies. Lanes 2,3,5,7,9,11 of lead acetate (Fig. 3A ▶) and RNase A (Fig. 3B ▶) footprinting gels were used for the analysis. Band intensities of each lane were normalized to G61 band intensity in lead acetate footprinting gel (Fig. 3A ▶) and to G42 in RNase A footprinting gel (Fig. 3B ▶). The ratio of intensities of each band to the intensities of the corresponding band in the control lane (lane 2) were calculated. When the difference in the band intensities compared with the control exceeded 10%, they were plotted. Positive bars indicate the protection observed at the particular nucleotide, whereas negative bars stand for increase of cleavage at the corresponded nucleotide, due to a conformational change in the RNA molecule induced by ligand binding. The sites on RRE IIB, which appear protected in both lead acetate and RNase A footprinting patterns are assumed to be the binding sites for the ligands.
As seen from Figure 4 ▶, A and B, both L-Rev and D-Rev produce very similar footprinting patterns when complexed with RRE IIB. Lead acetate footprinting reveals possible conformational destabilization in the lower stem, as alterations in cleavage are observed at nucleotides 40–46. The upper part of the bulge and the adjacent upper stem region (nucleotides 49–53), appear protected when probed by both lead acetate and RNase A. Protection with L-Rev and D-Rev is also observed at A59 in the apical loop. Figure 5 ▶, A and B, presents a summary of footprinting results for the binding of D- and L-Rev peptides. All three footprinting methods used in this study indicate that L- and D-Rev peptides both bind to almost exactly the same site on RRE IIB. The site is formed by G46 (RNase T1 results), C49-A52 and G68-A69.
FIGURE 5.

Summary of RRE IIB footprinting studies. (Open circles) Nucleotides that were found to be protected in two different footprinting methods. Protection observed in lead acetate and RNAse A footprinting experiments. (Filled circles) Nucleotides appeared protected in all three footprinting methods. Protection observed in RNAse T1 footprinting experiments. (Arrows) Enhancement of cleavage observed in lead acetate and RNAse A footprinting experiments.
Figure 4 ▶, C–E, presents data on the footprinting pattern of D- and L-RevErse peptides, and the aminoglycoside (neomycin B), bound to the RRE IIB construct. Neomycin B was used as a control, as the RRE IIB-binding site for aminoglycosides is clearly distinct from the binding site for L-Rev (Lacourciere et al. 2000). The results are summarized schematically in Figure 5 ▶, C and D, as the binding sites for these ligands on RRE IIB. L-RevErse appears to bind in a similar fashion to L-Rev or D-Rev, but the binding interactions are farther from the bulge and closer to the apical loop. In addition, a much less pronounced protection pattern is observed here. D-RevErse appears to bind differently than L- and D-Rev do. Its footprint pattern resembles a composite of the binding of both aminoglycoside (neomycin B) and L- or D-Rev. C49-C51 and A44, G46, and G64 are protected, but enhanced cleavage at C54, G68, and A69 suggests differences in the binding of D-RevErse to this site compared with L- or D-Rev.
Finally, protection experiments with neomycin B suggest that its binding site is located in the lower stem-bulge region of the RRE construct (Fig. 5E ▶), and spans nucleotides U44, G46, and G47. Increased cleavage at the upper stem region suggests a significant conformation change upon neomycin B binding. This result is in good accordance with previously published data (Lacourciere et al. 2000).
Differential CD spectra of L/D-Rev and L/D-RevErse
The fact that D- and L-Rev peptides are able to saturably bind to RRE IIB and with similar affinities suggests that they must bind to the RNA molecules with different conformations. It has been shown previously that L-Rev (34–50) binds to RRE IIB in an α-helical conformation, although the peptide is largely random coil in solution in the absence of RNA (Tan et al. 1993; Tan and Frankel 1994).
