

Abstract
Previous work on group I introns has suggested that a central base triple might be more important for the first rather than the second step of self-splicing, leading to a model in which the base triple undergoes a conformational change during self-splicing. Here, we use the well-characterized L-21 ScaI ribozyme derived from the Tetrahymena group I intron to probe the effects of base-triple disruption on individual reaction steps. Consistent with previous results, reaction of a ternary complex mimicking the first chemical step in self-splicing is slowed by mutations in this base triple, whereas reaction of a ternary complex mimicking the second step of self-splicing is not. Paradoxically, mechanistic dissection of the base-triple disruption mutants indicates that active site binding is weakened uniformly for the 5′-splice site and the 5′-exon analog, mimics for the species bound in the first and second step of self-splicing. Nevertheless, the 5′-exon analog remains bound at the active site, whereas the 5′-splice site analog does not. This differential effect arises despite the uniform destabilization, because the wild-type ribozyme binds the 5′-exon analog more strongly in the active site than in the 5′-splice site analog. Thus, binding into the active site constitutes an additional barrier to reaction of the 5′-splice site analog, but not the 5′-exon analog, resulting in a reduced reaction rate constant for the first step analog, but not the second step analog. This threshold model explains the self-splicing observations without the need to invoke a conformational change involving the base triple, and underscores the importance of quantitative dissection for the interpretation of effects from mutations.
Keywords: RNA structure, catalysis, threshold effects, conformational changes, base triple, group I intron
INTRODUCTION
Functional RNAs participate in a number of multistep processes that require conformational changes as part of a normal catalytic cycle. These processes include protein synthesis by the ribosome and pre-mRNA splicing by the spliceosome (e.g., Raghunathan and Guthrie 1998; Staley and Guthrie 1999; Pape et al. 2000; Ogle et al. 2002). Conformational changes are also required for the self-splicing reaction of group I and group II introns, and some evidence for such conformational transitions has been presented (Chanfreau and Jacquier 1996; Golden and Cech 1996). The self-splicing introns offer particularly appealing systems to study functional conformational transitions of RNA, as these systems are complex enough to require such rearrangements, but are far simpler than such multicomponent molecular machines as the ribosome or the spliceosome.
Using a combination of site-directed mutagenesis and kinetic measurements, Tanner and Cech (1997) showed that a base triple in the group I intron core was important for splicing activity. Because this triple [(G111·C209)·U305 in Tetrahymena, see Fig. 1 ▶] connects the P4/P6 domain with the P3/P7/P8 domain, it was suggested that this triple ties together the core of the ribozyme, thereby allowing formation of the exon-binding site (Michel and Westhof 1990; Tanner and Cech 1997). This notion was confirmed in the recent crystal structure of the Azoarcus intron (Adams et al. 2004). Additionally, it was suggested that the base triple is formed during the first, but not the second step of self-splicing, implying its involvement in a conformational change between the two chemical steps of self-splicing (Tanner et al. 1997). This proposal was based on the observation that disruption of the base triple resulted in disappearance of a splicing intermediate, suggesting that the first step of self-splicing was slowed more than the second step.
FIGURE 1.

Secondary structure of the L-21 ScaI group I ribozyme from Tetrahymena thermophila. This schematic representation shows the secondary structure in the topology of the tertiary structure (Golden et al. 1998). The (G111·C209)·U305 base triple is highlighted in bold, as are P1 and P9.0 formed in trans with oligonucleotide substrate S and UCG, respectively. The RNA core that is conserved between all subclasses of group I introns is shown in the gray-shaded area (Michel and Westhof 1990).
Here, we use the well-characterized Tetrahymena ribozyme system, in which splice site analogs are added in trans to further explore the role of the (G111·C209)·U305 base triple. The extensive prior kinetic and thermodynamic framework for this ribozyme renders it an ideal system for probing the role of particular interactions and the effects of mutations (Fig. 2 ▶; Strobel and Cech 1996; Narlikar and Herschlag 1998; Szewczak et al. 1998, 2002; Shan et al. 2001; Karbstein et al. 2002; Shan and Herschlag 2002). In addition, the ribozyme is fully active, so that kinetic results are not confounded by intron constructs with only fractional occupancy of the active state and potentially rate-limiting conformational changes from inactive to active forms (e.g., Walstrum and Uhlenbeck 1990; Woodson and Cech 1991; Emerick and Woodson 1993; Emerick et al. 1996).
FIGURE 2.

Thermodynamic framework for the group I ribozyme reaction (e.g., Herschlag and Cech 1990; Bevilacqua et al. 1992; McConnell et al. 1993). (Es)o and (Es)c [or (EP)o and (EP)c] refer to the open and closed conformations of the P1 duplex, in which tertiary interactions between the ribozyme core are absent and present, respectively.
We show herein that disruption of the base triple slows the forward ribozyme reaction but not the reverse reaction, analogous to the differential effects on the first and second step in self-splicing. Nevertheless, additional experiments provide strong evidence that the base triple is formed with both bound substrates and bound products. These paradoxical observations can be traced to a threshold effect on binding of reactants at the active site. Such thresholds allow uniform energetic contributions to have differential observed effects, and can thereby serve important biological functions while confounding interpretation of site-directed mutagenesis results.
RESULTS AND DISCUSSION
As noted in the Introduction, previous work on the group I self-splicing introns from Tetrahymena and Anabaena suggested the formation of a base triple that positions the P4–P6 domain of the ribozyme with respect to the P3–P7–P8 domain, and further suggested that this base triple was involved in mediating conformational changes between the two steps of self-splicing (Tanner and Cech 1997; Tanner et al. 1997; Adams et al. 2004). To further investigate and quantitate the effect from base-triple disruption on individual steps of the group I self-splicing reaction, we used the Tetrahymena L-21 ScaI ribozyme. The L-21 ScaI ribozyme is well suited for such a study, as individual reaction steps have been identified and their kinetics and thermodynamics have been quantitatively described (Fig. 2 ▶; Herschlag and Cech 1990; Bevilacqua et al. 1992, 1993; McConnell et al. 1993; Knitt et al. 1994; Narlikar et al. 1995; Narlikar and Herschlag 1996; Shan and Herschlag 1999; Karbstein et al. 2002). Furthermore, the ribozyme system, in contrast to the self-splicing system, is not confounded by slow exchange from inactive conformations. The experiments herein involve the same mutants used in the previous work (Tanner et al. 1997), except that they are investigated in the ribozyme instead of the self-splicing background; the wild-type base triple (G111·C209)·U305 is disrupted by a switch in the base pairing residues to (C111·G209) or a change in the third base from U305 to C305, and isosteric interactions are restored in the triple mutant (C111·G209)·C305 (Fig. 1 ▶; see Tanner et al. 1997 for a pictorial representation).
