

Abstract
Leishmania and other trypanosomatids are early eukaryotes that possess unusual molecular features, including polycistronic transcription and trans-splicing of pre-mRNAs. The spliced leader RNA (SL RNA) is joined to the 5′ end of all mRNAs, thus donating a 5′ cap that is characterized by complex modifications. In addition to the conserved m7GTP, linked via a 5′-5′-triphosphate bound to the first nucleoside of the mRNA, the trypanosomatid 5′ cap includes 2′-O methylations on the first four ribose moieties and unique base methylations on the first adenine and the fourth uracil, resulting in the cap-4 structure, m7 Gpppm36,6,2′Apm2′Apm2′ Cpm23,2′U, as reported elsewhere previously. A library of analogs that mimic the cap structure to different degrees has been synthesized. Their differential affinities to the cap binding proteins make them attractive compounds for studying the molecular basis of cap recognition, and in turn, they may have potential therapeutic significance. The interactions between cap analogs and eIF4E, a cap-binding protein that plays a key role in initiation of translation, can be monitored by measuring intrinsic fluorescence quenching of the tryptophan residues. In the present communication we describe the multistep synthesis of the trypanosomatid cap-4 structure. The 5′ terminal mRNA tetranucleotide fragment (pm3 6,6,2′Apm2′Apm2′ Cpm23,2′U) was synthesized by the phosphoramidite solid phase method. After deprotection and purification, the 5′-phosphorylated tetranucleotide was chemically coupled with m7GDP to yield the cap-4 structure. Biological activity of this newly synthesized compound was confirmed in binding studies with eIF4E from Leishmania that we recently cloned (LeishIF4E-1), using the fluorescence time-synchronized titration method.
Keywords: cap-4, chemical synthesis, Leishmania, eIF4E, fluorescence
INTRODUCTION
Leishmania and other trypanosomatids belong to the order Kinetoplastida and lead a digenetic life cycle, migrating between invertebrate vectors and mammalian hosts. They reside as promastigotes in the alimentary canal of sand fly females that transmit them to a mammalian host during their blood meal. Upon transmission, the parasites enter macrophages and cells of the immune system where they transform into amastigotes and become obligatory intracellular organisms. Leishmania parasites cause a wide spectrum of diseases, ranging from the self-healing skin lesions to the lethal visceral disease.
Trypanosomatids are early eukaryotic protozoans that possess unique molecular features. Their RNA pol II genes are transcribed polycistronically, and the pre-mRNAs are further processed to monocistronic molecules by trans-splicing and polyadenylation. During trans-splicing, a conserved capped spliced leader RNA (SL RNA) is donated to the 5′ end of all mRNAs. Cap biogenesis in trypanosomatids proceeds by posttranscriptional addition of the consensus m7GTP, and is followed by modifications on the first four transcribed nucleotides. These include the addition of 2′-O methyl groups on all four ribose moieties and unique base methylations on the first adenosine and the fourth uridine. The resulting cap, m7 Gpppm36,6,2′Apm2′Apm2′- Cpm23,2′U, is denoted cap-4 and is found on the 5′ end of all mRNA molecules in all trypanosomatids (Bangs et al. 1992).
The 5′ cap structure in eukaryotes, as well as in trypanosomatids, participates in a variety of biological mechanisms, including mRNA transport between the nucleus and the cytoplasm, control of mRNA stability, and formation of the translation initiation complex (Varani 1997). The general translation factor eIF4E binds the 5′ cap, and is part of a larger complex, eIF4F, that recruits the small ribosomal subunit (Gingras et al. 1999). Thus, the addition of synthetic m7GTP inhibits translation in an in vitro system very efficiently (Cai et al. 1999). The interaction between m7GTP and purified eIF4E can be followed by a biophysical method that monitors fluorescence quenching of Trp residues found in the cap-binding proteins (Carberry et al. 1989; Blachut-Okrasinska et al. 2000; Niedzwiecka et al. 2002). A library of cap analogs was synthesized and examined for their ability to bind eIF4E and to inhibit translation in vitro (Cai et al. 1999). Such compounds, once they penetrate the cells, could be used to inhibit translation in vivo. Synthetic analogs were also used to discriminate between the binding specificities of the different eIF4E isoforms in the nematode Caenorhabditis elegans (Miyoshi et al. 2002).
An eIF4E homolog from Leishmania was cloned and expressed in bacteria. Although the cloned gene showed only 43% similarity with the mouse eIF4E, the recombinant protein, LeishIF4E-1, bound the eukaryote m7GTP very efficiently, allowing its purification to homogeneity over m7GTP-Sepharose (Y. Yoffe, J. Zuberek, M. Lewdorowicz, Z. Zeira, Ch. Keasar, B. Shaanan, I. Orr-Dahan, M. Jankowska-Anyszka, E. Darzynkiewicz, and M. Shapira, in prep.). In an attempt to determine whether and how the modifications on the first four nucleotides of the Leishmania cap-4 structure affect its binding to the eIF4E isoforms, we prepared a synthetic cap-4 analog.
Chemical synthesis of the 5′-phosphorylated tetranucleotide mRNA fragment was achieved by the phosphoramidite solid phase method. For the final step of the capping reaction we developed a new strategy, which included coupling of 7-methylguanosine 5′-diphosphate imidazolide (Sawai et al. 1999; Jemielity et al. 2003) with a 5′-phosphorylated tetranucleotide, in the presence of zinc chloride as a catalyst. This coupling approach appeared to be more efficient than that one reported previously (Zuberek et al. 2002, 2003).