To evaluate the conformations of the peptides in complex with D- and L-RRE IIB, CD spectroscopic experiments were performed. Figure 6A ▶ shows CD spectra of D- and L-RRE IIB in the absence of ligands. The D-RRE IIB spectrum is typical for A-form RNA (Daly et al. 1995). As expected, the spectrum of L-RRE IIB appears as a mirror image of the D-RRE IIB spectrum. In the absence of RNA, all of the peptides were found to be unstructured (data not shown). The differential CD spectrum of L-Rev in the presence of an equimolar concentration of D-RRE IIB was obtained by the subtraction of the CD spectrum of D-RRE IIB from the spectra of peptide–RNA complexes. As expected, the spectrum suggests an α-helical conformation of L-Rev in complex with D-RRE IIB. A clear minimum at 208 nm is observed, along with a weak minimum at 223 nm. By way of comparison, the spectrum of the enantiomeric D-Rev peptide in complex with enantiomeric L-RRE IIB is shown in Figure 6B ▶. As expected, this spectrum appears as the mirror image of the L-Rev/D-RRE IIB spectrum.
FIGURE 6.

(A) CD spectra of D- and L-RRE IIB RNA. (B) Differential CD spectra of L-Rev peptide in complex with D-RRE IIB and D-Rev peptides in the complex with L-RRE IIB RNA. (C) Differential CD spectra of L- Rev in complex with L-RRE IIB and D- in complex with D-RRE IIB RNA. (D) Differential CD spectra of L- and D-RevErse peptides in the presence of D-RRE IIB RNA.
The CD spectrum of the D-Rev peptide in complex with D-RRE IIB (Fig. 6C ▶) does not reveal the presence of an α-helix. By way of comparison, Figure 6C ▶ also shows the spectrum of D-Rev peptide in the presence of D-RRE IIB. This spectrum appears as a mirror image of the L-Rev/L-RRE IIB spectrum. A positive band was observed in the D-Rev/D-RRE IIB spectrum at 225 nm and a negative band at 215 nm. Thus, although D-Rev peptide binds to the same site on D-RRE IIB as L-Rev, it clearly adopts a different conformation.
Further CD experiments were performed on the binding of D- and L-RevErse peptides to the D-RRE IIB RNA construct to extend the results described above. L-RevErse peptide was designed as an upside-down model of L-Rev, and D-RevErse was prepared as an enantiomeric control for L-RevErse. In the absence of RNA, both L- and D-RevErse peptides appear unstructured. The differential CD spectrum of L-RevErse peptide in complex with D-RRE suggests an α-helix, similar to that observed with L-Rev (Fig. 6D ▶). This result indicates that RRE IIB induces the formation of an α-helix in both L-Rev, and in its reverse sequence analog. However, as shown above, these two peptides do not bind to identical sites on RRE IIB, although their binding sites are very close. Finally, D-Rev in complex with D-RRE IIB (Fig. 6D ▶) forms what appears to be a random coil structure (Woody 1995).
To further study the specificity of the RNA interaction with peptides, similar CD experiments were performed on the M3 mutant construct. Interestingly, none of the peptides appeared helical when bound to the M3 (data not shown), suggesting a different mode of binding of Rev peptides to RRE M3 than to RRE IIB.
DISCUSSION
Intact HIV-1 Rev protein binds cooperatively to the 244-nucleotide full-length RRE with a stoichiometry of 4 Rev monomers per RRE (Van Ryk and Venkatesan 1999). Rev monomer binds initially to the high-affinity stem–loop region of RRE, followed by oligomerization of Rev mediated by protein–protein interactions (Van Ryk and Venkatesan 1999). A short α-helical peptide corresponding to the arginine-rich RNA-binding domain of Rev binds with high affinity to the stem–loop IIB of RRE, providing a good model for the initial process of Rev monomer recognition of the RRE stem–loop (Kjems et al. 1991, 1992; Tan et al. 1993; Tan and Frankel 1994; Frankel and Young 1998). Rev peptide binding to RRE occurs in two steps, an initial encounter, followed by isomerization of the RNA (Lacourciere et al. 2000). A variety of structural and binding studies are consistent with the notion that the interactions between Rev protein, or Rev peptide, and RRE RNA exhibit elements of specificity (Kjems et al. 1992). This is especially true for the intact Rev protein. However, there is also evidence suggesting a comparative lack of specificity in the interactions in the peptide series. For example, a 40-mer Rev peptide RP194, containing arginine-rich RRE-binding domain, failed to significantly discriminate with respect to binding affinities between native RRE, stem II, and a comparably sized antisense RRE construct, or even tRNA. Binding occurred in the nanomolar range in all cases (Daly et al. 1995). Whereas the interaction of complete Rev protein with RRE is relatively specific, a truncated Rev, lacking 20 amino-terminal residues, displays less ability to discriminate RRE from nonspecific RNA, yet still binds sense and anti-sense RNA species with high affinities (Daly et al. 1995). It is reasonable to suggest that only when the peptides are highly structured within the intact protein, will they display a high degree of binding specificity for a particular RNA target.
