

Abstract
Structural investigations of a GST (glutathione transferase), adGSTD4-4, from the malaria vector Anopheles dirus show a novel lock-and-key ‘Clasp’ motif in the dimer interface of the Delta class enzyme. This motif also appears to be highly conserved across several insect GST classes, but differs from a previously reported mammalian lock-and-key motif. The aromatic ‘key’ residue not only inserts into a hydrophobic pocket, the ‘lock’, of the neighbouring subunit, but also acts as part of the ‘lock’ for the other subunit ‘key’. The ‘key’ residues from both subunits show aromatic ring stacking with each other in a pi–pi interaction, generating a ‘Clasp’ in the middle of the subunit interface. Enzyme catalytic and structural characterizations revealed that single amino acid replacements in this ‘Clasp’ motif impacted on catalytic efficiencies, substrate selectivity and stability. Substitutions to the ‘key’ residue create strong positive co-operativity for glutathione binding, with a Hill coefficient approaching 2. The lock-and-key motif in general and especially the ‘Clasp’ motif with the pi–pi interaction appear to play a pivotal role in subunit communication between active sites, as well as in stabilizing the quaternary structure. Evidence of allosteric effects suggests an important role for this particular intersubunit architecture in regulating catalytic activity through conformational transitions of subunits. The observation of co-operativity in the mutants also implies that glutathione ligand binding and dimerization are linked. Quaternary structural changes of all mutants suggest that subunit assembly or dimerization basically manipulates subunit communication.
Keywords: Anopheles dirus, aromatic ring stacking, glutathione transferase, lock-and-key, pi–pi interaction, subunit interface
Abbreviations: ANS, 8-anilinonaphthalene-l-sulphonic acid; CDNB, 1-chloro-2,4-dinitrobenzene; CPK, Corey–Pauling–Koltun; DCNB, 1,2-dichloro-4-nitrobenzene; EA, ethacrynic acid; GST, glutathione transferase; PNBC, p-nitrobenzyl chloride; PNPB, p-nitrophenethyl bromide; TIM, triosephosphate isomerase
INTRODUCTION
Glutathione transferases (GST; EC 2.5.1.18) are dimeric enzymes playing a major role in detoxication mechanisms by nucleophilic addition of GSH (γ-glutamyl-L-cysteinyl-glycine) to the electrophilic centre of diverse non-polar endogenous and xenobiotic compounds, rendering them less reactive and more water-soluble substances [1–4]. Cytosolic GSTs are ubiquitous enzymes found in all organisms to date. The nomenclature of this enzyme has now been categorized to contain at least 13 classes, including seven mammalian classes (Alpha, Mu, Pi, Sigma, Theta, Omega and Zeta) and six non-mammalian classes (Beta, Delta, Epsilon, Lambda, Phi and Tau) [4,5]. In addition, members in this enzyme superfamily are increasing due to the massive growth of genomic information, which also yields a number of novel unclassified GSTs [6], a so-called ‘U’ class.
In general, all GSTs adopt the same highly conserved tertiary structure [2]. A dimeric quaternary structure is thought to be essential for a fully functional active site, since the active site is formed by amino acid residues from both subunits. The dimeric structure also has been shown to be involved in stabilization of tertiary structures of individual subunits [7], as well as to provide a non-substrate ligand-binding site at the dimer interface [8,9]. Studies of equilibrium folding revealed that dimeric formation of GSTs such as Alpha [10], Pi [11] and Sj26GST [12] has significant impact on stabilization of subunit tertiary structure, whereas the dimerization of GSTs such as Sigma [13] and Mu [14] has less influence on subunit stability, due to the presence of stable monomeric intermediates in an unfolding/refolding pathway [15]. Therefore, although the overall tertiary structure is conserved, structural variations occur that generate diversity between the GST classes.
Structural observations of GSTs demonstrate two distinct types of intersubunit interactions [3]. The first is a ‘ball-and-socket’ or so-called ‘lock-and-key’ hydrophobic interaction, involving an aromatic ‘key’ residue from domain I of one subunit that inserts into several hydrophobic ‘lock’ residues of domain II in the other subunit. This interaction was reported to be specific to mammalian Alpha/Mu/Pi classes [16–19]. The second type of intersubunit interaction is more hydrophilic and lacks the ‘lock-and-key’ motif, as in Theta/Sigma classes [20,21]. The class-specific intersubunit interactions result in different interface topologies and contribute to interface specificity. Accordingly, only subunits with the same interfacing type appear to be compatible for dimerization.
In the present study, an available refined crystal structure of adGSTD4-4, an insect Delta class GST from the mosquito malaria vector Anopheles dirus [22], was investigated for intersubunit interactions along the dimer interface. The enzyme was previously reported as adGST1-4 and later renamed to be in concord with a universal GST nomenclature [23]. This Delta GST was also observed to possess a lock-and-key motif. However, this lock-and-key ‘Clasp’ motif is found in the centre of a 2-fold axis. Therefore the motif appears to be different from those reported in mammalian Alpha/Mu/Pi classes. A striking characteristic of this motif involving the ‘key’ residue is that it not only inserts into a hydrophobic pocket of the neighbouring subunit, but also itself acts as part of the ‘lock’ for the other subunit ‘key’. In addition, the ‘key’ residues from both subunits ‘hook around’ each other in an aromatic pi–pi interaction, through slightly offset aromatic ring stacking, generating a ‘clasp’ in the middle of the subunit interface. To examine the contribution of the lock-and-key ‘Clasp’ motif to Delta class structure and function, site-directed mutagenesis was performed to Phe-104 (the ‘key’ residue) and Val-107 (one of the ‘lock’ residues). Mutant recombinant enzymes were characterized for both enzymatic and structural properties by enzyme steady-state kinetics, substrate specificity, thermal stability, far-UV CD, tryptophan intrinsic fluorescence and fluorescent dye binding.
EXPERIMENTAL
Construction of mutants
A pET3a plasmid containing the full coding sequence of the A. dirus Delta class GST isoform 4, adGSTD4-4 (GenBank® accession number AF273040), was derived from a previous study [24]. PCR-based site-directed mutagenesis following the method of Stratagene Quik Change™ Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, U.S.A.) was used to introduce point mutations to the amino acid 104 or 107 of the wild-type enzyme. A clone with double mutations at both positions was also generated. The entire full-length regions encoding all mutant adGSTD4-4 were verified, using BigDye™ terminator cycle sequencing ready reaction kit version 3.1, at least twice to confirm the absence of undesirable mutations elsewhere. V107M mutagenic plasmid was kindly provided by J. Piromjitpong (Institute of Molecular Biology and Genetics, Mahidol University).