RESULTS AND DISCUSSION
Chemical synthesis
The 5′-phosphorylated tetramer (pm3 6,6,2′Apm2′Apm2′- Cpm23,2′U, compound 13, where m denotes methyl groups within nuclotide units), the key intermediate for the synthesis of 5′-terminal cap of Leishmania (m7 Gpppm36,6,2′-Apm2′Apm2′ Cpm23,2′U, Cap-4, compound 15) was prepared by the standard phosphoramidite solid phase method (Beaucage and Caruthers 1981). However two of the four protected monomers, that is, N6,N6,O2′-trimethyladenosine and N3,O2′-dimethyluridine are not commercially available. Synthesis of N6,N6,O2′-tri-methyladenosine (compound 4) and its derivatization to the suitable building block (compound 6) for solid phase synthesis are shown in Figure 1. The synthetic route included (1) introduction of the N6,N6-dimethylamino group into adenosine following previously published protocols (Zemlicka and Sorm 1965; Morris et al. 1976), (2) O′-methylation (Robins et al. 1974), (3) 5′-O-dimethoxytritylation (Sproat and Lamond 1991), and (4) activation with a phosphoramidite reagent (Kierzek et al. 1986; Scaringe et al. 1990; Sproat and Lamond 1991). The other unique structure, N3,O2′-dimethyluridine (compound 9) was prepared in a one-pot methylation of uridine that included (1) the reaction of uridine with diazomethane to give N3-uridine (compound 7; Szer and Shugar 1968), (2) subsequent methylation of the O2′ and O3′ positions with the same reagent, after addition of stannous chloride as a catalyst (Robins et al. 1974), and (3) HPLC separation of N3,O2′-dimethyluridine and N3,O3′-dimethylridine mixture. The protection of compound 9 in the O5′ position with dimethoxytrityl group gave 5′-O-dimethoxytrityl-N3,O2′-dimethyluridine (compound 10), which was attached to the solid support using standard methodology (Kierzek et al. 1986; Sproat and Lamond 1991) based on a succinic linker (Fig. 2). The resulting resin was used as the starting material in the automated solid phase synthesis. After two subsequent coupling stages, which added O2′-methylcytidine and O2′-methyladenosine, the fourth nucleoside subunit (N6,N6,O2′-trimethyladenosine, compound 6) was attached. The last step of the solid phase synthesis involved phosphorylation of the tetranucleotide 5′ end with 2-cyanoethyl-3-(4,4′-dimethoxytrityloxy)-2,2-di(ethoxycarbonyl)-propyl-1-N,N-diisopropyl phosphoramidite (Guzaev et al. 1995). Conventional ammonolysis was used to cleave the RNA tetramer from the solid support, resulting with the simultaneous removal of the protecting groups from the nucleic bases and intranucleotide phosphates. The resulting product, still selectively protected at the 5′-phosphate, was separated from impurities by preparative reverse phase HPLC. Successive detritylation with 80% acetic acid and a brief treatment with methylamine generated an RNA tetramer with a free 5′-phosphate group, which was purified by preparative HPLC. The final capping reaction was performed by a method that was newly developed in our laboratory. It included coupling of the 5′-phosphorylated tetramer with an imidazolide derivative of 7-methylguanosine 5′-diphosphate [Im(m7GDP)], in the presence of anhydrous zinc chloride as a catalyst, dissolved in DMF (Fig. 3). Comparable yields of capped oligoribonucleotides were reported for a similar capping strategy that was performed in water and in the presence of manganese(II) ions (Sawai at al. 1999); however, a larger excess of Im(m7GDP) was used in that case. We also found the new coupling method to be superior in terms of yield and reproducibility over the previously reported method (Zuberek et al. 2002, 2003), which involved activation of a 5′-phosphorylated oligoribonucleotide with imidazole and its coupling with m7GDP.
FIGURE 1.

Chemical synthesis of 5′-O-dimethoxytrityl-N6,N6,O2′-trimethyladenosine-3′-O -[(2-cyanoethyl)-(N,N-diisopropyl)]phosphoramidite.
FIGURE 2.

Chemical synthesis of the uridine derivative attached to the resin.
FIGURE 3.

Chemical synthesis of cap-4.
To perform more profound structure–function studies we also synthesized the 5′-terminal dinucleoside triphosphate m7 Gpppm36,6,2′A, denoted cap-1 (compound 17; Fig. 4). The synthesis was carried out in solution, using a strategy that was similar to that developed previously (Stepinski et al. 2001; Jemielity et al. 2003). The synthetic route included preparation of N6,N6,O2′-trimethyladenosine (compound 4; Fig. 1) from inosine, its 5′-phosphorylation by the Yoshikawa procedure (Yoshikawa 1967), and a final coupling performed with Im(m7GDP) (Fig. 4).
FIGURE 4.

Chemical synthesis of cap-1.
The structure of the newly synthesized cap-4 was confirmed by NMR spectroscopy and mass spectrometry (ESI MS) as well as careful NMR analysis of all monomer units. The key intermediates of the synthesis were isolated and analyzed by NMR and MS (for details, see Materials and Methods).
LeishIF4E-1 binds m7GTP and cap-4
Purification of LeishIF4E-1 was based on its ability to bind m7GTP. The recombinant protein was purified by m7GTP-Sepharose affinity column chromatography, using a single-step procedure. Pellets of bacteria that were induced to express LeishIF4E-1 were loaded onto a column of m7GTP-Sepharose. Only washes containing free m7GTP and not GTP released LeishIF4E-1 from the column matrix (Fig. 5). For the fluorescence studies, elution was performed with high salt, rather than with the free ligand, resulting in a similar degree of purification.
FIGURE 5.

Chromatography and purification of LeishIF4E-1 on m7GTP-sepharose. Crude extracts from noninduced BL21 bacterial cells (crude) and the soluble fraction (Sup) following induction by IPTG were applied onto a column of m7GTP-Sepharose. The flow-through (FT) was collected and the column was washed (Wash) first with equilibration buffer and then with equilibration buffer containing GTP (100 μM). The protein was eluted by a wash containing m7GTP (200 μM).
To validate that the synthetically prepared cap-4 compound was biologically active, its ability to interact with the cap-binding protein from Leishmania was examined. Studies on the tertiary structure of eIF4E from different organisms illustrate the structural basis for cap recognition, and indicate a requirement for several conserved Trp residues (Marcotrigiano et al. 1997; Matsuo et al. 1997; Tomoo et al. 2003). Binding of the cap structure and its analogs results in quenching of the Trp fluorescence, and this can be used as a tool to calculate the Kas values (Carberry et al. 1989; Niedzwiecka et al. 2002). Because seven out of eight Trp residues are conserved between LeishIF4E-1 and eIF4E from mouse and yeast (Y. Yoffe, J. Zuberek, M. Lewdorowicz, Z. Zeira, Ch. Keasar, B. Shaanan, I. Orr-Dahan, M. Jankowska-Anyszka, E. Darzynkiewicz, and M. Shapira, in prep.), fluorescence assays were used to evaluate the binding affinity of LeishIF4E-1 with different cap structures.
Kas values for complexes of LeishIF4E-1 with the synthetic cap analogs were determined by the fast and accurate time-synchronized fluorescence titration method (TST; Niedzwiecka et al. 2002; Fig. 6; Table 1). This method takes into account the emission of the free cap analog and the concentration of the active protein, as free parameters of the numerical fitting. The Kas values determined for cap-4 and m7GTP were comparable, with a slight preference for cap-4 (Kas = 0.253 ± 0.003 μM−1 for cap-4, and Kas = 0.156 ± 0.005 μM−1 for m7GTP). These results emphasize that the synthetic cap-4 compound was efficiently recognized by the corresponding LeishIF4E-1. Examination of Kas values measured for analogs that represent intermediate compounds in cap-4 biosynthesis revealed that the binding affinity decreased by three- to fourfold, as compared to cap-4 (Kas = 0.0724 ± 0.002 μM−1 for m7GpppA and Kas =0.0654 ± 0.0041 μM−1 for m7 Gpppm36,6,2′A; Table 1). A reduced Kas for dinucleotide analogs was previously reported for other eIF4E proteins as well (Niedzwiecka et al. 2002). Multiple eIF4E homologs that exist in lower eukaryotes such as C. elegans show differential binding characteristics to m7GTP and the hypermethylated m32,2,7GTP (TMG). Some show exclusive binding with m7GTP (IFE-3) whereas others bind both structures (IFE-5; Miyoshi et al. 2002). LeishIF4E-1 does not bind TMG (Y. Yoffe, J. Zuberek, M. Lewdorowicz, Z. Zeira, Ch. Keasar, B. Shaanan, I. Orr-Dahan, M. Jankowska-Anyszka, E. Darzynkiewicz, and M. Shapira, in prep.), and binds both m7GTP and cap-4 with almost similar affinities, with a slight preference for the parasite cap-4. Thus it appears that m7GTP may play a key role in the interaction between LeishIF4E-1 and cap-4, and the remaining modified nucleotides possibly contribute to the additional specificity of binding.