Disrupting the (G111·C209)·U305 triple affects the forward but not the reverse reaction
To test whether base-triple disruption has an effect on the ribozyme reaction akin to the self-splicing reaction, we measured the rate constants (kcat/Km)G and (kcat/Km)GA for the forward and reverse reaction, respectively. The rate constants (kcat/Km)G and (kcat/Km)GA describe the reaction from E·S and free G (or from E·P and free GA, Eq. 1) and, therefore, include contributions from binding of G (or GA) and reactivity of the E·S·G (or E·P·GA) complex. These reactions are analogous to the first and second self-splicing steps, as described below (Scheme 1 ▶).
SCHEME 1.

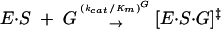 |
(1a) |
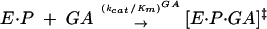 |
(1b) |
Disrupting the (G111·C209)·U305 triple decreases (kcat/Km) six- and 30-fold for the C305 and (C111·G209) mutants, respectively, although not affecting (kcat/Km)GA significantly (Table 1 ▶). Thereby, the ratio of (kcat/Km)G to (kcat/Km)GA is decreased five- and 50-fold for the C305 and (C111·G209) mutants, respectively. In the compensatory triple mutant (C111·G209)·C305, the wild-type value of (kcat/Km) G is restored and even exceeds the wild-type value by approximately sixfold (Table 1 ▶). Thus, binding of G and/or reactivity of the E·S·G complex is affected by changes in the base triple, but not binding of GA or reactivity of the E·P·GA ternary complex.
TABLE 1.
Effect of disruption of the base triple on the reaction equilibrium
| (kcat/Km)G | (kcat/Km)GA | (kcat/Km)G | |
| Ribozyme | (M−1 min−1) | ((kcat/Km)GA | |
| Wild type | 280 ± 60 | 1.9 ± 0.1 | 150 ± 24 |
| C305 | 50 ± 14 | 1.5 ± 0.1 | 33 ± 7 |
| (C111·G209) | 10 ± 5 | 3.0 ± 0.4 | 33 ± 1.6 |
| (C111 · G209) · C305) | 1820 ± 300 | 3.0 ± 1.5 | 610 ± 80 |
All experiments were performed at 30°C, pH 7.2. −1d,rSA and −1d,rP were used to ensure that the chemical step was rate-limiting in all cases.
In the previous self-splicing experiments, the base-triple disruptions were found to have a larger effect on the first step than on the second step. The results herein are analogous to the self-splicing results. This is because in the forward ribozyme reaction, a 5′-splice site analog, S, reacts with guanosine (G), to form a 5′-exon analog, P, and GA, analogous to the first step of self-splicing (Scheme 1 ▶, left). The reverse ribozyme reaction, between GA and the 5′-exon analog P, similarly resembles the second step of self-splicing (Scheme 1 ▶, center).
Disrupting the (G111·C209)·U305 base triple weakens binding of S
We next set out to determine which of the individual reaction steps was affected by disruption of the base triple. We first describe briefly the individual reaction steps of the ribozyme reaction (Fig. 2 ▶). Previous work has shown that S (and P) binding occurs in two steps, formation of a helix between S (or P) and the Internal Guide Sequence (IGS) of the ribozyme to give the P1 duplex bound in a so-called open complex [(E· S)o or (E·P)o in Fig. 2 ▶] and subsequent docking of the P1 duplex into tertiary interactions at the active site to give the so-called closed complex [(E·S)c or (E·P)c in Fig. 2 ▶; e.g., Pyle and Cech 1991; Bevilacqua et al. 1992; Herschlag 1992; Strobel and Cech 1993; Narlikar and Herschlag 1996; Bartley et al. 2003]. Binding of S and G (or P and GA) is not ordered and can occur through the top or bottom pathway, or the hybrid pathway in which G (or GA) binds to the (E·S)o [(or E·P)o] complex in Figure 2 ▶. For wild-type S [or P, i.e., with all ribose residues; note that a deoxyribose residue at the cleavage site does not affect docking (Narlikar et al. 1997); −1d,rSA, Chart 1 ▶] and wild-type ribozyme, docking of the P1 duplex is favorable, leading to accumulation of the (E·S)c [or (E·P)c] complex. However, certain modifications within the ribozyme or S strands of the P1 duplex disfavor the docked state, leading to stable accumulation of the (E·S)o complex (e.g., Herschlag et al. 1993; Strobel and Cech 1995, 1996; Narlikar and Herschlag 1996, 1998). Reaction can only proceed after formation of the closed, ternary complex, (E·S·G)c [or (E·P·GA)c], and the chemical step is preceded by loss of a proton, presumably from the attacking 3′-hydroxyl group of G in the forward reaction or the attacking 3′-hydroxyl group of P in the reverse reaction (Knitt and Herschlag 1996; Karbstein et al. 2002).
CHART 1.
Oligonucleotide substrates and their abbreviations and properties
| Abbreviation | Oligonucleotide | Attributes |
| 5′-Splice- site Analogs | ||
| Position: −3 −1 +1 | ||
| −1d,Rsa | rC rC rC rU rC dT rA | Wild type, with slow chemical step |
| −1d,−3m,rSA | rC rC rC mU rC dT rA rA4 | Undocked |
| −1d,rSA5 | rC rC rC mU rC dT rA rA4 | A5 tail interferes with P9.0 |
| 5′-Exon Analogs | ||
| Position: −3 −1 | ||
| −1d,rP | rC rC rC rU rC dT | Wild type, with slow chemical step |
Note: r = 2′-OH; d = 2′-H, m = 2′-OCH3
We first determined the effect of the base-triple mutation on the affinity for S. Effects on binding of S were determined by measuring dissociation rate constants, as S and P oligonucleotides have been shown to bind to ribozymes with essentially unchanged association rate constants, presumably because this association is limited by a simple duplex formation between the ribozyme’s IGS and the oligo-nucleotide (Herschlag and Cech 1990; Knitt and Herschlag 1996; Narlikar et al. 1999; Karbstein et al. 2002). For simplicity, the effects on S (and P, below) binding are reported as equilibrium dissociation constants with an assumed constant association rate constant of 108 M−1min−1 (Herschlag and Cech 1990; Knitt and Herschlag 1996; Narlikar et al. 1999; Karbstein et al. 2002).