The experiments described here were aimed at further exploring the level of the specificity, in particular, the binding stereospecificity, of the interactions between Rev 34–50 peptide and RRE. To address the question of stereospecificity of RRE–Rev interaction, the binding of L-Rev peptide and its D-enantiomer to the RRE IIB construct were studied. This RRE IIB construct is an A-form RNA duplex with a 5-nucleotide bulge (Scheme 1A; Tan et al. 1993). The binding of Rev peptide to RRE IIB induces a conformational change in the RNA, leading to the formation of noncanonical G47•A73 and G48•G71 base pairs along with groove widening at this site (PDB access code 1ETF, 1ETG; Bartel et al. 1991; Tan et al. 1993; Battiste et al. 1996; Scheme 1B ▶). The binding studies reported here show that both L-Rev and D-Rev peptides bind in a saturable fashion to RRE IIB, with similar affinities. This observation demonstrates a lack of stereospecificity of Rev–RRE interaction in the binding event. A similar observation, demonstrating lack of stereospecificity of Tat–TAR interaction, was reported previously with D-Tat (37–72) peptide and 59-mer TAR RNA (Huq et al. 1999).
These results were further extended by investigating the interactions of the enantiomeric L-RRE IIB construct with L-Rev. Enantiomeric L-RRE IIB, of course, has the same secondary structure as its wild-type D-enantiomer, but is the mirror-image. Binding studies show that the affinity of L-Rev to L-RRE IIB is the same as to D-RRE IIB, and, hence, that binding is again nonstereospecific. A similar observation demonstrating nonstereospecific RNA–peptide interactions was reported previously with TAR RNA L-enantiomer and Tat protein (Garbesi et al. 1998).
The binding studies could be construed to suggest a lack of stereospecificity in the interactions of Rev peptide and RRE IIB. However, there are clearly elements of specificity as well. For example, the reported binding events are saturable and consistent with the stoichiometric binding of Rev peptides to RNA. L-Rev peptide is also capable of discrimination between RRE IIB and M3 or M4 mutants. The observed difference in the affinities of L-Rev to RRE IIB and RRE mutants is ~5- to 10-fold. D-Rev peptide discriminates between the RRE IIB and M4 mutants, just like L-Rev does, although it does not discriminate between RRE IIB and M3. In addition, an N for A substitution in the D-Rev peptide produces a significant reduction in binding affinity to RRE IIB RNA. This result is similar to that described previously with the same L-Rev mutant (Tan et al. 1993). To further investigate the putative specificity of the binding events, footprinting experiments were performed with both the D- and L-Rev peptides.
Footprinting studies on the RRE IIB–peptide complexes suggest that the D- and L-Rev peptides bind to the same site in the bulge upper-stem portion of the RNA (Fig. 5 ▶). The site is formed by G4, C49-A52, and G68-A69. The enhanced cleavage at G70 observed in lead acetate footprinting, might indicate a conformational change at this site upon peptide binding. This region was reported previously as a specific binding site for Rev protein and L-Rev 33–50 peptide (Kjems et al. 1992).
The reverse sequence peptides, L- and D-RevErse, also bind to RRE IIB with high affinities, but bind to slightly different sites than do the Rev peptides. Whereas the site of L-RevErse binding to RRE may be similar to that of L- and D-Rev, the site of D-RevErse binding exhibits substantial differences.