FIGURE 6.

Fluorescence titration measurements on binding of LeishIF4E-1 to cap analogs. LeishIF4E-1 proteins, at a concentration of 0.3 μM, were titrated by the cap analogs: cap-4 (open squares), m7GTP (open circles), m7GpppA (black squares), m7 Gpppm36,6,2′A (open triangles), and GTP (black triangles), at 20°C in 50 mM HEPES/NaOH, 100 mM NaCl, 1 mM EDTA, 1 mM DTT. Protein fluorescence, presented as relative values, was excited at 295 nm and observed at 345 nm. The observed increasing fluorescence intensity at a higher concentration of ligand originates from the emission of the free cap analogs in solution.
TABLE 1.
The equilibrium association constants (Kas) for complexes of LeishIEF4-1 with cap-4 and other cap analogs at 20°C determined by fluorescence measurements
| Cap analogs | Kas (μM) −1 |
| m7GTP | 0.156 ± 0.005 |
| m7GpppA | 0.0724 ± 0.0020 |
| m7 Gpppm36,6,2′ A | 0.0654 ± 0.0041 |
| cap-4 | 0.253 ± 0.003 |
| GTP | <0.005 |
Synthetically prepared cap analogs provide an important research tool for establishing the structural basis of cap recognition by the corresponding binding proteins. To date, the role of the unique and complex cap found in trypanosomatids is only partially understood. Cap formation on the SL RNA proceeds in a 5′–3′ direction, as indicated by experiments performed with permeable trypanosomes (Ullu and Tschudi 1995). It appears that at least part of the modifications occur cotranscriptionally, as shown in studies using permeable trypanosomes (Mair et al. 2000) and that sequences within the SL RNA coding gene are required for obtaining the complete cap-4 structure (Ullu and Tschudi 1995; Zeiner et al. 2003). However, it was recently shown that the cytoplasmic stage is necessary for SL RNA biogenesis (Zeiner et al. 2003). It was further suggested that U4 modification of the SL RNA most probably serves as a signal for transport of snRNPs between the nucleus and the cytoplasm (Mandelboim et al. 2003). The availability of a synthetic and functional cap-4, as well as its intermediate compounds, is therefore of great importance. Such materials should also contribute to the identification of enzymes that participate in cap-4 biogenesis and in turn, could serve as a basis for the development of new therapeutic molecules, with antiparasitic activity.
The ability to chemically synthesize an analog of cap-4, a structure that is found on the 5′ end of mRNAs in all trypanosomatids, is of great importance. Trypanosomatids are known for their unusual molecular mechanisms, and it has been well established that differential gene expression in these organisms is achieved by posttranscriptional mechanisms such as processing and translation of mRNAs (Zilka et al. 2001). The 5′ cap has a key role in directing these processes in all eukaryotes including in trypanosomatids, but the latter possess a unique cap-4 structure that is found only in this group of organisms. Thus the availability of this synthetic structure as well as its intermediates is essential for identifying the components that participate in the biosynthetic pathway of cap-4. Once identified, such molecules, in principle, could be used as novel drug targets against Leishmania as well as other trypanosomatid parasites.
MATERIALS AND METHODS
Monitoring of the chemical synthesis
TLC analysis was performed on TLC plates coated with silicagel 60 F-254 (Merck), developed by chloroform-methanol-triethylamine 8.9:1:0.1 v/v/v (A), hexane-acetone-triethylamine 45:45:10 v/v/v (B), chloroform-methanol 8:2 v/v (C), dichloromethane-methanol 95:5 v/v (D).
Analytical HPLC was performed on a Spectra-Physics SP8800 apparatus, using a Supelcosil LC-18-T reverse phase column (4.6 × 250 mm, flow rate 1.3 mL/min) with various gradients of buffer A (0.05 M ammonium acetate at pH 5.9) and buffer B (methanol and buffer A, 1:1 v/v).
1H NMR and 13C NMR spectra were run on a Varian UNITY-plus spectrometer at 400 and 100.56 MHz, respectively, at ambient temperatures and at concentrations of ca. 2 mg/mL (1H NMR), and ca. 20 mg/mL (13C NMR) using tetramethylsilane (TMS) as the internal standard in CDCl3 and sodium 3-trimethylsilyl-[2,2,3,3-D4]-propinonate (TSP) in D2O.
Mass spectra were recorded on a Micromass QToF 1 MS spectrometer using electrospray ionization.
Preparative HPLC
Preparative HPLC was performed on a Waters 600E Multisolvent Delivery System apparatus, using a Waters HR-C-18 reverse phase column (19 × 300 mm, flow rate 5.0 mL/min). The mobile phase for this method was the same as for analytical HPLC (see above).
2′,3′,5′-tri-O-acetylinosine (compound1)
Inosine (2 g, 7.5 mmoles) and N,N-dimethylaminopyridine (0.150 g, 1.23 mmoles) were added to acetic anhydride (87 mL), and the mixture was stirred for 3 d (72 h) at room temperature. The progress of the reaction was monitored by HPLC (Rtsubstrate = 2.9 min, Rtproduct = 27 min, in buffer B). The solution was evaporated under reduced pressure at 30°C and the residue was coevaporated with three portions of ethanol (30 mL). The resulting colorless solid material was crystallized from 20 mL of hot ethanol to give 2′,3′,5′-tri-O-acetylinosine (compound 1; 2.382 g, 6 mmoles, 80%).
N6,N6-dimethyladenosine (compound2)
A mixture of 2′,3′,5′-tri-O-acetylinosine (compound 1; 4.5 g, 11.4 mmoles), (chloromethylene)dimethylammonium chloride (10 g, 78 mmoles) and chloroform (35 mL) was refluxed during 2 h. This procedure resulted in a reaction mixture that contained the main product, 2′,3′,5′-Tri-O-acetyl-6-chloro-9-(β-D-ribofuranosyl)purine, with only traces of the starting material. The progress of the reaction was monitored by HPLC (Rtsubstrate = 27 min, Rtproduct = 25 min, in buffer B). The mixture was allowed to cool and sodium hydrogen carbonate (6 g, 71 mmoles) in water (26 mL) was then added dropwise. The aqueous layer was extracted with chloroform (3 × 15 mL) and the chloroform layers were combined, dried over anhydrous sodium sulphate, filtered, and evaporated under reduced pressure. (Chloromethylene)dimethylammonium chloride (5.3 g, 41 mmoles) in chloroform (15 mL) was then added to this residue, and the resulting mixture was added drop-wise with cooling and stirring into a solution of methanolic ammonia. Solvents were evaporated under reduced pressure and the residue was washed with chloroform. The solid phase was filtered off and the filtrate was evaporated under reduced pressure. Methanolic ammonia solution was added to the residual syrup, and the mixture was kept at 0°C during 17 h. The resulting solution was then evaporated under reduced pressure and the residue was re-crystallized from ethanol to yield N6,N6-dimethyladenosine (compound 2; 2.2 g, 7.4 mmoles, 65%).