Protein expression and purification
Wild-type adGSTD4-4 and mutants were expressed in Escherichia coli BL21(DE3)pLysS as previously described [24]. The temperature of 25 °C was used during protein expression under 0.1 mM isopropyl β-D-thiogalactoside induction. Cells were harvested by centrifugation at 5000 g for 10 min. The cell pellet was treated with lysozyme to a final concentration of 0.4 mg/ml and then incubated on ice for 20 min. Cell lysate was additionally disrupted by sonication, followed by cell debris separation by centrifugation at 10000 g for 20 min at 4 °C. The soluble recombinant GST proteins were purified from the supernatant by using GSTrap™ FF affinity chromatography (Amersham Biosciences), except for those incapable of binding to the GSH matrix. In these cases, cation-exchange chromatography by HiTrap™ SP XL (Amersham Biosciences) and hydrophobic-interaction chromatography by HiTrap™ phenyl-Sepharose HP (Amersham Biosciences) were employed. The details of cationic and hydrophobic chromatography have been previously reported [25]. Purified recombinant proteins were homogeneous as judged by SDS/PAGE.
Enzyme characterization and protein assay
Enzyme activity conjugating GSH to the CDNB (1-chloro-2,4-dinitrobenzene) substrate was determined by monitoring the increase in absorbance (A) at 340 nm over time using a SpectraMax® 250 spectrophotometer following a previously described method [26]. The dependence of the initial rate on the substrate concentration was analysed according to the Michaelis–Menten equation (eqn 1), where V is the initial rate of the reaction, Vmax is the maximum velocity, [S] is the concentration of substrate and Km is the substrate concentration that gives the rate of the reaction equal to one-half of Vmax:
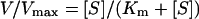 |
(1) |
The values for the kinetic parameters (Km and Vmax) were calculated by using GraphPad Prism® 4 software, version 4.01. The steady-state kinetics and substrate-specificity studies were performed as described previously [24]. Five different hydrophobic electrophilic compounds were used in a substrate-specificity study. The results shown are means±S.D. for at least three independent experiments. Protein concentration was determined by the Bradford method [27] using the Bio-Rad Laboratories (Hercules, CA, U.S.A.) protein reagent with BSA as the standard protein.
With evidence of co-operativity upon GSH binding, demonstrated by a sigmoidal curve instead of a hyperbolic curve on a Michaelis–Menten plot, a Hill equation (eqn 2) was used to fit the experimental kinetic data on the plot. K0.5 is the substrate concentration that gives the rate of reaction at one-half of Vmax, similar to the Km value for non-co-operative binding (h=1):
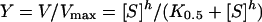 |
(2) |
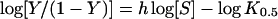 |
(3) |
A sigmoidal Hill equation was transformed into a linear rate equation (eqn 3), where Y is the fractional saturation, h is the Hill coefficient and K0.5 is an averaged binding constant at Y=0.5. A Hill plot, a plot between log[Y/(1−Y)] and log[S], was employed to determine the degree of co-operativity by the slope of the plot which yields the Hill coefficient (h) [28].
Thermal stability assay
Enzymes derived from the different engineered clones [each at a concentration of 0.1 and 1 mg/ml in 0.1 M phosphate buffer (pH 6.5) containing 5 mM dithiothreitol and 1 mM EDTA] were incubated at 45 °C. Enzymatic activity was measured as a function of time. An appropriate amount of incubated mixture (adjusted for the specific activity of each engineered enzyme) was taken to assay for residual GST activity at various time points ranging from 0 to 420 min. The log percentage of original activity was plotted versus preincubation time. Slopes from linear regression analysis using GraphPad Prism® 4 software were used for the half-life calculation [28], t1/2=−(0.693/2.3)/Slope.
CD
Far-UV CD measurements were carried out on a Jasco J715 spectropolarimeter. CD spectra of proteins at a concentration of 0.4 mg/ml in 0.1 M phosphate buffer (pH 6.5) with no additives were measured in a 0.2 cm path length quartz cell at room temperature (25 °C). Spectra were recorded from 190 to 260 nm with 1 nm steps, averaged over three scans at a scan rate of 50 nm/min and corrected by subtraction of solvent spectra under identical conditions.
Fluorescence measurements
Both intrinsic and ANS (8-anilinonaphthalene-l-sulphonic acid) fluorescence emission spectra were carried out on a Jasco model FP-6300 spectrofluorimeter in a 0.5 cm path length quartz cell. Excitation and emission bandwidths were kept at 2.5 nm. Fluorescence spectra of 2 μM enzyme in 0.1 M phosphate buffer (pH 6.5) with no additives were recorded at room temperature. All measurements were averaged over three scans at a scanning speed of 500 nm/min and corrected by subtraction of the solvent spectra under identical conditions.
Intrinsic fluorescence measurements were performed at an excitation wavelength of 295 nm and the emission spectra were recorded from 300 to 500 nm, whereas ANS fluorescence measurements were at an excitation wavelength of 395 nm and the emission spectra were recorded from 400 to 600 nm. The final concentration of 200 μM ANS was homogeneously mixed with enzyme solutions prior to the measurements.
RESULTS
Protein expression and purification
All recombinant proteins were effectively expressed at comparable levels in E. coli as soluble protein with a size of approx. 25 kDa. Generally, adGSTD4-4 was easily purified in a single chromatographic step using an immobilized GSH affinity chromatography column. It was found that the affinity for this matrix was strongly reduced for many mutants. Therefore an alternative method of cation-exchange and hydrophobic-interaction chromatography was employed. Protein purities from both techniques proved to be equivalent. All mutants were successfully purified and subjected to both enzymatic and structural characterizations.
Enzymatic properties
Enzyme kinetics
Steady-state kinetics was performed using CDNB as a co-substrate for GSH conjugation. Mutant recombinant enzymes with an amino acid substitution at Phe-104 possessed altered catalytic rates (kcat) ranging from approximately wild-type to 266-fold less than wild-type activity (Table 1). Substitutions with bulky (F104W) or polar/charged residues (F104H, F104Q, F104E and F104K) are catalytically unfavourable at position 104, as shown by a decrease of 5- to 266-fold in kcat. The binding affinity towards GSH (KmGSH) for all Phe-104 mutants was dramatically affected in the range of 4–29-fold less than the wild-type. These decreases in binding affinity were also accompanied by a change to positive co-operativity for GSH binding. For positive co-operativity, a sigmoidal curve was obtained from Michaelis–Menten plots instead of a hyperbolic curve. The degree of cooperativity as shown by the Hill coefficients illustrates that communication occurs between the active sites (Table 2). The sigmoidal-shaped curve for GSH binding signifies that the binding of one molecule of GSH in one active site increases the affinity of the protein for that substrate in the neighbouring active site of the other subunit, reflecting the role of the residue in communication between subunits to stabilize the GSH binding. In addition, the binding affinity towards CDNB substrate (KmCDNB) of the Phe-104 mutants varied slightly as well in the range of 1–3-fold, which most probably results from structural perturbation within the active-site pocket.