The similarity of the L- and D-Rev binding sites suggests that D-Rev in complex with RRE IIB must have a different structure from L-Rev in the complex. The differential CD spectra confirmed that the binding of L-Rev peptide to RRE IIB induces an α-helix (Kjems et al. 1991, 1992; Tan et al. 1993; Tan and Frankel 1994). Clearly, these differential spectra are biased by the spectrum of the RNA in the complex with peptide. However, in the range of 190–240 nm, the contribution of peptide conformation is mainly observed. The degree of helicity is proportional to the minimum at 222 nm, so in these experiments, the L-Rev peptide in the complex with RRE IIB appears as largely, but not completely, α-helical (Fig. 6B ▶). In contrast, D-Rev peptide in the complex with RRE IIB acquires a nonhelical conformation. The symmetry of the D-Rev/D-RRE IIB spectrum compared with the L-Rev/L-RRE IIB spectrum suggests similar conformations of these peptides in the complexes. After factoring in the influence introduced by RNA in the spectrum, the spectrum of the L-Rev peptide in the complex with L-RRE resembles a B-type CD spectrum of a β-turn type II structure (Hutchinson and Thornton 1994). This is characterized by a minimum at 220–230 nm and maximum at 205–210 nm (Woody 1995). It is concluded that L-Rev in the complex with L-RRE IIB acquires a conformation, which contains a β-turn portion. D-Rev in complex with D-RRE IIB appears as a mirror image of L-Rev in complex with L-RRE IIB, that is, a mirror-image structure containing a β-turn.
The CD experiments with enantiomeric L-RRE IIB construct also show that the geometry of the peptide interaction with L-RRE IIB is a mirror image of the geometry of the interaction with native D-RRE IIB. Thus, D-Rev in complex with L-RRE acquires an α-helical conformation, whereas L-Rev in the presence of L-RRE is probably structured as a β-turn type II. These experiments then show that the D- and L-Rev peptides can bind to the same site on RRE IIB because of the conformational flexibility of the Rev peptide. Molecular modeling can be used to frame a working hypothesis on how the D- and L-Rev peptides might bind to the same RNA site.
The conformations of the D- and L-Rev peptides in complex with RRE IIB are different, as shown by CD measurements. It is reasonable to suggest that in both cases, the peptide backbone structure produces a similar spatial arrangement of critical amino acid side residues, allowing for the same contacts with RNA. Assuming that specific RRE–Rev recognition demands a similar distribution of amino acid side chains in three dimensions as reported in studies on L-Rev (PDB access code 1ETF; Battiste et al. 1996), possible β-hairpin structures of D-Rev could be modeled that achieve the same spatial distribution of critical contacts. Two models are presented here. The first contains a β-turn induced at Arg 42 (model 1; Fig. 7B ▶), and the second contains a β-turn induced at Asn 40 (model 2; Fig. 7C ▶). The structures in model 1 are characteristic of a β-turn type II, whereas model 2 is characterized by a β-turn type II′ (Hutchinson and Thornton 1994). Because the CD spectrum of D-Rev is not a classic B-type spectrum (Woody 1995), the latter model seems more plausible. Deviations in the spectrum from a classic B-type spectrum could be due to the 4–amino acid peptide tail, which is not involved in the β-structure. Such a tail would probably be structured as a random coil, and produce a corresponding CD spectrum. This could contribute to a nonideal CD B-type spectrum of D-Rev in complex with RRE IIB.
FIGURE 7.

Model of D-Rev peptide in the complex with RRE IIB. Secondary structures of L-Rev peptide α-helix (A) in complex with RRE IIB (PDB access code 1ETF (8)) and D-Rev β-hairpin models (B,C) are shown as ribbons. D-Rev peptide modeled as a β-hairpin with the turn induced at Arg 42 (model 1, B) and Asn 40 (model 2, C). Residues in D-Rev models (B,C) are numbered accordingly to the positions in the native Rev protein. Residues not involved in RNA interaction are omitted in L-Rev structure (A). Amino acid residues important for RNA binding include Thr 34, Arg 35, Arg 38, Arg 39, Asn 40, or Arg 44, as defined by mutational analysis (Tan and Frankel 1994). The residues that are similarly positioned in L-Rev 34–50 (A) and in D-Rev models (B,C) are shown in color.