N6,N6,O2′-trimethyladenosine (compound4) and N6,N6,O3′-trimethyladenosine (compound3)
N6,N6-dimethyladenosine (compound 2) (2 g, 6.8 mmoles) and stannous chloride dihydrate (0.067 g, 0.3 mmoles) in methanol (540 mL) were stirred at room temperature. A solution of diazomethane in 1,2-dimethoxyethane (0.43 M; Robins et al. 1974) was slowly added into this mixture and the progress of the reaction was monitored by TLC. When the starting material disappeared (after about 3 h), the solvent was evaporated under reduced pressure. The residue was dissolved in 25% aqueous methanol and purified by column chromatography (50 × 2 cm, 110 mL, Dowex 1X2 in OH− form, fractions of 23 mL each were collected), performed in the same solvent. Fractions 33–47 contained N6,N6,O2′-trimethyladenosine (compound 4; 0.688 g, 2.2 mmoles, 34%, Rf = 0.1 in system A): 1H NMR (CDCl3, δ ppm): 8.31 (1H, H8 s), 7.79 (1H, H2 s), 5.85 (1H, H1′ d, J = 7.6 Hz), 4.67 (1H, H2′ q, J = 4.67 Hz), 3.65 (2H, H5′,5′ m), 4.57 (1H, H3′ d, J = 4.67 Hz), 3.45 (6H, N–(CH3)2 m), 4.33 (1H, H4′ s), 3.25 (3H, O–CH3 s).
Fractions 54–102 contained N6,N6,O3′-trimethyladenosine (compound 3; 1.1 g, 3.5 mmoles, 52%, Rf = 0.1 in system A): 1HNMR (CDCl3, δ ppm): 8.21 (1H, H2 s), 7.73 (1H, H8 s), 5.67 (1H, H1′ d, J = 7.6 Hz), 5.03 (1H, H2′ t, J = 6.2 Hz), 4.34 (1H, H4′ s), 4.06 (1H, H3′ d, J = 5.6 Hz), 3.80 (2H, H5′,5′ m), 3.53 (3H, O–CH3 s), 3.50 (6H, N–(CH3)2 broad s).
5′-O-dimethoxytrityl-N6,N6,O2′-trimethyladenosine (compound5)
A mixture of N6,N6,O2′-trimethyladenosine (compound 4; 0.460 g, 1.5 mmoles), 4,4′-dimethoxytrityl chloride (1.5 g, 4.4 mmoles) and triethylamine (0.587 mL) in anhydrous pyridine (8 mL) was stirred for 2 h at room temperature, and the progress of the reaction was monitored by TLC. The mixture was quenched with methanol (1.5 mL) and evaporated under reduced pressure at room temperature. The residual syrup was dissolved in chloroform (30 mL) and washed with two 32-mL portions of 1 M aqueous sodium hydrogen carbonate solution. The chloroform layer was dried over anhydrous sodium sulphate, filtered, and evaporated under reduced pressure. The residual pyridine was removed by coevaporation with three 3-mL portions of toluene. The crude product was purified by column chromatography over silicagel, and was eluted with a stepwise gradient system (0.25%–2%) of methanol in dichloromethane to give 5′-O-dimethoxytrityl-N6,N6,O2′-trimethyladenosine (compound 5; 0.73 g, 1.2 mmoles, 81%, Rf = 0.3 in A).
5′-O-dimethoxytrityl-N6,N6,O2′-trimethyladenosine-3′-O-[(2-cyanoethyl)-(N,N-diisopropyl)]phosphoramidite (compound6)
A mixture of 5′-O-dimethoxytrityl-N6,N6,O2′-trimethyladenosine (compound 5; 0.7 g, 1.14 mmoles), N,N-diisopropylethylamine (0.3 mL, 1.71 mmoles), and 2-cyanoethoxy N,N-diisopropylaminochlorophosphine (0.33 mL, 0.350 g, 1.48 mmoles) in anhydrous acetonitrile (5 mL) was stirred at room temperature for 1 h. The progress of the reaction was monitored by TLC. The crude product was purified using column chromatography (silicagel, eluting with a stepwise gradient system of ethyl acetate in hexane containing 1% triethylamine [from 5% to 50% ethyl acetate]) to give 5′-O-dimethoxytrityl-N6,N6,O2′-trimethyladenosine-3′-O-[(2-cy-anoethyl)-(N,N-diisopropyl)]phosphoramidite (compound 6; 0.68 g, 0.84 mmoles, 73%, Rf = 0.4 in system B): 1H NMR (CDCl3, δ ppm): 8.40 (1H, H2 s), 7.87 (1H, H8 d), 7.20 (9H, DMTr m), 6.80 (4H m, DMTr), 6.16 (1H, H1′ t, J = 5.1 Hz), 4.65 (1H, H2′ m), 4.55 (1H, H3′ m), 4.27 (1H, H4′ m), 3.90 (2H, H5′,5′ m), 3.78 (6H s, DMTr), 3.60 (6H m, N–(CH3)2), 3.50 (3H s, O–CH3), 2.68 (2H, O–CH2–CH2–CN t, J = 6.4 Hz), 2.63 (2H t, O–CH2–CH2–CN, J = 6.4 Hz), 1.30 (2H m, (CH3)2–CH–N–CH–(CH3)2), 1.20 (12H m, (CH3)2–CH–N–CH–(CH3)2).
N3,O2′-dimethyluridine (compound9) and N3,O3′-dimethyluridine (compound8)
A mixture of uridine (3 g, 12 mmoles) and methanol (80 mL) was stirred in a round-bottom flask at room temperature. A solution of diazomethane in 1,2-dimethoxyethane (0.43 M; Robins et al. 1974) was then slowly added into this mixture and the progress of the reaction was monitored by TLC. After 1 h all the starting material disappeared and N3-methyluridine (compound 7) (Rf = 0.2 in system C) dominated the reaction mixture. Stannous chloride dihydrate (0.061 g, 0.27 mmoles) and the second portion of an ethereal solution of diazomethane were then slowly added and the reaction was completed after 1 h. Two regioisomers: N3,O2′-dimethyluridine (compound 9) and N3,O3′-dimethyluri-dine (compound 8) were separated by preparative HPLC (Rt8 = 35 min, Rt9 = 38 min, using a linear 0%–100% gradient of buffer B in buffer A within 40 min) to yield 47% of N3,O2′-dimethyluri-dine (compound 9) and 50% of N3,O3′-dimethyluridine (compound 8).