Native gel-pulse chase experiments were used to obtain dissociation rate constants and affinities for S for the base-triple ribozyme variants. Disruption of the base triple by either mutation weakened S affinity by 15- to 20-fold, and the compensatory base-triple mutant restores wild-type affinity to within threefold (Table 2 ▶). The affinity for S in the disrupting mutants is consistent with S bound in the open complex (Kd,open = 2.0 nM and Kd,closed = 0.18 nM for wild-type ribozyme; Kd,obs = 3.4 and 2.8 nM for the C305 and the mutants, respectively, (Table 2 ▶; data not (C111·G209) shown). This observation suggests that disrupting the base triple disrupts interactions required for P1 docking, consistent with the suggested role of the triple in aligning the P4–P6 domain with respect to the P3–P7–P8 domain, the two regions that appear to provide the docking interface for P1 (Michel and Westhof 1990; Tanner and Cech 1997; Golden et al. 1998; Szewczak et al. 1998; Adams et al. 2004).
TABLE 2.
Effect of disruption of the base triple on binding of 5′-splice site and 5′-exon analogs (S and P)
| Ribozyme | KSd(nM) | kc(min−1) | KdP (nM) | K−c (min−1) |
| Wild type | 0.18 ± 0.02 | 0.036 ± 0.06 | 8.8 ·10−3± 2 3 ·10−3 | 0.22 ± 0.03 |
| C305 | 3.4 ± 0.2 | 0.013 ± 0.05 | 0.19 ± 0.03 | 0.14 ± 0.01 |
| (C111 ·G209) | 2.8 ± 0.2 | 0.0070 ± 0.003 | 0.94 ± 0.06 | 0.25 ± 0.01 |
| (C111 · G209) ·C305 | 0.51 ± 0.01 | 0.019 ± 0.02 | 8.4 ·10−3± 9 3 ·10−4 | 0.18 ± 0.02 |
All experiments were performed at 30°C and pH 7.2 using −1d,rSA and −1d,rP. Dissociation constant for S and P were calculated from the dissociation rate constants determined in gel-shift experiments and the association rate constant of 108 M−1 min−1 (Herschlag and Cech, 1990; Knitt and Herschlag 1996; Karbstein et al 2002). kc values were obtained from the plateau of reaction rate constant dependences on G concentration and represent the rate constant at saturating G and S. k−c values were obtained from the plateau of the rate constant of the reverse reaction at high AUCGA concentration and represent the rate constant at saturating AUCGA and P. The faster rate constants for the reverse reaction relative to the forward reaction can be explained by the use of AUCGA. Control experiments show that AUCG reacts approximately fivefold faster than G (data not shown).
Disrupting the (G111·C209)·U305 base triple does not affect G affinity
We compared G binding to wild-type and mutant (E·S)o complexes to determine whether disruption of the base triple affected G binding. Binding of G to the (E·S)o complexes was the same, within twofold, for the wild-type and mutant ribozymes (Table 3 ▶), providing no indication of an effect of the triplex on the G binding site.
TABLE 3.
Guanosine affinity with wild-type and undocked substrate
| KGd(μM) | KGd,(E·S·G)o | ||
| Ribozyme | (E·S·G)c | (E·S·G)o | KGd(E·S·G)c |
| Wild type | 128 ± 30 | 520 ± 60 | 4.0 ± 0.5 |
| C305 | — | 400 ± 160 | — |
| (C111 · G209) | — | 220 ± 40 | — |
| (C111 · G209) · C305 | 12 ± 4 | 350 ± 20 | 30 ± 8 |
All experiments were performed at 30°C using −1d,rSA or−1d,−3m,rSA5 for binding in the closed and open complex, respectively. Note that no measurement of G binding to the C305 and(C111· G209) mutants in the closed complex is reported because the−1d,rSA substrate does not bind to these mutants in the closed complex. Dissociation constants for G were determined from dependences of the rate constant for reaction of −1d,rSA and −1d,−3m,rSA5 on G concentration.
Binding of G to the wild-type ribozyme is approximately fourfold stronger to the (E·S)c complex than to free E or the (E·S)o complex. This differential binding represents thermodynamic coupling between G binding and S docking (McConnell et al. 1993; Karbstein et al. 2002; Shan and Herschlag 2002). The similar dissociation constants for G binding to the (C111·G209) and C305 mutants with bound −3m,−1d,rSA5, which ensures formation of the open complex, and with bound −1d,rSA, the normal 5′-exon analog, provide further evidence for binding of the normal 5′-exon analog in the open rather than closed complex when (C111·G209) and C305 ribozymes are used (Kd,G = 220 ± 8 and 400 ± 160 μM using −1d,rSA and −1d, −3m,rSA5 substrates with the C305 mutant and Kd,G = 402 ± 130 and 220 ± 40 μM using −1d,rSA and −1d, −3m,rSA5 substrates with the (C111·G209) mutant (Table 3 ▶; data not shown). The coupling that is absent for the (C111·G209) and C305 mutant ribozymes is restored in the compensatory mutant in which the base triple is reformed and binding in the closed complex is restored (Table 3 ▶); coupling for this variant is even stronger than for wild type (30-fold instead of fourfold; Table 3 ▶).
Direct comparison of the wild-type and mutants for G binding to the closed complex of E and S was not possible, because S does not stably dock to the (C111·G209) and the C305 mutant ribozymes, as noted above.
Disrupting the (G111·C209)·U305 base triple does not affect P9.0 formation
Whereas in the first step of self-splicing, G binds solely via interactions with the G binding site, the G at the 3′-splice site (′G) is preceded by a short sequence that can form a helix, termed P9.0, which helps to position ′G at the active site (Burke 1989; Michel et al. 1989; Moran et al. 1993; Russell and Herschlag 1999b; Karbstein et al. 2002; Fig. 1 ▶; Eq. 1). Thus, whereas the chemical transformations in the two steps of self-splicing are simply the reverse of one another, some of the surrounding interactions are different, including the absence or presence of the P9.0 helix. To further relate the results herein to the self-splicing reaction, we determined whether formation of the (G111·C209)·U305 base triple affected the energetic contribution from the P9.0 helix.
In the ribozyme reaction, P9.0 can be formed in trans, using the analog AUCG and, for the reverse reaction, AUCGA (Moran et al. 1993; Russell and Herschlag 1999b). Table 4A ▶ compares the values for (kcat/Km)AUCG and (kcat/Km)G for the wild-type and mutant ribozymes. The ratio of (kcat/Km)AUCG and (kcat/Km)G is the same within threefold for the mutant ribozymes. Similarly, the ratio of AUCG to G-binding affinities to the (E·S)o complex is the same within twofold for the wild-type and base-triple disruption ribozymes, indicating that P9.0 provides the same energetic contribution. Surprisingly, binding of AUCG is especially strong for the compensatory triple mutant, thereby leading to a fivefold increase in the binding ratio (Table 4A ▶); we do not understand the origin of this effect. The contribution of P9.0 in the reverse reaction with AUCGA was the same within threefold for all ribozyme forms (Table 4B ▶).