It is clear that a high-resolution technique, such as NMR structural analysis, is required to elucidate the specific nature of the interactions between D-Rev peptides and RRE IIB RNA. However, inspection of our model-building exercise with respect to the Rev 34–50 structure (Fig. 7A ▶), shows that it is indeed possible to mimic the critical distribution of essential amino acid side chains when D-Rev peptide is folded as a β-hairpin. The amino acid residues known to be essential for RRE IIB binding, Thr 34, Arg 35, Arg 38, Arg 39, Asn 40, or Arg 44 (Tan and Frankel 1994), are presented in similar three-dimensional arrays in the models (Fig. 6 ▶). These models are certainly just two examples of a variety of possible structures that meet the general requirement that the key Arg, Asn, and Gln moieties be displayed similarly in space in both D- and L-Rev peptides. Note that the Glu and Trp side chains are also positioned similarly in the L-Rev and modeled D-Rev peptides.
It is also possible to suggest a mode of L-RevErse binding to RRE IIB. The differential CD spectrum of L-RevErse is, at least in part, consistent with an α-helical conformation of the peptide (Fig. 6 ▶). Footprinting studies suggest that L-RevErse and L-Rev bind to very close sites on RRE IIB (Fig. 5 ▶). Taken together, these observations indicate that L-RevErse binds to RRE IIB in a similar fashion to L-Rev, whereas binding in upside-down mode on the RRE IIB.
In conclusion, L- and D-Rev peptides are able to bind to RRE IIB with similar affinities because of their conformational flexibility. There are clear elements of specificity in these binding interactions, although not to the level found in intact Rev. This could have general implications for the design of small molecule drugs intended to target specific RNA molecules.
MATERIALS AND METHODS
Materials
Peptides
L-Rev, D-Rev, L-RevErse, D-RevErse, and D-RevMut1 were synthesized at the Harvard Medical School peptide synthesis facility by Dr. Charles Dahl. The sequence of D- and L-Rev peptides corresponds to Rev 34–50: (d/l)-TRQARRNRRRRWRERQR. D- and L-RevErse peptides contain reversed amino acid sequence: (d/l)-RQRERWRRRRNRRAQRT; D-RevMut1 (N|ZyA): (d)-TRQAR RARRRRWRERQR.
Fluorescent tracers
Rhodamine-labeled L-Rev peptide (RevRh) was synthesized by reacting L-Rev peptide with 5-carboxytetramethylrhodamine succinyimidyl ester (Molecular Probes) in a water-acetonitrile mixture (1:1) at pH 7.0 (200 mM phosphate buffer). The conjugate was purified by HPLC on 250 × 4.6-mm C-18 YMC AQ column (pore size 200 Å) in a 30-min water-acetonitrile gradient (0%–50%) at a flow rate of 1 mL/min. The RevRh peak was eluted at 20 min, 54 sec. The compound was analyzed by mass-spectroscopy (MALDI-TOF MS). A molecular weight of 2854.02 (calculated 2852) was found.
RNA constructs
RRE IIB (wild type); RRE M3; RRE M4 (Scheme 1) were synthesized chemically by Dharmacon Co. The RNA samples are supplied in a protected form. The samples were deprotected by the manufacturer’s instructions prior to use.
The enantiomeric L-RRE IIB construct of the same sequence as RRE IIB wild type was prepared from DMT-L-ribo phosphoamidates (ChemGenes) and L-riboC CPG solid support on an Expedite oligonucleotide synthesizer using 1 μmole RNA cycle with a coupling time of 600 sec. After deblocking of the product with ethanolic ammonia, the L-RNA was deprotected using neat triethylamine trihydrofluoride, precipitated with n-butanol and desalted on a Nick Sephadex G-50 (Amersham-Pharmacia) column. The L-RNA oligonucleotide was purified by gel-electrophoresis on a 12% polyacrylamide/7 M urea gel.
All stock solutions were prepared in nuclease-free water, and were diluted with the appropriate buffers prior to use. Concentrations of RNA and peptides were determined spectrophotometrically, by absorption at 260 and 280 nm, respectively. The RNA molecules were annealed by heating to 95°C, followed by cooling to room temperature. The secondary structures of the mutants, predicted by Mfold (Walter et al. 1994), are shown in Scheme 1 ▶.