1H NMR for N3,O2′-dimethyluridine (compound 9): (D2O, δ ppm): 7.90 (1H, H6 d, J = 8.02 Hz), 6.00 (1H, H1′ d, J = 3.67 Hz), 5.98 (1H, H5 d, J = 8.02 Hz), 4.30 (1H, H3′ t, J = 6.01 Hz), 4.11 (1H, H4′ m), 4.00 (1H, H2′ t, J = 3.68 Hz), 3.85 (2H, H5′,5′ m), 3.54 (3H, N–CH3), 3.30 (3H s, O–CH3).
5′-O-dimethoxytrityl-N3,O2′-dimethyluridine (compound10)
A mixture of N3,O2′-dimethyluridine (compound 9; 0.5 g, 1.8 mmoles), 4,4′-dimethoxytrityl chloride (1.2 g, 3.5 mmoles) and triethylamine (0.6 mL) in anhydrous pyridine (7 mL) was stirred for 2 h at room temperature, and the progress of the reaction was monitored by TLC. The reaction was quenched with methanol (1.5 mL) and evaporated under reduced pressure at room temperature. The residual syrup was dissolved in chloroform (33 mL) and washed with two 33-mL portions of 1 M aqueous sodium hydrogen carbonate solution. The chloroform layer was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The residual pyridine was removed by coevaporation with 3 × 3 mL of toluene. The crude product was purified by column chromatography (silicagel, eluted by a stepwise gradient of methanol in dichloromethane [ranging from 0.25% to 2% methanol]), to give 5′-O-dimethoxytrityl-N3,O2′-dimethyluridine (compound 10) (0.84 g, 1.5 mmoles, 83%, Rf = 0.7 in system A): 1H NMR (CDCl3, δ ppm): 8.01 (1H, H6 d, J = 8.00 Hz), 7.40 (9H m, DMTr), 6.82 (4H m, DMTr), 5.97 (1H, H1′ s), 5.35 (1H, H5 d, J = 8.00 Hz), 4.46 (1H, H3′ q), 3.99 (1H, H2′ t, J = 7.89 Hz), 3.79 (6H s,–O–CH3, DMTr), 3.77 (1H, H4′ s), 3.67 (3H s, N–CH3), 3.55 (2H, H5′,5′ m), 3.32 (3H s, O–CH3) 13C NMR (CDCl3, δ ppm): 162.81 (1C, C(4)=O), 158.67, 144.32, 135.31, 130.15, 129.09, 113.25, 87.00 (19C, DMTr), 150.89 (1C, C2=O), 137.62 (1C, C6), 101.42 (1C, C5), 87.60, 83.97, 83.06, 68.25, 61.05, 58.63 (6C, C1′, C2′, C3′, C4′, C5′, O2′–CH3), 55.20 (2C, O–CH3, DMTr), 27.45 (1C, N–CH3).
5′-O-dimethoxytrityl-N3,O2′-dimethyluridine-3′-O-pentachlorophenylsuccinate (compound11)
A mixture of 5′-O-dimethoxytrityl-N3,O2′-dimethyluridine (compound 10; 0.534 g, 0.92 mmoles), succinic anhydride (0.101 g, 1.01 mmoles), and N,N-dimethylaminopyridine (0.134 g, 1.1 mmoles) in dichloromethane (5 mL) was stirred for 2 h at room temperature. The progress of the reaction was monitored by TLC (Rf = 0.3 in system D). N,N-dicyclohexylocarbodiimide (0.246 g, 1.2 mmoles) and pentachlorophenol (0.270 g, 1.1 mmoles) were then added. The mixture was allowed to stand overnight and the precipitate was filtered off. The filtrate was extracted with a saturated aqueous solution of sodium dihydrogen phosphate and purified by column chromatography (silicagel). Elution was performed with a stepwise gradient (1%–2%) of methanol in dichloromethane, to give 5′-O-dimethoxytrityl-N3,O2′-dimethyluridine-3′-O-penta-chlorophenylsuccinate (compound 11; 0.798 g, 0.86 mmoles, 93%, Rf = 0.8 in system D): 1H NMR (CDCl3, δ ppm): 7.87 (1H, H6 d, J = 7.99 Hz), 7.40 (9H m, DMTr), 7.30 (9H m, DMTr), 6.82 (4H m, DMTr), 6.00 (1H, H1′ d, J = 3.20 Hz), 5.42 (1H, H5 d, J = 7.99 Hz), 4.26 (1H, H3′ q), 4.10 (1H, H2′ m, J = 3.10 Hz), 3.78 (6H s, –O–CH3, DMTr), 3.78 (1H, H4′ s), 3.52 (2H, H5′,5′ m), 3.50 (3H, N–CH3 s, N3-methyluracyl ring), 3.32 (3H s, O2′–CH3), 2.90 (4H m,–C(O)–CH2–CH2–C(O)–).
5′-O-dimethoxytrityl-N3,O2′-dimethyluridine-3′-O-succinamide attached to the resin (compound12)
A mixture of 5′-O-dimethoxytrityl-N3,O2′-dimethyluridine-3′-O-pentachlorophenylsuccinate (compound 11; 0.150 g, 0.16 mmoles), triethylamine (0.4 mL), and Fractosil 500 (1 g; Merck) in dimethylformamide (4 mL) was shaken overnight at room temperature. The resin was then washed successively with four portions of dichloromethane and methanol, filtered, and dried. Free NH2-groups on the resin were acetylated with acetic anhydride (1 mL), N,N-dimethylaminopyridine (0.010 g, 0.082 mmoles) in anhydrous pyridine (5 mL). The mixture was shaken for 1 h and then the resin was filtered off, washed successively with four portions of dichloromethane, and methanol. Afterward the resin was dried.
Solid phase synthesis
The RNA tetramer was synthesized on an Applied Biosystems 392 synthesizer using a 1 μmole scale protocol with 10 min coupling for each step. The starting material (compound 12) was placed in a standard reaction vessel and, successively, building blocks were coupled for elongation of the ribonucleic chain. N-Benzoyl-5′-dimethoxytrityl-2′-O-methyl-cytidine-3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite and 5′-dimethoxytrityl-2′-O-methyl-N-phenoxyacetyladenosine-3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite were commercial products (Glen Research). 5′-Dimethoxytrityl-N6,N6,O2′-trimethyladenosine-3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite was synthetically prepared (compound 6). The 5′-phosphorylating reagent 2-cyanoethyl-3-(4,4′-dimethoxytrityloxy)-2,2-di(ethoxycarbonyl)propyl-1-N,N-diisopropyl phosphoramidite, which was employed in the last step of the automated synthesis, was purchased from Glen Research.