Table 1. Steady-state kinetic constants of engineered GSTs compared with adGSTD4-4 (wild-type).
The results shown are the means±S.D. for at least three independent experiments.
| Km (mM) | kcat/Km (mM−1·s−1) | |||||
|---|---|---|---|---|---|---|
| Enzyme | Vmax (μmol·min−1·mg−1) | kcat (s−1) | GSH | CDNB | GSH | CDNB |
| Wild-type | 44.7±2.3 | 18.60 | 0.59±0.06 | 0.73±0.07 | 31.53 | 25.49 |
| F104A | 23.3±1.7 | 9.67 | 11.99±0.20 | 2.27±0.31 | 0.85 | 4.26 |
| F104L | 49.3±1.8 | 20.59 | 3.55±0.40 | 0.69±0.15 | 6.32 | 29.85 |
| F104M | 42.1±1.9 | 17.51 | 2.21±0.11 | 0.82±0.06 | 10.81 | 21.36 |
| F104Y | 42.7±1.2 | 17.86 | 6.96±0.31 | 0.57±0.05 | 2.76 | 31.34 |
| F104W | 3.9±0.3 | 1.63 | 15.26±0.54 | 1.49±0.12 | 0.13 | 1.09 |
| F104H | 1.8±0.1 | 0.74 | 16.97±1.54 | 0.79±0.05 | 0.04 | 0.94 |
| F104Q | 9.5±0.4 | 3.97 | 15.35±1.21 | 1.32±0.11 | 0.29 | 3.00 |
| F104E | 4.6±0.3 | 1.91 | 16.54±1.34 | 1.86±0.14 | 0.12 | 1.03 |
| F104K | 0.17±0.01 | 0.07 | 6.62±0.14 | 0.85±0.13 | 0.01 | 0.08 |
| F104A/V107A | 15.3±0.6 | 6.34 | 10.06±0.58 | 2.07±0.18 | 0.63 | 3.06 |
| V107A | 50.6±1.5 | 21.04 | 0.73±0.13 | 0.56±0.02 | 28.82 | 37.57 |
| V107L | 46.9±1.7 | 19.53 | 0.40±0.03 | 0.47±0.05 | 48.83 | 41.56 |
| V107M | 49.9±0.8 | 20.89 | 0.64±0.10 | 0.79±0.04 | 32.64 | 26.44 |
| V107N | 37.2±2.2 | 15.49 | 0.45±0.09 | 0.68±0.09 | 34.43 | 22.78 |
| V107D | 13.5±0.4 | 5.62 | 1.23±0.16 | 0.51±0.03 | 6.32 | 11.02 |
| V107K | 0.29±0.01 | 0.12 | 8.88±0.19 | 0.71±0.05 | 0.02 | 0.17 |
Table 2. Hill coefficients (h) for the binding of GSH substrate.
The results shown are the means±S.D. for at least three independent experiments. ANOVA with Dunnett multiple comparisons test was applied with wild-type enzyme as control.
| Enzyme | h |
|---|---|
| Wild-type | 1.02±0.14 |
| F104A | 1.67±0.09* |
| F104L | 1.44±0.12* |
| F104M | 1.23±0.08 |
| F104Y | 1.73±0.08* |
| F104W | 1.33±0.15* |
| F104H | 2.10±0.05* |
| F104Q | 1.64±0.11* |
| F104E | 1.99±0.15* |
| F104K | 1.58±0.05* |
| F104A/V107A | 0.99±0.09 |
| V107A | 0.96±0.07 |
| V107L | 0.99±0.14 |
| V107M | 0.93±0.09 |
| V107N | 0.99±0.11 |
| V107D | 1.34±0.13* |
| V107K | 1.44±0.11* |
* Significantly different (P<0.01).
Amino acid replacements at the position Val-107 demonstrated that a charged residue substitution would impact upon the catalytic efficiency of the enzyme by reducing kcat and increasing KmGSH, in addition to inducing positive co-operativity for GSH binding. In contrast, substitutions with polar, uncharged or non-polar amino acid residues with various sized side chains show little impact on catalytic properties. Kinetic constants of the double mutant F104A/V107A also demonstrated that the disruption at the Phe-104 position has a major influence on catalysis, as shown by the kinetic properties of the single alanine mutations to either Phe-104 or Val-107. However, this double-mutation enzyme displayed a different characteristic from F104A by possessing non-co-operative binding for GSH substrate (Table 2). This demonstrates that the alanine substitution at Val-107 confers a different structural arrangement that affects the communication between subunits.
Substrate specificity
Five different hydrophobic substrates were used to investigate the changes in substrate specificities of the mutant recombinant enzymes (Figure 1). Each engineered GST displayed a different pattern of substrate specificity. The engineered GSTs with low affinity towards GSH (high KmGSH) also appeared to possess low activity towards most hydrophobic substrates tested, including CDNB. In this regard, the decrease in activity appears to result from a major disturbance to the G-site (GSH-binding site). This disturbance produces active-site atom movements as well as electrostatic field rearrangement, causing substrate orientation to be in less suitable configurations resulting in catalytic changes. Interestingly, F104L, F104M and F104Y possess comparable activity and specificity with the wild-type enzyme towards CDNB, as shown by kcat and (kcat/Km)CDNB. However, these enzymes displayed higher specific activity with DCNB (1,2-dichloro-4-nitrobenzene), lower specific activity with PNPB (p-nitrophenethyl bromide) and PNBC (p-nitrobenzyl chloride), but a variable activity with EA (ethacrynic acid) as the substrate. This evidence suggests that the replacement residues in the G-site Phe-104 position have significant consequences on the H-site (hydrophobic substrate-binding site) of the enzyme. Therefore diverse effects on the enzyme activities towards various substrates demonstrate that the different enzyme–substrate interactions that occur are affected in distinctly different ways.
Figure 1. Plots of the changes in substrate specificity for engineered GSTs relative to the wild-type enzyme.

The change in specific activity of each enzyme for each substrate is shown as fold change relative to the wild-type enzyme. The experiment was performed in triplicate and the results shown are means±S.D.; n.d., no detectable activity.