Fluorescence measurements
Fluorescence anisotropy measurements were performed as described previously (Wang et al. 1997; Hamasaki and Rando 1998; Lacourciere 2000) on a Perkin Elmer LS-50B luminescence spectrometer equipped with a thermostat accurate to ±0.1°C. In the study of D/L-RRE IIB and RRE mutant binding, samples were excited at 550 nm and monitored at 580 nm for RevRh. The integration time was 4 sec. For every single point, 10–15 measurements were made, and their average values were used for calculation. Measurements were performed at 20°C in buffer solution A containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 20 mM HEPES (pH 7.50). Tracer concentrations were determined spectroscopically at 550 nm using a molar extinction coefficient of 6.00 × 104 M−1 cm−1 (Hamasaki and Rando 1998).
Determination of dissociation constants
The following equation (eq. 1) was used for the determination of the dissociation constants (Kd) for the interactions between RNA and the fluorescently labeled tracer, RevRh (Wang et al. 1997):
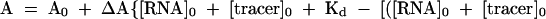 |
(1) |
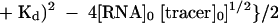 |
in which A and A0 are the fluorescence anisotropy in the presence and absence of RNA, respectively, and ΔA is the difference between the fluorescence anisotropy of 1 nM of the tracer (RevRh) in the presence of an infinite concentration of RNA minus the fluorescence anisotropy in the absence of RNA. [RNA]0 and [tracer]0 are the initial concentrations of RNA and tracer.
In the competitive binding assay, equation 2 is used for the calculation of the KD values (Wang et al. 1997):
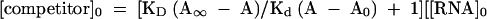 |
(2) |
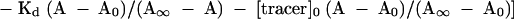 |
in which KD is the dissociation constant for dissociation of the RNA and the competitor peptides. [competitor]0 is the initial concentration of the competitor A∞-anisotropy of completely bound tracer, A0–anisotropy of free tracer, A-measured anisotropy value. Both Kd and KD were determined by nonlinear curve fitting, using the equations described above, and are presented as a mean value of three independent measurements.
RNA footprinting
For the footprinting experiments, RRE IIB RNA was purified by gel-electrophoresis on 12% polyacrylamide/7 M urea gel. The purified RNA was radioactively labeled at its 5′ end with 1 μL of [γ32P]ATP (6000 Ci/mmole, New England Nuclear) per 1 nmole of RNA using T4 polynucleotide kinase (Ambion) in a buffer containing 70 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 5 mM DTT. The labeled RNA was extracted successively with water-saturated phenol, phenol/chloroform (1:1), and chloroform, and precipitated from 75% ethanol. All RNA samples were annealed by heating them to 95°C for 3 min, followed by cooling to room temperature in a 10 mM HEPES buffer (pH 7.0).
RNase A footprinting
In a typical experiment, 10 μL of 10 mM HEPES buffer (pH 7.0) containing 50 mM KCl and 50–100 ng of 5′-32P-labeled RRE IIB (~0.3–0.6 μM) was incubated with 200 pM RNase A (Sigma) at room temperature for 10 min in the presence of Rev peptides and neomycin B at various concentrations. After the incubation, 10 μL of formamide/bromphenol blue/xylene cyanol loading buffer (Ambion) was added to the samples. The samples were heated to 90°C for 2 min and were resolved by electrophoresis on a 40 cm × 0.75 mm 16.7% polyacrylamide denaturing gel (7 M urea) for 3.5 h at 45–50 W constant power. The results were visualized by exposing the wet gels to Molecular Dynamics PhosphorImager plates, which were read on a Personal FX PhosphorImager (Bio-Rad) and quantitated using QuantityOne software.
RNase T1 footprinting
In a typical experiment, 10 μL of 10 mM HEPES buffer (pH 7.0), containing 50 mM KCl and 50–100 ng of 5′-32P-labeled RRE IIB (~0.3–0.6 μM) was incubated with 0.02 U RNase T1 (Sigma) at 37°C for 20 min in the presence of Rev peptides and neomycin B at various concentrations. After the incubation, 10 μL of formamide/bromphenol blue/xylene cyanol loading buffer was added to the samples. The samples were heated to 90°C for 2 min, and were resolved by electrophoresis and quantitated as described above.