Unprotected RNA fragment (pm36,6,2′Apm2′Apm2′Cpm23,2′U) (compound13)
A 5′-blocked RNA fragment (obtained from the solid phase synthesis) was detritylated for 2 h with 80% acetic acid. After 2 h acetic acid was evaporated under reduced pressure and then methylamine (6 mL) was added. After 15 min, the methylamine was removed using P4O10 in a vacuum desiccator and the product was purified by preparative HPLC (Rtproduct = 16.9 min, linear 0–100% gradient of buffer B in buffer A over 50 min) to give 0.021 g (0.0143 mmoles) of pm36,6,2′Apm2′Apm2′ Cpm23,2′U (compound 13) as an ammonium salt (m/z 1384.6 by ESI-MS).
P2-imidazolide 7-methylguanosine 5′-diphosphate, Im(m7GDP) (compound14)
A mixture of triethylammonium salt of 7-methylguanosine 5′-diphosphate (0.1 g, 0.131 mmoles), imidazole (0.178 g, 2.63 mmoles), 2,2′-dithiodipyridine (0.230 g, 1.048 mmoles), triethylamine (18 μL), and triphenylphosphine (0.275 g, 1.05 mmoles) in dimethylformamide (1.5 mL) was stirred overnight at room temperature. A solution of sodium perchlorate (0.09 g, 0.74 mmoles) in anhydrous acetone (4 mL) was then added. The pure product that appeared as a sodium salt of P2-imidazolide 7-methylguanosine 5′-diphosphate (compound 14) was filtered off, washed with four portions of acetone, and dried over P4O10 (0.063 g, 0.11 mmoles, 84%).
Cap-4 (m7Gpppm36,6,2′Apm2′Apm2′Cpm23,2′U) (compound15)
A mixture of the ammonium salt of pm36,6,2′Apm2′Apm2′- Cpm23,2′U (compound 13; 10 mg, 6.8 μmoles), the sodium salt of P2-imidazolide 7-methylguanosine 5′-diphosphate Im(m7GDP) (compound 14; 18 mg, 34.3 μmoles) and ZnCl2 (46 mg, 340 μmoles) in dimethylformamide (0.08 mL) was stirred for 16 h at room temperature. The progress of the reaction was monitored by HPLC (Rtsubstrate = 17.8 min, Rtproduct = 16.5 min, using a linear 0%–100% gradient of buffer B in buffer A over 20 min). The mixture was then diluted with water (3.5 mL). Preparative HPLC purification (linear 0%–100% gradient of buffer B in buffer A over 50 min) gave m7 Gpppm36,6,2′Apm2′A pm2′ Cpm23,2′U (compound 15) as ammonium salt (5.14 mg, 2.64 μmoles, 40%): 1H NMR (D2O, δ ppm): 8.23 (1H, H8 s, adenine), 8.05 (1H, H8 s, N6,N6-dimethyladenine), 8.04 (1H, H2 s, adenine), 7.67 (1H, H2 s, N6,N6-dimethyladenine), 7.82 (1H, H6 d, cytidine) 7.50 (1H, H6, s, N3-methyluracyl), 5.98 (2H, broad s, H1′ N6,N6,O2′-trimeth-yladenosine, H1′ O2′-methyladenosine), 5.90 (1H, H1′ d, O2′-methylcytidine), 5.79 (2H, t, H5 cytidine, H5 N3-methyluracyl), 5.62 (1H, H1′ s, 7-methylguanine), 5.39 (1H, H1′ d, N3,O2′-dimethyluridine), 4.90–3.80 (25H, H2′, H3′, H4′, H5′,5′ m), 3.96 (3H, s N7CH3), 3.71, 3.58, 3.51, 3.24 (15H m, N6,N6-dimethyl-adenosine O2′–CH3, adenosine O2′–CH3, cytosine O2′–CH3, N3,O2′-dimethyluridine O2′–CH3, and N3–CH3); 4.8 and 3.3 (6H, broad s N–(CH3)2 N6,N6-dimethyladenine; m/z 1825.5 by ESI-MS).
N6,N6,O2′-trimethyladenosine 5′-monophosphate (compound16)
A mixture of N6,N6,O2′-trimethyladenosine (compound 4; 0.058 g, 0.188 mmoles) and trimethyl phosphate (1.55 mL) was stirred in an ice bath for 3.5 h. Into this mixture phosphorus oxychloride (0.044 mL) was added and the progress of the reaction was monitored by analytical HPLC (Rtsubstrate = 24.3 min, Rtproduct = 17.3 min, linear 0%–100% gradient of buffer B in buffer A, over 20 min). The mixture was diluted with water (50 mL), neutralized by the addition of 1.4 M solution of triethylammonium bicarbonate (TEAB), and purified by column chromatography (DEAE-Sephadex, A-25, HCO3− form) using a linear gradient of 0–0.7 M TEAB to obtain N6,N6,O2′-trimethyladenosine 5′-monophosphate as tri-ethylammonium salt (compound 16; 0.067 g, 0.113 mmoles, 60%).
Cap-1 (m7Gpppm36,6,2′A) (compound17)
A mixture of triethylammonium salt of N6,N6,O2′-trimethyladenosine 5′-monophosphate (compound 16; 67 mg, 113 μmoles), a sodium salt of P2-imidazolide 7-methylguanosine 5′-diphosphate Im(m7GDP) (compound 14; 63 mg, 109 μmoles), and ZnCl2 (160 mg, 1200 μmoles) in dimethylformamide (1.2 mL) was stirred for 2 h at room temperature. The progress of the reaction was monitored by HPLC (Rtsubstrate = 17.8 min, Rtproduct = 14.2 min, linear 0%–100% gradient of buffer B in buffer A over 20 min). The mixture was then diluted with water (60 mL), and EDTA (704 mg, 1.9 μmoles) was added. Then the solution was neutralized with NaHCO3. The crude product was purified by column chromatography (DEAE-Sephadex, using a linear gradient of 0–1.2 M TEAB) to give (compound 17) (m7 Gpppm36,6,2′A, cap-1) as a triethylammonium salt (15 mg, 13 μmoles, 12%): 1H NMR (D2O, δ ppm): 8.32 (1H, H8 s, N6,N6,O2′-adenine), 8.09 (1H, H2 s, N6,N6-adenine), 6.05 (1H, H1′ s, N6,N6,O2′-trimeth-yladenosine), 5.74 (1H, H1′ s, 7-methylguanosine), 4.63 (1H, H2′ t, N6,N6,O2′-trimethyladenosine), 4.34 (10H broad m, H2′ 7-methylguanosine, H3′, H4′, H5′, H5′ 7-methylguanosine and N6,N6,O2′-trimethyladenosine), 3.97 (3H s, N7–CH3) 3.42 (9H broad s, N6,N6,O2′-trimethyladenosine O–CH3 and N6–CH3).