The positively charged replacement V107K performed poorly with all hydrophobic substrates tested, whereas the other mutants showed variable activity towards the different substrates. The double mutant F104A/V107A had low specificity for most of the hydrophobic substrates tested. However, the enzyme demonstrated EA activity comparable with the wild-type, although no detectable EA activity was shown by either of the alanine single-mutation enzymes. Accordingly, the properties of F104A/V107A are not additive influences of F104A and V107A, but result from a particular rearrangement of the enzyme active site that occurs upon packing changes induced by the double mutation.
Structural properties
Enzyme stability
Thermal stability was monitored to demonstrate that amino acid substitutions at either Phe-104 or Val-107, residues involved in the lock-and-key ‘Clasp’ motif, have a role in structural stability of adGSTD4-4 (Figure 2). Most of the mutants showed a concentration-dependent thermal stability in which increasing concentration showed greater stability. The engineered enzymes appeared to undergo structural changes more rapidly at low concentrations compared with the wild-type, which displayed similar stability at both concentrations used. The results suggest that residue 104 is involved in regulating structural integrity of the enzyme, while substitutions to residue 107 appeared to enhance structural maintenance as shown by an increase in half-life.
Figure 2. Thermal stability of engineered GSTs compared with the wild-type adGSTD4-4.

Half-life determinations were from at least three independent experiments at 45 °C. Enzyme concentrations for heat inactivation are 0.1 and 1 mg/ml respectively. The bar graph represents mean±S.D. (An asterisk indicates that enzyme activity was undetectable upon heat inactivation, which precluded half-life determination.)
CD
CD was used to monitor for protein secondary-structure disruption. The ellipticity spectra measured for the Phe-104 mutants were similar to the spectrum of the wild-type enzyme, with ellipticity minima at 208 and 222 nm. This is characteristic of an α- helix predominant structure and suggests no overall secondary-structure change (results not shown).
Intrinsic fluorescence
Tryptophan intrinsic fluorescence (excitation at 295 nm) was used as a tertiary-structure probe. Each adGSTD4-4 subunit possesses two tryptophan residues located at positions 64 and 191. All mutants showed roughly the same emission maximum of 339±2 nm (Figure 3). As expected, the F104W enzyme, with one additional tryptophan residue, also showed the same emission maximum (Figure 3A), demonstrating that residue 104 is buried in the interior core of the GST dimer interface. Although all substitutions in the present study had no impact on solvent exposure of the tryptophan residues, the variable fluorescent intensities from the different mutants indicate that differences occur in local quenching by the surrounding amino acids such as cysteine, histidine, glutamine and asparagine [29].
Figure 3. Intrinsic fluorescence of engineered GSTs compared with wild-type adGSTD4-4.

F104X, F104W and V107X represent spectra of replacements of (A) Phe-104 with one of the amino acids in the present study, replacement of Phe-104 with Trp, and (B) replacements of Val-107 with one of the amino acids in the present study respectively. Fluorescence intensity was measured in arbitrary units. Additional lines illustrate variation between all the engineered GSTs.
Fluorescent dye binding
ANS (extrinsic fluorophore) binding was used as a quaternary structure probe. Upon binding, ANS fluorescence was significantly enhanced and its emission maximum wavelength shifted from 515 nm to approx. 479 nm. Most Phe-104 and Val-107 mutants showed a reduction of fluorescence intensity compared with wild-type (Figure 4). Only F104W slightly increased the fluorescent signal (Figure 4C). Both quenching and enhancement of the fluorescent signals demonstrate conformational changes in the non-substrate ligand-binding site for ANS.
Figure 4. Fluorescence spectrophotometry of ANS binding.

(A) Fluorescence spectra of Phe-104 mutants. (B) Fluorescence spectra of Val-107 mutants. (C) A bar graph representation of relative fluorescence intensity compared with the wild-type enzyme. Fluorescence intensity was measured in arbitrary units. Additional lines illustrate variation between all the engineered GSTs.
DISCUSSION
The lock-and-key ‘Clasp’ motif shows some similar characteristics to previously investigated lock-and-key motifs in the GST classes Alpha [30], Mu [31] and Pi [32], for example, stabilization of the dimeric structure through intersubunit interactions. However, the ‘Clasp’ motif is situated in a different location along the dimer interface and possesses a unique feature (Figure 5). Using the completed Anopheles gambiae genome project, an African mosquito malarial vector, members of the GST supergene family have been identified as insecticide-resistant-associated enzymes to enable the monitoring of resistance status of mosquitoes [33]. Putative GST genes were confirmed at the transcriptional level and 12 out of 28 putative genes were classified as insect-specific Delta class [6]. Primary sequences of A. gambiae GSTs were aligned with available A. dirus Delta class GSTs. Equivalent amino acid residues involved in the ‘Clasp’ motif of Anopheline GSTs are shown in Table 3, as well as residues of the Australian sheep blowfly (Lucilia cuprina) GST. The amino acid residues involved in the motif are shown to be highly conserved across several insect GST classes (Table 3). Available X-ray crystal structures of six Delta class GSTs show their structural conservation in the quaternary structure. Three-dimensional superimposition of equivalent residues in each of the Delta class enzymes shows conserved residues in similar environments (Figure 6). Several of the lock residues have been previously studied by alanine scanning in adGSTD3-3, which is an alternatively spliced product derived from the same gene as adGSTD4-4 [34]. The corresponding residue to adGSTD4-4 Glu-65 (Glu-64 in adGSTD3-3) was suggested to be involved in an initial folding step of the enzyme and this residue is highly conserved among GST classes. The corresponding residue to adGSTD4-4 Arg-67 (Arg-66 in adGSTD3-3) revealed a significant impact on the ionization process of GSH, as well as structural stabilization. However, the corresponding residue to adGSTD4-4 Val-107 (Met-101 in adGSTD3-3) showed no significant functional or structural effects. Leu-103 has previously been characterized in adGSTD4-4 and shown to be important for GSH binding, with evidence for positive co-operativity in some mutants, as well as being involved in structural stabilization [35]. In an attempt to characterize the contribution of the lock-and-key ‘Clasp’ motif, the ‘key’ residue Phe-104 and the ‘lock’ residue Val-107 of adGSTD4-4 were investigated.
Figure 5. Two types of intersubunit lock-and-key motifs found in different GST classes.

(A) Lock-and-key ‘Clasp’ motif in adGSTD4-4 (PDB id: 1JLW). (B) Lock-and-key motif in human GST class Pi, GSTP1-1 (PDB id: 1GSS). Subunits are distinguished by colouring in grey and dark grey, amino acid residues involved in the ‘lock’ are shown by stick representation, whereas the ‘key’ residues, Phe-104 in adGSTD4-4 or Tyr-50 in human GSTP1-1, are shown in CPK (Corey–Pauling–Koltun) space-filling mode.
Table 3. Lock-and-key ‘Clasp’ residues in insect GST classes.