Lead (II) acetate footprinting
In a typical experiment, 10 μL of 10 mM HEPES buffer (pH 7.0), containing 50 mM KCl, was incubated with 50–100 ng of 5′-32P-labeled RRE IIB RNA (~0.3–0.6 μM) in the presence of varying concentrations of Rev peptides and neomycin B at room temperature for 5 min. Cleavage reactions were initiated by addition of Pb(OAc)2 to final concentrations of 0.1 mM. After 20-min incubation at room temperature, 10 μL of formamide/bromphenol blue/xylene cyanol loading buffer, containing 10 mM EDTA, was added to the samples. The samples were heated to 90°C for 2 min, and were resolved by electrophoresis and quantitated as described above.
CD spectroscopy
CD spectra were recorded on an Aviv 202 spectrometer at 4°C in a buffer containing 20 mM sodium phosphate (pH 7.5) and 100 mM NaF. Spectra were analyzed using CDS software.
Molecular modeling
The structure of D-Rev peptide was modeled using SGI Octane computer (Silicon Graphics Inc.) and InsightII (MSI Inc.) software. Several initial model structures of D-Rev peptide were built with a β-turn induced at different positions. Each of the structures was energy minimized and dynamic simulations were performed using the Discover module of MSI and the consistent valence force field CVFF (Dauber-Osguthorpe et al. 1988). The models were further subjected to molecular dynamic simulations using the CHARMm module and the CHARMm force field (Brooks et al. 1983). All simulations were preceded by 0.3 ps heating from 0 to 300 K, followed by 2.7 ps equilibration period. Each dynamic stage included 5000 steps of 1 fs, followed by a minimization stage, until the derivatives were smaller than 0.001 kcal/mole Å. The resulting β-hairpin structures were used for further modeling.
Acknowledgments
The work described here from the authors’ laboratory was supported by the U.S. Public Health Service NIH grant EY-12375.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RRE, Rev responsive element
HIV, Human Immunodeficiency virus
mRNA, messenger RNA
NMR, nuclear magnetic resonance
rRNA, ribosomal RNA
RevRh, carboxymethyl rhodamine-modified L-Rev peptide
MALDI-TOF MS, matrix assisted laser desorption/ionization-time of flight mass spectroscopy
CPG, controlled pore glass
DMT, dimethoxytrityl
CD, circular dichroism
EDTA, ethylene diamine tetraacetic acid
HEPES, 4-(2-hydroxyethyl)piperazine-1-sulfonic acid
CHARMm, Chemistry at HARvard Molecular Mechanics forcefield
CVFF, consistent valence forcefield
PDB, Protein Data Bank
KD, Kd, dissociation constants
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2172103.
REFERENCES
- Bartel, D.P., Zapp, M.L., Green, M.R., and Szostak, J.W. 1991. HIV-1 Rev regulation involves recognition of non-Watson-Crick base pairs in viral RNA. Cell 67: 529–536. [DOI] [PubMed] [Google Scholar]
- Battiste, J.L., Mao, H., Rao, N.S., Tan, R., Muhandiram, D.R., Kay, L.E., Frankel, A.D., and Williamson, J.R. 1996. α helix-RNA major groove recognition in an HIV-1 Rev peptide-RRE RNA complex. Science 273: 547–1551. [DOI] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4: 187–217. [Google Scholar]
- Chen, L. and Frankel, A.D. 1995. A peptide interaction in the major groove of RNA resembles protein interactions in the minor groove of DNA. Proc. Natl. Acad. Sci. 92: 5077–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly, T.J., Doten, R.C., Rusche, J.R., and Auer, M. 1995. The amino terminal domain of HIV-1 Rev is required for discrimination of the RRE from nonspecific RNA. J. Mol. Biol. 253: 243–258. [DOI] [PubMed] [Google Scholar]