Cloning and expression of the Leishmania eIF4E-1 in bacteria for protein purification
The eIF4E-1 isoform from Leishmania major was amplified by PCR, using genomic DNA as template. The forward primer corresponded to positions 1–21 GGAATTCCATATGTCAGCCCC GTCTTCAGTT-3′ and the reverse primer corresponded to positions 625–645 5′-CGCGGATCCCTAAGACGCCTCGCCGTGC TT-3′. All primers contained anchor sequences that incorporated the NdeI and BamHI restriction sites at the amino (fwd primer) and carboxy (rev primer) termini, respectively (the restriction sites are underlined). The amplified fragments were digested with NdeI and BamHI and further cloned into the corresponding site of the pHis-parallel vector (Sheffield et al. 1999), resulting in plasmid pLeishIF4E-1. This cloning strategy eliminated the His-tag and generated a recombinant protein that initiated from its own first methionine. The nucleotide sequences of the amplified fragments were compared to the genomic sequence obtained from the L. major genome project.
The recombinant pLeishIF4E-1 was introduced into Escherichia coli BL21 cells and expression was induced at 20°C in cultures grown to OD660 0.5 for 3 h, by the addition of 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). Cells were washed once in sonication buffer (20 mM HEPES at pH 7.6, 1 mM DTT and 2 mM EDTA, 5% glycerol), harvested, and disrupted by sonication during 2 min on ice, using pulses of 20 sec interspaced by intervals of 20 sec. The cell extract was clarified by centrifugation during 30 min at 20,000g and the supernatant was loaded on m7GTP-Sepharose, equilibrated in the sonication buffer. The protein was eluted in the presence of 150 μM of the free cap analog m7GTP (Sigma). Alternatively, for fluorescence assays the protein was eluted by high salt (200 mM NaCl).
Fluorescence measurements and data analysis
Freshly prepared protein samples were used for the fluorescence assays, after removal of the high salt by filter centrifugation over 4 mL Biomax 5K NMWL columns (Millipore). Protein concentrations were determined by spectrophotometry, assuming ɛ280 = 47,500 M−1cm−1, which were calculated from the amino acid composition (Gill and von Hippel 1989). Fluorescence measurements were performed on a LS-50B Spectrofluorometer (Perkin Elmer Co.), in a quartz semi-micro cuvette (Hellma) with optical lengths 4 mm and 10 mm for absorption and emission, respectively. Fluorescence time-synchronized titrations (TST; Niedzwiecka et al. 2002) were performed in 50 mM HEPES/NaOH (pH 7.2), 100 mM NaCl, 1 mM EDTA, and 1 mM DTT at 20°C by adding 1-μL aliquots of the cap analog solution to 1.4 mL of 0.1 or 0.3 μM protein solution of LeishIF4E-1. Each titration consisted of 40–65 data points. Fluorescence intensities (excited at 280 or 295 nm and observed at 337 or 345 nm) were corrected for sample dilution (<5%) and for the inner filter effect, using empirical calibration curve (Lakowicz 1999). The theoretical curve for the fluorescence intensity (F) as a function of [L] was fitted to the experimental data points according to the following equation:
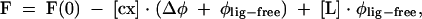 |
where the equilibrium concentration of the cap-LeishIF4E complex [cx] is given by
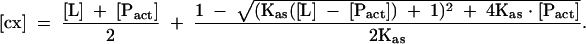 |
The parameters to be extracted from the fit were as follows: Kas, the association constant; [Pact], the concentration of the active protein; Δφ= φP-act-free − φcx, the difference between the fluorescence efficiencies of the apo-protein and the complex; φlig-free, the fluorescence efficiency of the free cap analog in the solution; F(0), the initial fluorescence intensity (Niedzwiecka et al. 2002). The final Kas was calculated as a weighted average of two to five independent titrations; numerical least-squares nonlinear regression analysis was performed using ORIGIN 6.0 (Microcal Software Inc.).
Acknowledgments
We are indebted to Dr. Nahum Sonenberg from McGill University, Canada, for his generous gift of the eIF4E(28–217) plasmid. This research was supported by the State Committee for Scientific Research (Grants PBZ-KBN 059/T09/10, PBZ-KBN 059/T09/14, and KBN 3 P04A 021 25 to E.D., R.K., and R.S., accordingly), and grants from the German-Israel Foundation (GIF, 728-23.2/2002) and Israel Ministry of Health 5440 (to M.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7510504.
REFERENCES
- Bangs, J.D., Crain, P.F., Hashizume, T., McClosky, J.A., and Boothroyd, J.C. 1992. Mass spectrometry of mRNA cap 4 from Trypanosomatids reveals two novel nucleosides. J. Biol. Chem. 267: 9805–9815. [PubMed] [Google Scholar]
- Beaucage, S.L. and Caruthers, M.H. 1981. Deoxynucleotide phosphoramidites—A new class of key iintermediates for deoxypoly-nucleotide synthesis. Tetrahedron Lett. 22: 1859–1862. [Google Scholar]
- Blachut-Okrasinska, E., Bojarska, E., Niedzwiecka, A., Chlebicka, L., Darzynkiewicz, E., Stolarski, R., Stepinski, J., and Antosiewicz, J.M. 2000. Stopped-flow and Brownian dynamics studies of electrostatic effects in the kinetics of binding of 7-methyl-GpppG to the protein eIF4E, Eur. Biophys. J. 29: 487–498. [DOI] [PubMed] [Google Scholar]
- Cai, A., Jankowska-Anyszka, M., Centers, A., Chlebicka, L., Stepinski, J., Stolarski, R., Darzynkiewicz, E., and Rhoads, R.E. 1999. Quantitative assessment of mRNA cap analogues as inhibitors of in vitro translation. Biochemistry 38: 8538–8547. [DOI] [PubMed] [Google Scholar]
- Carberry, S.E., Rhoads, R.E., and Goss, D.J. 1989. A spectroscopic study of the binding of m7GTP and m7GpppG to human protein synthesis initiation factor 4E. Biochemistry 28: 8078–8083. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182: 319–326. [DOI] [PubMed] [Google Scholar]
- Gingras, A.C., Raught, B., and Sonenberg, N. 1999. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68: 913–963. [DOI] [PubMed] [Google Scholar]
- Guzaev, A., Salo, H., Azhayev, A., and Lonnberg, H. 1995. A new approach for chemical phosphorylation of oligonucleotides at the 5′-terminus. Tetrahedron 51: 9375–9384. [Google Scholar]
- Jemielity, J., Fowler, T., Zuberek, J., Stepinski, J., Lewdorowicz, M., Niedzwiecka, A., Stolarski, R., Darzynkiewicz, E., and Rhoads, R.E. 2003. Novel “anti-reverse” cap analogs with superior translational properties. RNA 9: 1108–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierzek, R., Caruthers, M.H., Longfellow, C.E., Swinton, D., Turner, D.H., and Freier, S.M. 1986. Polymer-supported RNA synthesis and its application to test the nearest-neighbor model for duplex stability. Biochemistry 25: 7840–7846. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Instrumentation for fluorescence spectroscopy. In Principles of fluorescence spectroscopy, 2nd ed. pp. 25–61. Kluwer Academic/Plenum, New York.