Numbering of residues is based on adGSTD4 sequence. *adGST, agGST and LcGST represent GSTs from A. dirus, A. gambiae and L. cuprina respectively. D is Delta, u is unclassified, E is Epsilon, T is Theta, S is Sigma, Z is Zeta and O is Omega GST class. GenBank® accession numbers: adGSTD1 (AF273041), adGSTD2 (AF273038), adGSTD3 (AF273039), adGSTD4 (AF273040), adGSTD5 (AF251478), adGSTD6 (AY014406), agGSTD1-3 (Protein ID AAC79992), agGSTD1-4 (Protein ID AAC79994), agGSTD1-5 (Protein ID AAC79993), agGSTD1-6 (Protein ID AAC79995), agGSTD2 (Z71480), agGSTD3 (AF513638), agGSTD4 (AF513635), agGSTD5 (AF513634), agGSTD6 (AF513636), agGSTD7 (AF071161), agGSTD8 (AF316637), agGSTD9 (AY255857), agGSTD10 (AF515527), agGSTD11 (AF513637), agGSTD12 (AF316638), agGSTu1 (AF515521), agGSTu2 (AF515523), agGSTu3 (AF515524), agGSTE1 (AF316635), agGSTE2 (AF316636), agGSTE3 (AY070234), agGSTE4 (AY070254), agGSTE5 (AY070255), agGSTE6 (AY070256), agGSTE7 (AF491816), agGSTE8 (AY070257), agGSTT1 (AF515526), agGSTT2 (AF515525), agGSTS1-1 (L07880), agGSTS1-2 (AF513639), agGSTZ1 (AF515522), agGSTO1 (AY255856) and LcGST (P42860). ‡104a and 104b indicate that the residue at this position is from different subunits. ‡agGSTD6 and agGSTD9 were suggested to be pseudogenes [6].
| Key residue | Lock residue | ||||||
|---|---|---|---|---|---|---|---|
| Enzyme* | 104a† | 65 | 67 | 68 | 103 | 104b† | 107 |
| adGSTD4 | F | E | R | A | L | F | V |
| adGSTD1 | F | E | R | A | L | F | M |
| adGSTD2 | Y | E | R | A | L | Y | M |
| adGSTD3 | Y | E | R | A | L | Y | M |
| adGSTD5 | H | E | R | V | L | H | L |
| adGSTD6 | F | E | Y | A | L | F | I |
| agGSTD1-3 | F | E | R | A | L | F | V |
| agGSTD1-4 | Y | E | R | A | L | Y | M |
| agGSTD1-5 | Y | E | R | A | M | Y | M |
| agGSTD1-6 | Y | E | R | A | L | Y | M |
| agGSTD2 | F | E | R | A | L | F | A |
| agGSTD3 | F | E | Y | A | L | F | I |
| agGSTD4 | F | E | C | A | L | F | V |
| agGSTD5 | F | E | Y | A | L | F | I |
| agGSTD6‡ | F | E | S | A | L | F | I |
| agGSTD7 | H | E | R | V | L | H | L |
| agGSTD8 | F | E | R | A | L | F | H |
| agGSTD9‡ | L | E | G | A | L | L | C |
| agGSTD10 | F | E | Y | A | L | F | N |
| agGSTD11 | Y | E | R | A | L | Y | M |
| agGSTD12 | F | E | Y | A | L | F | S |
| agGSTu1 | C | E | N | A | L | C | L |
| agGSTu2 | H | E | R | A | L | H | L |
| agGSTu3 | Y | E | K | A | L | Y | S |
| agGSTE1 | H | E | H | A | L | H | S |
| agGSTE2 | H | E | H | A | L | H | S |
| agGSTE3 | H | D | H | A | L | H | S |
| agGSTE4 | H | D | H | A | L | H | S |
| agGSTE5 | H | D | H | A | L | H | S |
| agGSTE7 | H | A | H | A | L | H | A |
| agGSTE6 | F | D | H | A | L | F | S |
| agGSTE8 | C | D | H | A | L | C | N |
| agGSTT1 | E | E | V | A | L | E | H |
| agGSTT2 | S | E | V | A | L | S | H |
| agGSTS1-1 | D | Q | L | A | V | D | N |
| agGSTS1-2 | D | Q | V | A | V | D | N |
| agGSTZ1 | E | E | V | S | C | E | A |
| agGSTO1 | E | E | L | V | I | E | A |
| LcGST | Y | E | R | A | L | Y | M |
Figure 6. Structural representations of highly conserved lock-and-key ‘Clasp’ motifs in various Delta class GSTs.

(a) A. dirus adGSTD4; (b) A. dirus adGSTD3; (c) A. dirus adGSTD5; (d) A. dirus adGSTD6; (e) L. cuprina LcGST; and (f) A. gambiae agGSTD1-6. Subunits are differentiated by grey and dark grey colours. Lock-and-key ‘Clasp’ residues are shown in CPK.
Enzymatic properties of the Phe-104 mutants demonstrated a major impact for this particular position on GSH binding affinity, as shown by increased KmGSH values for all of the Phe-104 mutants. The altered side chains at position 104 in both subunits would impact on their interface interaction as well as causing a G-site topology change through rearrangement of ‘lock’ residues, several of which are in the G-site. Replacements with hydrophobic amino acids generated enzymes with varying catalytic rates. Reduced catalytic rates were observed for both F104A and F104W, indicating that not only hydrophobic packing in the region but also the size of the replaced residue is important for efficient subunit dimerization. Structural investigation reveals that the ‘Clasp’ motif resides within the interior core of the dimer interface that connects the G-site of one subunit to the other (Figure 7A). The communication between subunits through this Clasp motif was illustrated by catalytic co-operativity for GSH binding in all Phe-104 mutants, as shown by Hill coefficients (Table 2). In this regard, the h value of 2.10±0.05 upon GSH binding for F104H is likely to be statistically similar (95% confidence interval) to the maximum number of interacting sites of 2, which demonstrates extremely strong positive co-operativity or complete co-operativity between subunits. Therefore it is reasonable to state that the ‘key’ residue Phe-104 plays a pivotal role in the dimeric structure of adGSTD4-4 in modulation of GSH binding for both subunits.
Figure 7. Interactions of amino acids in the lock-and-key ‘Clasp’ motif of adGSTD4-4.