- Dauber-Osguthorpe, P., Roberts, V.A., Osguthorpe, D.J., Wolff, J., Genest, M., and Hagler, A.T. 1988. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins, Struct. Funct. Genet. 4: 31–47. [DOI] [PubMed] [Google Scholar]
- Frankel, A.D. and Young, J.A.T. 1998. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 67: 1–25. [DOI] [PubMed] [Google Scholar]
- Garbesi, A., Hamy, F., Maffini, M., Albrecht, G., and Klimkait, T. 1998. TAR-RNA binding by HIV-1 Tat protein is selectively inhibited by its L-enantiomer. Nucleic Acids Res. 26: 2886–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad, J., Kotra, L.P., Llano-Sotelo, B., Kim, C., Azucena Jr., E.F., Liu, M., Vakulenko, S.B., Chow, C.S., and Mobashery, S. 2002. Design of novel antibiotics that bind to the ribosomal acyltransfer site. J. Am. Chem. Soc. 124: 3229–3237. [DOI] [PubMed] [Google Scholar]
- Hamasaki, K. and Rando, R.R. 1998. A high-throughput fluorescence screen to monitor the specific binding of antagonists to RNA targets. Anal. Biochem. 261: 183–190. [DOI] [PubMed] [Google Scholar]
- Henderson, B.R. and Percipalle, P. 1997. Interactions between HIV Rev and nuclear import and export factors: The Rev nuclear localisation signal mediates specific binding to human importin-β. J. Mol. Biol. 274: 693–707. [DOI] [PubMed] [Google Scholar]
- Huq, I., Ping, Y-H., Tamilarasu, N., and Rana, T.M. 1999. Controlling human immunodeficiency virus type 1 gene expression by unnatural peptides. Biochemistry 38: 5172–5177. [DOI] [PubMed] [Google Scholar]
- Hutchinson, E.G. and Thornton, J.M. 1994. A revised set of potentials for β-turn formation in proteins. Protein Sci. 3: 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn, J. 1991. Control of human immunodeficiency virus replication by the tat, rev, nef and protease genes. Curr. Opin. Immunol. 3: 526–536. [DOI] [PubMed] [Google Scholar]
- Kjems, J., Frankel, A.D., and Sharp, P.A. 1991. Specific regulation of mRNA splicing in vitro by a peptide from HIV-1 Rev. Cell 67: 169–178. [DOI] [PubMed] [Google Scholar]
- Kjems, J., Brown, M., Chang, D.D., and Sharp, P.A. 1992. Specific binding of a basic peptide from HIV-1 Rev. EMBO J. 11: 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacourciere, K.A., Stivers, T.J., and Marino, J.P. 2000. Mechanism of neomycin and Rev peptide binding to the Rev responsive element of HIV-1 as determined by fluorescence and NMR spectroscopy. Biochemistry 39: 5630–5641. [DOI] [PubMed] [Google Scholar]
- Pollard, V.W. and Malim, M.H. 1998. The HIV-1 Rev protein. Annu. Rev. Microbiol. 52: 491–532. [DOI] [PubMed] [Google Scholar]
- Tan, R. and Frankel, A.D. 1994. Costabilization of peptide and RNA structure in an HIV Rev peptide-RRE complex. Biochemistry 48: 14579–14585. [DOI] [PubMed] [Google Scholar]
- Tan, R., Chen, L., Buettner, J.A., Hudson, D., and Frankel, A.D. 1993. RNA recognition by an isolated α helix. Cell 73: 1031–1040. [DOI] [PubMed] [Google Scholar]
- Van Ryk, D.I. and Venkatesan, S. 1999. Real-time kinetics of HIV-1 Rev-Rev response element interactions. Definition of minimal binding sites on RNA and protein and stoichiometric analysis. J. Biol. Chem. 274: 17452–17463. [DOI] [PubMed] [Google Scholar]
- Walter, A.E., Turner, D.H., Kim, J., Lyttle, M.H., Muller, P., Mathews, D.H., and Zuker, M. 1994. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc. Natl. Acad. Sci. 91: 9218–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Hamasaki, K., and Rando, R.R. 1997. Specificity of aminoglycoside binding to RNA constructs derived from the 16S rRNA decoding region and the HIV-RRE activator region. Biochemistry 36: 768–779. [DOI] [PubMed] [Google Scholar]
- Woody, R.W. 1995. Circular dichroism. Methods Enzymol. 246: 34–71. [DOI] [PubMed] [Google Scholar]