- Mair, G., Ullu, E., and Tschudi, C. 2000. Cotranscriptional Cap 4 formation on the Trypanosoma brucei Spliced Leader RNA. J. Biol. Chem. 275: 28994–28999. [DOI] [PubMed] [Google Scholar]
- Mandelboim, M., Barth, S., Biton, M., Liang, X.H., and Michaeli, S. 2003. Silencing of Sm proteins in Trypanosoma brucei by RNA interference captured a novel cytoplasmic intermediate in spliced leader RNA biogenesis. J. Biol. Chem. 278: 51469–51478. [DOI] [PubMed] [Google Scholar]
- Marcotrigiano, J., Gingras, A.C., Sonenberg, N., Burley, S.K. 1997. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89: 951–961. [DOI] [PubMed] [Google Scholar]
- Matsuo H., Li, H., McGuire, A.M., Fletcher, C.M., Gingras, A.C., Sonenberg, N., Wagner, G. Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat. Struct. Biol. 4: 717–724. [DOI] [PubMed]
- Miyoshi, H., Dwyer, D.S., Keiper, B.D., Jankowska-Anyszka, M., Darzynkiewicz, E., and Rhoads, R.E. 2002. Discrimination between mono- and trimethylated cap structures by two isoforms of Caenorhabditis elegans eIF4E. EMBO J. 21: 4680–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, M.J., MacCoss, M., Naik, S.R., and Ramani, G. 1976. Nucleic acid related compounds. 21. Direct fluorination of uracil and citizen bases and nucleosides using trifluoromethyl hypofluorite. Mechanism, stereochemistry, and synthetic applications. J. Am. Chem. Soc. 98: 7381–7390. [DOI] [PubMed] [Google Scholar]
- Niedzwiecka, A., Marcotrigiano, J., Stepinski, J., Jankowska-Anyszka, M., Wyslouch-Cieszynska, A., Dadlez, M., Gingras, A.-C., Mak, P., Darzynkiewicz, E., Sonenberg, N., et al. 2002. Biophysical studies of eIF4E cap-binding protein: Recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP proteins. J. Mol. Biol. 319: 615–635. [DOI] [PubMed] [Google Scholar]
- Robins, M.J., Naik, S.R., and Lee, A.S.K. 1974. Nucleic acid related compounds. 12. The facile and high-yield stannous chloride catalyzed monomethylation of the cis-glycol system of nucleosides by diazomethan. J. Org. Chem. 39: 1891–1899. [DOI] [PubMed] [Google Scholar]
- Sawai, H., Wakai, H., and Nakamura-Ozaki, A. 1999. Synthesis and reaction of nucleoside 5′-diphosphate imidazolide. A nonenzymatic capping agent for 5′-monophosphorylated oligoribonucleotides in aqueous solution. J. Org. Chem. 64: 5836–5840. [Google Scholar]
- Scaringe, S.A., Francklyn, C., and Usman, N. 1990. Chemical synthesis of biologically active oligoribonucleotides using β-cyanoethyl protected ribonucleoside phosphoramidite. Nucleic Acid Res. 18: 5433–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield, P., Garrard, S., and Derewenda, Z. 1999. Overcoming expression and purification problems of RhoGDI using a family of “parallel expression vectors.” Protein Expr. Purif. 15: 34–39. [DOI] [PubMed] [Google Scholar]
- Sproat, B.S. and Lamond, A.I. 1991. 2′-Methyloligoribonucleotides: Synthesis and applications. In Oligonucleotides and Analogues (ed. F. Eckstein), pp. 49–86. Oxford University Press, New York.
- Stepinski, J., Waddell, C., Stolarski, R., Darzynkiewicz, E., and Rhoads, R.E. 2001. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogues 7-methyl(3′-O-methyl)GpppG and 7-methyl (3′-deoxy)GpppG. RNA 7: 1486–1495. [PMC free article] [PubMed] [Google Scholar]
- Szer, W. and Shugar, D. 1968. 2′,3′-O-Isopropylidene-3-methyluridine. In Synthetic procedures in nucleic acid chemistry, Vol. 1 (eds. W.W. Zorbach and R.S. Tipson), pp. 433–435. Wiley, New York.
- Tomoo, K., Shen, X., Okabe, K., Nozoe, Y., Fukuhara, S., Morino, S., Sasaki, M., Taniguchi, T., Miyagawa, H., Kitamura, K., et al. 2003. Structural features of human initiation factor 4E, studied by X-ray crystal analyses and molecular dynamics simulations. J. Mol. Biol. 328: 365–383. [DOI] [PubMed] [Google Scholar]
- Ullu, E. and Tschudi, C. 1995. Accurate modification of the trypanosome spliced leader cap structure in a homologous cell-free system. J. Biol. Chem. 270: 20365–20369. [DOI] [PubMed] [Google Scholar]
- Varani, G. 1997. A cap for all occasions. Structure 5: 855–858. [DOI] [PubMed] [Google Scholar]
- Yoshikawa, M., Kato, T., and Takenishi, T. 1967. A novel method for phosphorylation of nucleosides to 5′-nucleotides. Tetrahedron Lett. 50: 5065. [DOI] [PubMed] [Google Scholar]
- Zeiner, G.M., Sturm, N.R., and Campbell, D.A. 2003. Exportin 1 mediates nuclear export of the Kinetoplastid Spliced Leader RNA. Eukaryot. Cell 2: 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemlicka, J. and Sorm, F. 1965. The reaction of dimethylchloro-methyleneammoniumchloride with 2′,3′,5′-tri-O-acetylinosine. A new synthesis of 6-chloro-9-(β-D-ribofuranosil)purine. Collection Czech. Chem. Commun. 30: 1880–1889. [Google Scholar]
- Zilka, A., Garlapati, S., Dahan, E., Yavelski, V., and Shapira, M. 2001. Developmental regulation of HSP83 in Leishmania: 3′ processing and mRNA stability control transcript abundance and translation is directed by a determinant in the 3′ untranslated region. J. Biol. Chem. 276: 47922–47929. [DOI] [PubMed] [Google Scholar]
- Zuberek, J., Stepinski, J., Niedzwiecka, A., Stolarski, R., Salo, H., Lonnberg, H., and Darzynkiewicz, E. 2002. Synthesis of tetraribo-nucleotide cap analogue m7GpppAm2′pUm2′pAm2′ and its interaction with eukaryotic initiation factor eIF4E. Collection Symposium Series. 5: 399–403. [Google Scholar]
- Zuberek, J., Wyslouch-Cieszynska, A., Niedzwiecka, A., Dadlez, M., Stepinski, J., Augustyniak, W., Gingras, A.-C., Zhang, Z., Burley, S.K., Sonenberg, N., et al. 2003. Phosphorylation of eIF4E attenuates its interaction with mRNA 5′ cap analogs by electrostatic repulsion: Intein-mediated protein ligation strategy to obtain phosphorylated protein. RNA 9: 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]