(A) Stereo view of the lock-and-key ‘Clasp’ motif connecting two active site pockets of adGSTD4-4. The surface of the dimeric enzyme is shown in grey, GSH in each active site in yellow, the active- site amino acids in green and the Clasp residues in blue. Five of the six Clasp amino acids are also active-site residues. The top panel is looking down on to the 2-fold axis at the two active sites, which are diagonal to each other. The arrow in the top panel points to the interface groove that runs across the protein. The middle panel shows the GST rotated 90° in the direction indicated and the bottom panel shows the GST rotated a further 90° in the same direction. (B) Stereo view showing communication of GSH binding from one active site to the other through residues involved in the Clasp motif. Crystal structure of adGSTD4-4 (PDB id: 1JLW) is superimposed on adGSTD3-3 (PDB id: 1JLV) by fitting 1616 backbone atoms, with an R.M.S.D. (root mean square deviation) of 0.82 Å (1 Å=0.1 nm). Distances between centroids of aromatic ‘key’ residues are monitored from adGSTD4-4, whereas distances of GSH-interacting residues are monitored from the adGSTD3-3 enzyme complex. Amino acid labelling is for adGSTD4-4. Amino acid residues of adGSTD4-4 and adGSTD3-3 are coloured in blue and green respectively. The surface representation is of ‘lock’ residues from the subunit partner.
The co-operativity event upon GSH binding has been previously reported for several GST studies. Interestingly, co-operativity from point mutations of a human GSTP1-1 at position Gly-41 [36], Cys-47 [37] and Lys-54 [38] was suggested to be a consequence of structural perturbation of helix α2, which in turn was transmitted to Tyr-50 and passed to the neighbouring subunit through the intersubunit lock-and-key motif, where Tyr-50 is the ‘key’ residue. Likewise, a single point mutation at His-38 located in helix α2 of adGSTD4-4 also revealed positive co-operativity for GSH binding [25]. In this case, three-dimensional structure suggests that His-38 is a G-site residue in close contact with the glycine moiety of GSH. Therefore an alteration at His-38 may be transmitted to the lock-and-key ‘Clasp’ motif by the glutamyl moiety of GSH (Figure 7B). For subunit interface residues, co-operativity for GSH binding was evident in studies of Tyr-50 of human GSTP1-1 [32] as well as co-operativity for GSH conjugation of 4-hydroxynonenal through Arg-69 of murine GSTA4-4 [39] and Asp-101 of human GSTA1-1 [40]. As a whole, although the co-operativity mechanism upon substrate binding is unclear, the subunit communication is apparently triggered from one substrate binding site to the other neighbouring subunit through an existing subunit interface interaction that varies among the GST classes, and the Clasp motif seems to possess a major role in Delta class GSTs.
The Val-107 position exhibited the greatest influence on enzymatic properties when substituted with charged residues. This suggests that a hydrophobic amino acid is not necessary at this position, as shown by V107N. However, decreases in catalytic rates and affinity towards GSH with co-operativity on GSH binding of V107D and V107K suggest that these replacements might create additional electrostatic interactions to residues in close proximity, such as Glu-65 or Arg-67, that subsequently cause structural perturbation to the G-site. Particularly, the Val-107 position is of interest because almost all substitutions seemed to enhance structural stability of the enzyme, especially V107A. It is very unusual for mutants to be significantly more stable than the wild-type. The Clasp motif is composed of hydrophobic residues that can stabilize the structure through hydrophobic force as well as polar/charged residues that would maintain structural integrity by ionic interactions. The changes at the Val-107 position appear to contribute stabilizing forces between subunits and weaken conformational strain of the dimerization. V107A displayed appreciably improved stability while maintaining functionality.
There was no secondary or tertiary structural changes detected by CD or intrinsic fluorescence measurements of tryptophan. However, intensity differences of ANS binding indicate conformational alterations at the quaternary level. Blue shift of the fluorescence emission maximum of bound ANS indicates that the ANS-binding site in adGSTD4-4 is similar to hGSTA1-1 [30] and pGSTP1-1 [41] with an emission maximum of approx. 480 nm, but more hydrophobic than in squid GSTS1-1 [13], hGSTM1-1 [14] and rGSTM1-1 [31] which have emission maxima in the range 490–500 nm. In hGSTA1-1, ANS binding was suggested to be at the cleft in the dimer interface binding one molecule per subunit in a native protein [42]. Assuming that ANS binding of adGSTD4-4 is also located along the subunit interface, the present study shows that the Clasp motif influenced ANS binding. Since ANS fluorescence is quenched by water, the reduction of fluorescence intensity of protein-bound ANS in mutants illustrates greater exposure of ANS to solvent. This suggests that amino acid substitution to either Phe-104 or Val-107 alters the binding-site topology/solvent exposure for ANS.
The aromatic stacking of the ‘key’ residues suggests aromatic residues to be important for enzyme stability, as shown by half-life data for F104Y, F104W and F104H, although these replacements are not always favourable for catalytic function. The presence of aromatic residues protruding from one subunit to the other was suggested to be advantageous for increasing interface affinity, hence gluing protein subunits together [43]. However, different aromatic residues employed in diverse Delta class GSTs would generate isoform-specific subunit interfaces. Therefore substitutions with ‘key’ residues from other Delta class GSTs such as in F104Y and F104H appear to induce conformation changes that impact upon catalytic function.
Phenylalanine residues interacting across a 2-fold axis also were shown at the dimer interface of the TIM (triosephosphate isomerase) enzyme family. The interface between TIM subunits is mainly composed of loops 1–3 from both monomers. At the tip of loop 2 in trypanosomal TIM, a phenylalanine residue (Phe-45) forms a hydrophobic contact across the dimer interface to the equivalent residue on the other subunit. In Escherichia coli TIM, the residue was replaced by a glutamate residue (Glu-46) [44]. The Glu-46 also interacts with its corresponding residue on the neighbouring subunit contributing extensive van der Waals contacts with its local environment, as a result inducing conformational changes to the remaining intersubunit contacts which are different from that observed in other TIMs [44]. This implies that intersubunit residue interactions of oligomeric enzymes are to stabilize quaternary structure or dimerization. With similar features to GSTs, all TIMs are active only in the dimeric form, although each monomer has its own active site [45]. In view of an important glycolytic function, TIMs are of major interest in recent years as a potential target for therapeutic drug design, principally by disturbing some of the intersubunit contacts of the protein. For example, disruption of subunit assembly through effects on pi–pi interactions of two aromatic clusters in the interface of trypanosomal TIM [46,47] and Plasmodium falciparum TIM [48,49] was investigated for anti-trypanosomal agents and antimalarial drug design.