TABLE 4A.
Effect from disruption of the base triple on P9.0 formation in the forward reaction
| Kd (μM) | Ratio | (kcat/Km) (M−1 min−1) | Ratio | |||
| Ribozyme | AUCG | G | G/AUCG | AUCG | G | AUCG/G |
| Wild type | 14.4 ± 5.2 | 520 ± 60 | 36 ± 9 | 9.8 · 104 ± 3000 | 280 ± 60 | 350 ± 65 |
| C305 | 8.0 ± 2.0 | 400 ± 160 | 50 ± 8 | 7.5 · 103 ± 170 | 50 ± 14 | 150 ± 38 |
| (C111 · G209) | 19.7 ± 1.4 | 220 ± 40 | 11 ± 1 | 3.7 · 103 ± 300 | 10 ± 5 | 370 ± 150 |
| (C111 · G209) · C305 | 2.2 ± 0.2 | 350 ± 10 | 159 ± 10 | 2.0 · 105 ± 7000 | 1820 ± 300 | 110 ± 14 |
All experiments were performed at 30°C, pH 7.2. Kd values were determined with −1d,−3m,rSA5 to ensure formation of the open complex. −1d,rSA was used to determine (kcat/Km) values.
TABLE 4B.
Effect from disruption of the base triple on P9.0 formation in the reverse reaction
| (kcat/Km) (M −1 min−1) | Ratio | ||
| Ribozyme | AUCGA | GA | AUCGA/GA |
| Wild type | 3.9 · 103 ± 1000 | 1.9 ± 0.1 | 2052 ± 418 |
| C305 | 4.7 · 103 ± 500 | 1.5 ± 0.1 | 3133 ± 124 |
| (C111 · G209) | 7.1 · 103 ± 1300 | 3.0 ± 0.4 | 2367 ± 354 |
| (C111 · G209) · C305 | 1.8 · 104 ± 3000 | 3.0 ± 1.5 | 6000 ± 800 |
All experiments were performed at 30°C, pH 7.2. −1d,rP was used to ensure that the chemical step was rate-limiting in all cases.
Disrupting the (G111·C209)·U305 base triple has little or no effect on the chemical step
To investigate whether the base triple affected the actual transformation of the ternary complex of bound substrates or products, we directly determined these single turnover reaction rate constants. There were modest effects for reaction of E·G·S upon disruption of the base triple, and this effect was rescued upon restoration of the base triple (Table 2 ▶). However, as noted above, the mutants with disrupted base triples bind S in the open, rather than closed complex that is required for reaction. Thus, the modest observed effects of three- and fivefold could result from an additional energetic barrier to form the reactive, closed complex (see Herschlag et al. 1993; Knitt et al. 1994; Narlikar et al. 1999; Engelhardt et al. 2000). For the reverse reaction, which starts from (E·P·AUCGA)c for the wild-type and mutant ribozymes (see below), there is no effect of the mutations on the reaction rate (Table 2 ▶). This observation strongly suggests that disruption of the base triple does not affect the chemical-transition state, so that disruption of docking interactions can account for the observed effects on (kcat/Km)G and kc. Support for this conclusion is presented in the next section.
Disrupting the (G111·C209)·U305 base triple weakens binding of P
The absence of an effect of the mutations on the reverse reaction of E·P with GA and of the E·P·AUCGA ternary complex was consistent with an absence of a role of the base triple in the reverse ribozyme reaction, analogous to the proposed absence of an effect on the second self-splicing step (see Scheme 1 ▶ and above). However, direct investigation of the binding affinity of the 5′-exon analog P revealed an effect of the base-triple mutations analogous to that observed with the 5′-splice site analog S.
Binding of P was determined from the association rate constant of 108 M−1min−1, as noted above, and from the dissociation rate constants obtained in native gel-pulse chase experiments. Binding of P to free E is weakened ~20-and 100-fold in the C305 and (C111·G209) mutants, respectively, and wild-type affinity is restored in the compensatory triple mutant (Table 2 ▶). Not only is binding of the 5′-exon analog weakened, but the effect is larger than that for binding of S. Nevertheless, reactivity of bound S was affected more strongly than reactivity of bound P (see above). Below, we describe a model that reconciles these paradoxical observations and quantitatively accounts for their origin.
A threshold model accounts for the differential effects of base-triplet disruption on the first and second step of self-splicing
The weaker binding of S and P and the modest effects on binding of G with a docked but not an undocked substrate for the (C111·G209) and the C305 mutants suggests that disrupting the (G111·C209)·U305 base triple weakens docking of the P1 duplex into tertiary interactions with the ribozyme core. This conclusion is consistent with the importance of the (G111·C209)·U305 base triple for alignment of the interface between P4/P6 and P3/P7/P9 for P1 docking (Tanner and Cech 1997; Szewczak et al. 1998; Adams et al. 2004). Below, we show that such an effect on docking can quantitatively account for all of the effects observed with the base-triple mutants according to the free-energy reaction profile in Figure 3 ▶.
FIGURE 3.

Quantitative free-energy profile for the group I reaction with wild-type and (C111·G209) ribozyme. The free-energy profiles were calculated at 30°C (pH 7.2) and for a standard state of 1 mM G and GA. In the case of the reaction with wild-type ribozyme (continuous line), the reaction described by (kcat/Km)G is from (E·S)c + G and the transition state for the chemical step (‡). For the (C111·G209) mutant (broken line), the reaction described by (kcat/Km)G is from (E·S)o + G, and the transition state of the chemical step (‡), and thus includes the additional barrier for docking. The free-energy profile was calculated as outlined in the Materials and Methods using the values listed in Table 6 ▶.
The data of Table 2 ▶ suggest that docking is destabilized sufficiently to change the ground state for bound S from the closed to the open state [(E·S)c to (E·S)o; Fig. 3 ▶, solid vs.broken line]. We have shown directly that base-triple disruption does not affect G binding, so this step is drawn the same for both the wild-type and mutant ribozymes in Figure 3 ▶. The results above for reaction of the E·P·AUCGA complex indicate that the chemical reactivity is not affected in the reverse reaction;2 on the basis of this observation, we assume that the same holds for reaction of the E·S·G closed complex, and this assumption is supported by the self-consistency of the data for the forward and reverse reactions in the model of Figure 3 ▶, as described below. We therefore show the free energy difference between the (E·S)c complex and the transition state for the chemical step (‡) to be the same for the wild-type and mutant ribozymes (Fig. 3 ▶). For the mutant, however, there is an additional barrier to reaction of the E·S complex, because the starting ground state is the (E·S)o complex instead of the (E·S)c complex. This additional energetic cost is responsible for the observed slower reaction of the mutant ribozymes. According to Figure 3 ▶, the size of this additional barrier is determined by the detrimental effect of the triple mutation (ΔGmut) and the equilibrium for docking in the wild-type case (ΔGdock).
Because the equilibrium for docking of P is larger than the equilibrium for docking of S (Bevilacqua et al. 1994; Narlikar et al. 1995) (ΔGPdock < ΔGSdock), the reverse reaction with bound P is predicted to be affected less than that for reaction of bound S from destabilized docking. If the perturbation is less than the overall docking energy (i.e., if the absolute value |ΔGmut| < |ΔGdock|), then no significant effect will be observed on reaction of bound P; the entire energetic effect, ΔGmut, will instead be manifest on P affinity, as observed herein where P binding is weakened more than binding of S, whereas its reactivity is unaffected (Tables 1 ▶, 2 ▶). According to this model (Fig. 3 ▶), the uniform destabilization of the (E·S)c and (E·P)c complexes leads, paradoxically, to differential effects in the forward and reverse reaction because of the different threshold for docking of S and P.
To show that the model of uniform destabilization of the docked state can account for the data herein, the destabilization of the docked state with S and P is compared (Table 5 ▶). P remains docked on the mutant ribozymes as suggested by the lack of an effect on reactivity of the E·P·AUCGA complex (Tables 2 ▶, 4B ▶), and as also suggested by the affinity of P, which is slightly stronger than the affinity for S, even though P lacks stacking interactions that strengthen binding of S relative to P (Narlikar et al. 1995; Table 2 ▶). Because P remains docked, the difference in affinity of P between wild-type and mutant ribozymes provides a direct readout for the amount of destabilization of docking from the mutants. As shown in Table 2 ▶, this destabilization amounts to 22-and 107-fold for the C305 and the (C111·G209) mutants, respectively. Because S undocks, the equilibrium constant for docking of S, Kdock, to wild-type ribozyme provides a lower limit for the amount of destabilization of S. The value for Kdock of 22 (Bartley et al. 2003) and the approximately three- and fivefold effects on kc for the C305 and the (C111·G209) mutants, respectively, can be combined to obtain an estimate for the amount of destabilization of S docking (Table 5 ▶; see Materials and Methods for details of the calculation). For the (C111·G209) mutant, these calculations give a uniform destabilization of docking of S and P of 107-and 90-fold. For the C305 mutant, the calculations show a twofold difference, which could arise from a small additional effect or from combined errors in measurements with small differences. We conclude that the threshold model provides a reasonable description of the effects from these mutations.
TABLE 5.
Uniform effects from mutations on docking of S and P
| Ribozyme | KPmut | KSmut |
| C305 | 22 | 40 |
| (C111 · G209) | 107 | 90 |
KmutS and KmutP represent the fold-effect from the base triple mutations on the docking equilibrium for S and P, respectively; for both mutants cases the docking equilibrium is destabilized. Values for KmutP were obtained directly from the difference in affinity of P to wild type and mutant ribozymes (Table 2 ▶). Values for KmutS were obtained from the equilibrium constant for docking of substrate to wild-type ribozyme, Kdock,wt and the difference in reaction rate constant kc between wild-type and mutant ribozymes (Table 2 ▶) as described in the Materials and Methods.
Implications for self-splicing
Reaction of S with G resembles the first step of self-splicing, whereas reaction of P with AUCGA resembles the second step of self-splicing (Eq. 1). Thus, our results predict that during self-splicing, the base-triple disruptions slow the first step of self-splicing, for which there is now an additional barrier, although not affecting the second step of self-splicing. Previous work has shown exactly this behavior (Tanner et al. 1997). Whereas it was suggested in the previous work that this observation was due to a conformational change that disrupted this base triple, our data show that such a model does not need to be invoked, and suggest that, instead, the effects on splicing are the result from the different threshold for undocking of S and P as described above.
General implications
Threshold effects result from the occurrence of distinct binding modes, either as a result of mutagenesis or substrate alterations, as shown here for the group I intron, or as part of their functional reaction cycle. Examples for the latter include nonspecific binding modes of DNA-binding proteins, such as restriction endonucleases and aminoacyl-tRNA binding to the ribosome prior to accommodation in the active site. In these cases, the distinct binding modes are used as components in systems evolved to achieve high specificity. Appreciating the balance between these highly evolved states is therefore critical not only for interpreting experimental observations, as shown herein, but also for a full understanding of the functioning of biological machines and systems.
MATERIALS AND METHODS
Materials
L-21 ScaI ribozyme and its mutants were transcribed and purified over an RNeasy column (QIAGEN) or via gel-purification as described previously (Russell and Herschlag 1999b; Karbstein et al. 2002). Ribozyme mutants were constructed via the Quikchange method (Stratagene). Clones were sequenced to verify the correct sequence.
RNA oligonucleotides were purchased from Dharmacon Research, Inc. Chart 1 ▶ lists the oligonucleotides used in these studies. RNA oligonucleotides were 5′-end-labeled using γ[32P]ATP and T4 polynucleotide kinase and were gel-purified using standard procedures (Zaug et al. 1988). RNA oligonucleotides used unlabeled were purified by anion exchange HPLC (NucleoPak, Dionex) and desalted on Sep-Pak C-18 columns (Waters).
General kinetic methods
All reactions were single turnover and, unless otherwise noted, were carried out with L-21 ScaI ribozyme at 30°C in 50 mM Na-MOPS (pH 7.2) and 10 mM MgCl2. Prior to reaction, ribozyme was preincubated for 30 min at 50°C (pH 6.0) with 10 mM MgCl2 to allow folding to the active state (Herschlag and Cech 1990; Russell and Herschlag 1999a). Reaction aliquots were removed at specified times to be quenched with 2 vol of a solution containing 20 mM EDTA and 85% formamide. Radiolabeled oli-gonucleotides were separated by denaturing gel electrophoresis (7 M urea, 20% acrylamide) and quantitated using PhosphorImager analysis (Molecular Dynamics) with ImageQuant quantitation software. For slow reactions, rate constants were obtained from initial rates assuming endpoints of 95%. All other reactions were followed to completion, and good first-order fits to the data with endpoints of ≥90% were obtained in all cases (R2 ≥0.98) (Kaleidagraph, Synergy Software).
Measurement of (kcat/Km)(AUC)G and (kcat/Km)(AUC)GA values
(kcat/Km)G values were determined with three subsaturating concentrations of G and E saturating with respect to S (2–20 μM G, 0.5–2 μM G for the (C111·G209)·C305 mutant, 70 nM E). Rate constants were plotted as a function of G concentration to yield (kcat/Km)G from the slope. (kcat/Km)AUCG values were determined analogously using 0.05–0.2 μM AUCG for wild-type ribozyme, or 0.2–1 μM for the C305 mutant, 0.4–2 μM for the (C111·G209) mutant, and 0.04–0.06 μM AUCG for the (C111·G209)·C305 mutant. In all cases, the −1d,rSA substrate was used to ensure that the chemical step was rate limiting.