We have shown that an intersubunit lock-and-key ‘Clasp’ motif of adGSTD4-4 demonstrates functional and structural significance of the amino acids that interact across the dimer interface. Although residues of this motif do not provide the only interaction between subunits of adGSTD4-4, it is noteworthy that the unique characteristic of aromatic stacking in the middle of the dimer interface in fact connects the two active sites, enabling them to work in a co-operative manner. Evidence of allosteric effects suggests an important role for this particular intersubunit architecture in regulating catalytic activity through conformational transitions of subunits. Quaternary structural changes of all mutants detected by ANS dye binding suggest that subunit assembly or dimerization basically manipulates subunit communication. The observation of co-operativity in the mutants also implies that GSH ligand binding and dimerization are linked, consistent with the ANS-indicated quaternary structural changes; consequently, GSTs are catalytically active only in their dimeric forms. Correlations between oligomerization and ligand binding have also been shown for many other enzymes, such as dimerization of pyrophosphatase subunits improving its ligand binding, and vice versa with active-site adjustments upon metal cofactor or substrate binding increasing the stability of the dimer [50].
In conclusion, for GSTs, the lock-and-key motif in general, and especially the ‘Clasp’ motif with the pi–pi interaction, appear to play a pivotal role in subunit communication between active sites as well as in stabilizing the quaternary structure. Continued elucidation of the mechanism of positive co-operativity between subunits of oligomeric proteins would provide useful information for protein engineering.
Acknowledgments
This work was funded by the Thailand Research Fund. J. W. was supported by a Royal Golden Jubilee scholarship.
References
- 1.Armstrong R. N. Glutathione S-transferases: reaction mechanism, structure, and function. Chem. Res. Toxicol. 1991;4:131–140. doi: 10.1021/tx00020a001. [DOI] [PubMed] [Google Scholar]
- 2.Wilce M. C. J., Parker M. W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta. 1994;1205:1–18. doi: 10.1016/0167-4838(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong R. N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 4.Hayes J. D., Flanagan J. U., Jowsey I. R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 5.Sheehan D., Meade G., Foley V. M., Dowd C. A. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Y., Ortelli F., Rossiter L. C., Hemingway J., Ranson H. The Anopheles gambiae glutathione transferase supergene family: annotation, phylogeny and expression profiles. BMC Genomics. 2003;4:35–50. doi: 10.1186/1471-2164-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erhardt J., Dirr H. Native dimer stabilizes the subunit tertiary structure of porcine class pi glutathione S-transferase. Eur. J. Biochem. 1995;230:614–620. [PubMed] [Google Scholar]
- 8.Le Trong I., Strenkamp R. E., Ibarra C., Atkins W. M., Adman E. T. 1.3-Å resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins Struct. Funct. Genet. 2002;48:618–627. doi: 10.1002/prot.10162. [DOI] [PubMed] [Google Scholar]
- 9.Yassin Z., Ortiz-Salmerón E., García-Maroto F., Barón C., García-Fuentes L. Implications of the ligandin binding site on the binding of non-substrate ligands to Schistosoma japonicum-glutathione transferase. Biochim. Biophys. Acta. 2004;1698:227–237. doi: 10.1016/j.bbapap.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Wallace L. A., Sluis-Cremer N., Dirr H. W. Equilibrium and kinetic unfolding properties of dimeric human glutathione transferase A1-1. Biochemistry. 1998;37:5320–5328. doi: 10.1021/bi972936z. [DOI] [PubMed] [Google Scholar]
- 11.Dirr H. W., Reinemer P. Equilibrium unfolding of class Pi glutathione S-transferase. Biochem. Biophys. Res. Commun. 1991;180:294–300. doi: 10.1016/s0006-291x(05)81291-4. [DOI] [PubMed] [Google Scholar]
- 12.Andújar-Sánchez M., Clemente-Jiménez J. M., Rodríguez-Vico F., Las Heras-Vazquez F. J., Jara-Pérez V., Cámara-Artigas A. A monomer form of the glutathione S-transferase Y7F mutant from Schistosoma japonicum at acidic pH. Biochem. Biophys. Res. Commun. 2004;314:6–10. doi: 10.1016/j.bbrc.2003.12.067. [DOI] [PubMed] [Google Scholar]
- 13.Stevens J. M., Hornby J. A. T., Armstrong R. N., Dirr H. W. Class sigma glutathione transferase unfolds via a dimeric and a monomeric intermediate: impact of subunit interface on conformational stability in the superfamily. Biochemistry. 1998;37:15534–15541. doi: 10.1021/bi981044b. [DOI] [PubMed] [Google Scholar]
- 14.Hornby J. A. T., Luo J.-K., Stevens J. M., Wallace L. A., Kaplan W., Armstrong R. N., Dirr H. W. Equilibrium folding of dimeric class Mu glutathione transferases involves a stable monomeric intermediate. Biochemistry. 2000;39:12336–12344. doi: 10.1021/bi000176d. [DOI] [PubMed] [Google Scholar]
- 15.Dirr H. Folding and assembly of glutathione transferases. Chem. Biol. Interact. 2001;133:19–23. [Google Scholar]
- 16.Reinemer P., Dirr H. W., Ladenstein R., Schäffer J., Gallay O., Huber R. The three-dimensional structure of class Pi glutathione S-transferase in complex with glutathione sulfonate at 2.3 Å resolution. EMBO J. 1991;10:1997–2005. doi: 10.1002/j.1460-2075.1991.tb07729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji X., Zhang P., Armstrong R. N., Gilliland G. L. The three-dimensional structure of a glutathione S-transferase from the Mu gene class. Structural analysis of the binary complex of isoenzyme 3-3 and glutathione at 2.2-Å resolution. Biochemistry. 1992;31:10169–10184. doi: 10.1021/bi00157a004. [DOI] [PubMed] [Google Scholar]
- 18.Reinemer P., Dirr H. W., Ladenstein R., Huber R., Lo Bello M., Federici G., Parker M. W. Three-dimensional structure of class Pi glutathione S-transferase from human placenta in complex with S-hexylglutathione at 2.8 Å resolution. J. Mol. Biol. 1992;227:214–226. doi: 10.1016/0022-2836(92)90692-d. [DOI] [PubMed] [Google Scholar]