(kcat/Km)GA measurements were performed using 5–20 μM GA (Kd ≈ 1 mM) (Bevilacqua et al. 1996; data not shown) and E saturating with respect to P (70 nM E). Rate constants were plotted as above. (kcat/Km)AUCGA measurements were performed analogously using 1–4 μM AUCGA. The 5′-exon analog −1d,rP was used in these experiments to ensure that the chemical step remained rate limiting under all conditions.
Measurement of G and AUCG affinities
To determine the affinities of ribozyme complexes for G or AUCG, the rate constant for reaction of *S was determined as a function of G (or AUCG) concentration using concentrations of G (or AUCG) ranging from 10-fold below to 10-fold above the Kd. The ribozyme concentration was 25 nM to ensure that S was fully bound to the ribozyme. The observed rate constant for cleavage of *S was plotted as a function of G concentration and fit to Equation 2.
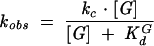 |
(2) |
The −1d,rSA (or −1d,rSA5 in the case of G) was used in these experiments to ensure that the chemical step was rate limiting and that the observed KG1/2 equals the KGd (McConnell et al. 1993). To determine the KGd in the open complex, a substrate with a 2′-methoxy substitution at position −3 was used (Narlikar and Herschlag 1996; Bartley et al. 2003) with 50 nM ribozyme.
The affinity of AUCGA for the E·P complex was determined analogously using −1d,rP and AUCGA concentrations ranging from 10-fold below to 10-fold above the Kd value with ribozyme saturating with respect to P [25 nM; Kd = 0.01 – 1 nM for wild-type and mutant ribozymes (Table 2 ▶)].
Dissociation constants for S and P
Because binding of S and P is very tight, it was not possible to measure dissociation constants for S and P by varying ribozyme concentration. This is because at ribozyme concentrations sufficiently low to be subsaturating, nonspecific losses to the tube walls occur (Karbstein et al. 2002). Equilibrium dissociation constants for S and P were instead calculated from dissociation rate constants and the association rate constant of ~108 M−1 min−1 that appears to hold for binding of all oligonucleotide substrates under a range of conditions (Herschlag and Cech 1990; Knitt et al. 1994; Karbstein et al. 2002).
The dissociation rate constants for S (or P) from E·S (or E·P) were obtained in pulse-chase gel-shift experiments (Mei and Herschlag 1996; Karbstein et al. 2002). Trace *S (or *P) was bound to saturating amounts of ribozyme (120 nM) and a large excess of unlabeled P (>10-fold over ribozyme) was then added. At specified times, aliquots were loaded onto a running native gel in THEM buffer (33 mM Tris, 67 mM HEPES, 1 mM EDTA, 10 mM MgCl2). The remaining fraction of bound oligonucleotide was plotted against the time of the chase and fit to an exponential decay. Control experiments confirmed the efficiency of the chase and full binding prior to addition of the chase.
Calculations for the free-energy profile in Figure 3 ▶
Free energies were calculated from equilibrium constants K or from rate constants k at 30°C and a standard state of 1 mM G and GA according to Equations 3 and 4,
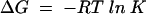 |
(3) |
 |
(4) |
where R is the Boltzmann constant, T = 303 K and ν = 6.212·1012 s−1. Table 6 ▶ lists the rate and equilibrium constants and their sources used to calculate the free energy profile.
TABLE 6.
Rate and equilibrium constants used to calculate the free energy profile in Figure 3 ▶
| Constant | Wild type | C111 · G209 | Reference |
| Kdocks,S | 22 | 0.07a | Bartley et al. 2003 |
| kdock,S (min−1) | 162 | 162b | Bartley et al. 2003 |
| Kd,G (μM) | 128 | 220 | Table 3 ▶ |
| kon,G (M−1 min−1) | 6 · 105 | 6 · 105 | Karbstein and Herschlag 2003 |
| Keq | 0.7 | 0.7 | K. Karbstein and D. Herschlag, unpubl. data |
| kc (min−1) | 0.03 | 0.03c | Table 2 ▶ |
| Kd,GA (mM) | 1d | 1d | Data not shown |
| kon,GA (M−1 min−1) | 6 · 105 | 6 · 105 | Karbstein and Herschlag 2003 |
| Kdock,P | 880 | 2.8e | Narlikar et al. 1995 |
| kdock,P (min−1) | 162b | 162b | Bartley et al. 2003 |
aThis value for Kdock was calculated from the 28-fold reduction in (kcat/Km)G and the twofold weaker G binding for this mutant, assuming that the rate constant for the chemical step was unaffected.
bMany substitutions have been shown to affect Kdock through kundock but not kdock; we therefore assume that kdock,S remains un-affected (Bartley et al. 2003).
cThe lower observed rate constant for reaction of the E · S · G ternary complex is due to unfavorable docking and not to a lower intrinsic reaction rate constant (See “A Threshold Model to Explain the Different Effects of Triplet Disruption on the First and Second Step of Self-splicing”).
dAs GA binding is weak (Kd > 1 mM, Bevilacqua et al. 1996 and data not shown), this value could not be reliably measured. However, the value of 1 mM, is obtained from the affinity of UCG to E · P of 88 μM and the 14-fold stronger binding of UCG than G (Karbstein et al., 2002).
eCalculated from Kdock,S and the 40-fold stronger docking of P relative to S (Narlikar et al. 1995).
Calculations of the effects on docking from the mutations in Table 5 ▶
KSmut represents the amount by which the docking equilibrium is destabilized by the mutant construct relative to the wild-type construct (Eq. 5).
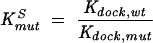 |
(5) |
Kdock,mut and Kdock,wt represent the equilibrium constant for docking for the mutant and wild-type ribozymes, respectively. The value for Kdock,wt of 22 was obtained from Bartley et al. (2003),and the value for Kdock,mut was obtained from Equation 6, which was derived with the assumptions that the wild-type and mutant ribozymes have the same reaction rate from their closed complexes and that the wild-type rate constant, but not the mutant-rate constant, is that for the fully docked case; this latter approximation does not significantly affect the calculated values for mutants, such as those herein, in which docking is substantially destabilized. kc,mut is the observed rate constant of cleavage by the mutant ribozyme with saturating substrates (G and −1d,rSA) and kc,wt is the corresponding rate constant for cleavage by the wild-type ribozyme.
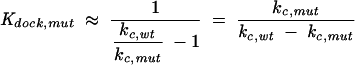 |
(6) |
Acknowledgments
This work was supported by NIH grant GM49243. We thank members of the Herschlag lab for comments on the manuscript. K.K. was supported in part by a graduate fellowship from the Boehringer Ingelheim Foundation.
Abbreviations
S, 5′-splice site analog without specifying the identity of the 2′-substituents, which can be a hydrogen atom, a hydroxyl group, or a methoxy group (see Chart 1 ▶ for the specific oligonucleotides used)
P, 5′-exon analog (see Chart 1 ▶)
*S, radioactively labeled S
*P, radioactively labeled P
G, guanosine
E, L-21 ScaI ribozyme (mutant or wild type)
HPLC, high pressure liquid chromatography
HEPES, 4-(2-hydroxy-ethyl)-piperazine-1-ethane-sulfonic acid
MOPS, 3-(N-morpholino)-propane-sulfonic acid
EDTA, ethylenediaminetetraacetic acid, Tris, Tris[hydroxymethyl]aminomethane
IGS, internal guide sequence
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7118104.
Footnotes
AUCGA was used instead of GA, as GA binds weakly and the additional P9.0 interactions allow saturation to be achieved (Moran et al. 1993; Russell and Herschlag 1999b; Karbstein et al. 2002).
REFERENCES
- Adams, P.L., Stahley, M.R., Kosek, A.B., Wang, J., and Strobel, S.A. 2004. Crystal structure of a self-splicing group I intron with both exons. Nature 430: 45–50. [DOI] [PubMed] [Google Scholar]
- Bartley, L.E., Zhuang, X., Das, R., Chu, S., and Herschlag, D. 2003. Exploration of the transition state for tertiary structure formation between an RNA helix and a large structured RNA. J. Mol. Biol. 328: 1011–1026. [DOI] [PubMed] [Google Scholar]
- Bevilacqua, P.C., Kierzek, R., Johnson, K.A., and Turner, D.H. 1992. Dynamics of ribozyme binding of substrate revealed by fluorescence-detected stopped-flow methods. Science 258: 1355–1357. [DOI] [PubMed] [Google Scholar]
- Bevilacqua, P.C., Johnson, K.A., and Turner, D.H. 1993. Cooperative and anticooperative binding to a ribozyme. Proc. Natl. Acad. Sci. 90: 8357–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua, P.C., Li, Y., and Turner, D.H. 1994. Fluorescence-detected stopped-flow With a pyrene-labeled substrate reveals that guano-sine facilitates docking of the 5′ cleavage site into a high free-energy binding mode in the Tetrahymena ribozyme. Biochemistry 33: 11340–11348. [DOI] [PubMed] [Google Scholar]
- Bevilacqua, P.C., Sugimoto, N., and Turner, D.H. 1996. A mechanistic framework for the second step of splicing catalyzed by the Tetrahymena ribozyme. Biochemistry 35: 648–658. [DOI] [PubMed] [Google Scholar]
- Burke, J.M. 1989. Selection of the 3′-splice site in group I introns. FEBS Lett. 250: 129–133. [DOI] [PubMed] [Google Scholar]
- Chanfreau, G. and Jacquier, A. 1996. An RNA conformational change between the two chemical steps of group II self-splicing. EMBO J. 15: 3466–3476. [PMC free article] [PubMed] [Google Scholar]
- Emerick, V.L. and Woodson, S.A. 1993. Self-splicing of the Tetrahymena pre-ribosomal-RNA is decreased by misfolding during transcription. Biochemistry 32: 14062–14067. [DOI] [PubMed] [Google Scholar]
- Emerick, V.L., Pan, J., and Woodson, S.A. 1996. Analysis of rate-determining conformational changes during self-splicing of the Tetrahymena intron. Biochemistry 35: 13469–13477. [DOI] [PubMed] [Google Scholar]
- Engelhardt, M.A., Doherty, E.A., Knitt, D.S., Doudna, J.A., and Her-schlag, D. 2000. The P5abc peripheral element facilitates preorganization of the Tetrahymena group I ribozyme for catalysis. Biochemistry 39: 2639–2651. [DOI] [PubMed] [Google Scholar]
- Golden, B.L. and Cech, T.R. 1996. Conformational switches involved in orchestrating the successive steps of group I RNA splicing. Biochemistry 35: 3754–3763. [DOI] [PubMed] [Google Scholar]
- Golden, B.L., Gooding, A.R., Podell, E.R., and Cech, T.R. 1998. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science 282: 259–264. [DOI] [PubMed] [Google Scholar]
- Herschlag, D. 1992. Evidence for processivity and two-step binding of the RNA substrate from studies of J1/2 mutants of the Tetrahymena ribozyme. Biochemistry 31: 1386–1399. [DOI] [PubMed] [Google Scholar]
- Herschlag, D. and Cech, T.R. 1990. Catalysis of RNA cleavage by the Tetrahymena-thermophila ribozyme. 1. Kinetic description of the reaction of an RNA substrate complementary to the active-site. Biochemistry 29: 10159–10171. [DOI] [PubMed] [Google Scholar]
- Herschlag, D., Eckstein, F., and Cech, T.R. 1993. Contributions of 2′-hydroxyl groups of the RNA substrate to binding and catalysis by the Tetrahymena ribozyme: An energetic picture of an active-site composed of RNA. Biochemistry 32: 8299–8311. [DOI] [PubMed] [Google Scholar]
- Karbstein, K. and Herschlag, D. 2003. Extraordinarily slow binding of guanosine to the Tetrahymena group I ribozyme: Implications for RNA structure and preorganization. Proc. Natl. Acad. Sci. 100: 2300–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbstein, K., Carroll, K.S., and Herschlag, D. 2002. Probing the Tetrahymena group I reaction in both directions. Biochemistry 41: 11171–11183. [DOI] [PubMed] [Google Scholar]
- Knitt, D.S. and Herschlag, D. 1996. pH dependencies of the Tetrahymena ribozyme reveal an unconventional origin of an apparent pKa. Biochemistry 35: 1560–1570. [DOI] [PubMed] [Google Scholar]
- Knitt, D.S., Narlikar, G.J., and Herschlag, D. 1994. Dissection of the role of the conserved G·U pair in group I RNA self-splicing. Biochemistry 33: 13864–13879. [DOI] [PubMed] [Google Scholar]
- McConnell, T.S., Cech, T.R., and Herschlag, D. 1993. Guanosine binding to the Tetrahymena ribozyme: Thermodynamic coupling with oligonucleotide binding. Proc. Natl. Acad. Sci. 90: 8362–8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, R. and Herschlag, D. 1996. Mechanistic investigations of a ribozyme derived from the Tetrahymena group I intron: Insights into catalysis and the second step of self-splicing. Biochemistry 35: 5796–5809. [DOI] [PubMed] [Google Scholar]
- Michel, F. and Westhof, E. 1990. Modeling of the three-dimensional architecture of group I catalytic introns based on comparative sequence-analysis. J. Mol. Biol. 216: 585–610. [DOI] [PubMed] [Google Scholar]
- Michel, F., Hanna, M., Green, R., Bartel, D.P., and Szostak, J.W. 1989. The guanosine binding-site of the Tetrahymena ribozyme. Nature 342: 391–395. [DOI] [PubMed] [Google Scholar]
- Moran, S., Kierzek, R., and Turner, D.H. 1993. Binding of guanosine and 3′ splice site analogs to a group I ribozyme: Interactions with functional groups of guanosine and with additional nucleotides. Biochemistry 32: 5247–5256. [DOI] [PubMed] [Google Scholar]
- Narlikar, G.J. and Herschlag, D. 1996. Isolation of a local tertiary folding transition in the context of a globally folded RNA. Nat. Struct. Biol. 3: 701–710. [DOI] [PubMed] [Google Scholar]
- ———. 1998. Direct demonstration of the catalytic role of binding interactions in an enzymatic reaction. Biochemistry 37: 9902–9911. [DOI] [PubMed] [Google Scholar]
- Narlikar, G.J., Gopalakrishnan, V., McConnell, T.S., Usman, N., and Herschlag, D. 1995. Use of binding energy by an RNA enzyme for catalysis by positioning and substrate destabilization. Proc. Natl. Acad. Sci. 92: 3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar, G.J., Khosla, M., Usman, N., and Herschlag, D. 1997. Quantitating tertiary binding energies of 2′ OH groups on the P1 duplex of the Tetrahymena ribozyme: Intrinsic binding energy in an RNA enzyme. Biochemistry 36: 2465–2477. [DOI] [PubMed] [Google Scholar]
- Narlikar, G.J., Bartley, L.E., Khosla, M., and Herschlag, D. 1999. Characterization of a local folding event of the Tetrahymena group I ribozyme: Effects of oligonucleotide substrate length, pH and temperature on the two substrate binding steps. Biochemistry 38: 14192–14204. [DOI] [PubMed] [Google Scholar]
- Ogle, J.M., Murphy, F.V., Tarry, M.J., and Ramakrishnan, V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732. [DOI] [PubMed] [Google Scholar]
- Pape, T., Wintermeyer, W., and Rodnina, M.V. 2000. Conformational switch in the decoding region of 16S rRNA during aminoacyl-tRNA selection on the ribosome. Nat. Struct. Biol. 7: 104–107. [DOI] [PubMed] [Google Scholar]
- Pyle, A.M. and Cech, T.R. 1991. Ribozyme recognition of RNA by tertiary interactions with specific ribose 2′-OH groups. Nature 350: 628–631. [DOI] [PubMed] [Google Scholar]
- Raghunathan, P.L. and Guthrie, C. 1998. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 8: 847–855. [DOI] [PubMed] [Google Scholar]
- Russell, R. and Herschlag, D. 1999a. New pathways in folding of the Tetrahymena group I RNA enzyme. J. Mol. Biol. 291: 1155–1167. [DOI] [PubMed] [Google Scholar]
- ———. 1999b. Specificity from steric restrictions in the guanosine binding pocket of a group I ribozyme. RNA 5: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, S. and Herschlag, D. 1999. Probing the role of metal ions in RNA catalysis: Kinetic and thermodynamic characterization of a metal ion interaction with the 2′-moiety of the guanosine nucleophile in the Tetrahymena group I ribozyme. Biochemistry 38: 10958–10975. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Dissection of a metal-ion-mediated conformational change in Tetrahymena ribozyme catalysis. RNA 8: 861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, S., Kravchuk, A.V., Piccirilli, J.A, and Herschlag, D. 2001. Defining the catalytic metal ion interactions in the Tetrahymena ribozyme reaction. Biochemistry 40: 5161–5171. [DOI] [PubMed] [Google Scholar]
- Staley, J.P. and Guthrie, C. 1999. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol. Cell 3: 55–64. [DOI] [PubMed] [Google Scholar]
- Strobel, S.A. and Cech, T.R. 1993. Tertiary interactions with the internal guide sequence mediate docking of the P1 helix into the catalytic core of the Tetrahymena ribozyme. Biochemistry 32: 13593–13604. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Minor-groove recognition of the conserved G·U pair at the Tetrahymena ribozyme reaction site. Science 267: 675–679. [DOI] [PubMed] [Google Scholar]
- ———. 1996. Exocyclic amine of the conserved G·U pair at the cleavage site of the Tetrahymena ribozyme contributes to 5′-splice site selection and transition-state stabilization. Biochemistry 35: 1201–1211. [DOI] [PubMed] [Google Scholar]
- Szewczak, A.A., Ortoleva-Donnelly, L., Ryder, S.P., Moncoeur, E., and Strobel, S.A. 1998. A minor-groove RNA triple-helix within the catalytic core of a group-I intron. Nat. Struct. Biol. 5: 1037–1042. [DOI] [PubMed] [Google Scholar]
- Szewczak, A.A., Kosek, A.B., Piccirilli, J.A., and Strobel, S.A. 2002. Identification of an active site ligand for a group I ribozyme catalytic metal ion. Biochemistry 41: 2516–2525. [DOI] [PubMed] [Google Scholar]
- Tanner, M.A. and Cech, T.R. 1997. Joining the two domains of a group I ribozyme to form the catalytic core. Science 275: 847–849. [DOI] [PubMed] [Google Scholar]
- Tanner, M.A., Anderson, E.M., Gutell, R.R., and Cech, T.R. 1997. Mutagenesis and comparative sequence analysis of a base triple joining the two domains of group I ribozymes. RNA 3: 1037–1051. [PMC free article] [PubMed] [Google Scholar]
- Walstrum, S.A. and Uhlenbeck, O.C. 1990. The self-splicing RNA of Tetrahymena is trapped in a less active conformation by gel purification. Biochemistry 29: 10573–10576. [DOI] [PubMed] [Google Scholar]
- Woodson, S.A. and Cech, T.R. 1991. Alternative secondary structures in the 5′ exon affect both forward and reverse self-splicing of the Tetrahymena intervening sequence RNA. Biochemistry 30: 2042–2050. [DOI] [PubMed] [Google Scholar]
- Zaug, A.J., Grosshans, C.A., and Cech, T.R. 1988. Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme: Enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme substrate complexes. Biochemistry 27: 8924–8931. [DOI] [PubMed] [Google Scholar]