- 19.Sinning I., Kleywegt G. J., Cowan S. W., Reinemer P., Dirr H. W., Huber R., Gilliland G. L., Armstrong R. N., Ji X., Board P. G., et al. Structure determination and refinement of human Alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J. Mol. Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 20.Ji X., Von Rosenvinge E. C., Johnson W. W., Tomarev S. I., Paitigorsky J., Armstrong R. N., Gilliland G. L. Three-dimensional structure, catalytic properties, and evolution of a sigma class glutathione transferase from squid, a progenitor of the lens S-crystallins of cephalopods. Biochemistry. 1995;34:5317–5328. doi: 10.1021/bi00016a003. [DOI] [PubMed] [Google Scholar]
- 21.Rossjohn J., McKinstry W. J., Oakley A. J., Verger D., Flanagan J., Chelvanayagam G., Tan K.-L., Board P. G., Parker M. W. Human theta class glutathione transferase: the crystal structure reveals a sulfate-binding pocket within a buried active site. Structure. 1998;6:309–322. doi: 10.1016/s0969-2126(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 22.Oakley A. J., Harnnoi T., Udomsinprasert R., Jirajaroenrat K., Ketterman A. J., Wilce M. C. J. The crystal structures of glutathione S-transferases isozymes 1-3 and 1-4 from Anopheles dirus species B. Protein Sci. 2001;10:2176–2185. doi: 10.1110/ps.ps.21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chelvanayagam G., Parker M. W., Board P. G. Fly fishing for GSTs: a unified nomenclature for mammalian and insect glutathione transferases. Chem. Biol. Interact. 2001;133:256–260. [Google Scholar]
- 24.Jirajaroenrat K., Pongjaroenkit S., Krittanai C., Prapanthadara L., Ketterman A. J. Heterologous expression and characterization of alternatively spliced glutathione S-transferases from a single Anopheles gene. Insect Biochem. Mol. Biol. 2001;31:867–875. doi: 10.1016/s0965-1748(01)00032-7. [DOI] [PubMed] [Google Scholar]
- 25.Vararattanavech A., Ketterman A. Multiple roles of glutathione binding-site residues of glutathione S-transferase. Protein Pept. Lett. 2003;10:441–448. doi: 10.2174/0929866033478654. [DOI] [PubMed] [Google Scholar]
- 26.Habig W. H., Pabst M. J., Jakoby W. B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 27.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 28.Segel I. H. New York: John Wiley & Sons; 1993. Enzyme Kinetics. Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. [Google Scholar]
- 29.Gore M. G. Oxford: Oxford University Press; 2000. Spectrophotometry and Spectrofluorimetry. [Google Scholar]
- 30.Sayed Y., Wallace L. A., Dirr H. W. The hydrophobic lock-and-key intersubunit motif of glutathione transferase A1-1: implications for catalysis, ligandin function and stability. FEBS Lett. 2000;465:169–172. doi: 10.1016/s0014-5793(99)01747-0. [DOI] [PubMed] [Google Scholar]
- 31.Hornby J. A. T., Codreanu S. G., Armstrong R. N., Dirr H. W. Molecular recognition at the dimer interface of a class Mu glutathione transferase: role of a hydrophobic interaction motif in dimer stability and protein function. Biochemistry. 2002;41:14238–14247. doi: 10.1021/bi020548d. [DOI] [PubMed] [Google Scholar]
- 32.Stenberg G., Abdalla A.-M., Mannervik B. Tyrosine 50 at the subunit interface of dimeric human glutathione transferase P1-1 is a structural key residue for modulating protein stability and catalytic function. Biochem. Biophys. Res. Commun. 2000;271:59–63. doi: 10.1006/bbrc.2000.2579. [DOI] [PubMed] [Google Scholar]
- 33.Ranson H., Claudianos C., Ortelli F., Abgrall C., Hemingway J., Sharakhova M. V., Unger M., Collins F. H., Feyereisen R. Evolution of supergene families associated with insecticide resistance. Science. 2002;298:179–181. doi: 10.1126/science.1076781. [DOI] [PubMed] [Google Scholar]
- 34.Winayanuwattikun P., Ketterman A. J. Catalytic and structural contributions for glutathione binding residues in a delta class glutathione S-transferase. Biochem. J. 2004;382:751–757. doi: 10.1042/BJ20040697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wongsantichon J., Harnnoi T., Ketterman A. J. A sensitive core region in the structure of glutathione S-transferases. Biochem. J. 2003;373:759–765. doi: 10.1042/BJ20030394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo Bello M., Nuccetelli M., Chiessi E., Lahm A., Mazzetti A. P., Parker M. W., Tramontano A., Federici G., Ricci G. Mutations of Gly to Ala in human glutathione transferase P1-1 affect helix 2 (G-Site) and induce positive cooperativity in the binding of glutathione. J. Mol. Biol. 1998;284:1717–1725. doi: 10.1006/jmbi.1998.2270. [DOI] [PubMed] [Google Scholar]
- 37.Ricci G., Lo Bello M., Caccuri A. M., Pastore A., Nuccetelli M., Parker M. W., Federici G. Site-directed mutagenesis of human glutathione transferase P1-1. Mutation of Cys-47 induces a positive cooperativity in glutathione transferase P1-1. J. Biol. Chem. 1995;270:1243–1248. doi: 10.1074/jbc.270.3.1243. [DOI] [PubMed] [Google Scholar]
- 38.Lo Bello M., Battistoni A., Mazzetti A. P., Board P. G., Muramatsu M., Federici G., Ricci G. Site-directed mutatgenesis of human glutathione transferase P1-1: spectral, kinetic, and structural properties of Cys-47 and Lys-54 mutants. J. Biol. Chem. 1995;270:1249–1253. [PubMed] [Google Scholar]
- 39.Xiao B., Singh S. P., Nanduri B., Awasthi Y. C., Zimniak P., Ji X. Crystal structure of a murine glutathione S-transferase in complex with a glutathione conjugate of 4-hydroxynon-2-enal in one subunit and glutathione in the other: evidence of signaling across the dimer interface. Biochemistry. 1999;38:11887–11894. doi: 10.1021/bi990468i. [DOI] [PubMed] [Google Scholar]
- 40.Lien S., Gustafsson A., Andersson A.-K., Mannervik B. Human glutathione transferase A1-1 demonstrates both half-of-the-sites and all-of-the-sites reactivity. J. Biol. Chem. 2001;276:35599–35605. doi: 10.1074/jbc.M103789200. [DOI] [PubMed] [Google Scholar]
- 41.Bico P., Erhardt J., Kaplan W., Dirr H. Porcine class Pi glutathione S-transferase: anionic ligand binding and conformational analysis. Biochim. Biophys. Acta. 1995;1247:225–230. doi: 10.1016/0167-4838(94)00236-a. [DOI] [PubMed] [Google Scholar]
- 42.Sayed Y., Hornby J. A. T., Lopez M., Dirr H. Thermodynamics of the ligandin function of human class Alpha glutathione transferase A1-1: energetics of organic anion ligand binding. Biochem. J. 2002;363:341–346. doi: 10.1042/0264-6021:3630341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones S., Thornton J. M. Protein-protein interactions: a review of protein dimer structure. Prog. Biophys. Mol. Biol. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 44.Noble M. E. M., Zeelen J. Ph., Wierenga R. K., Mainfroid V., Goraj K., Gohimont A.-C., Martial J. A. Structure of triosephosphate isomerase from Escherichia coli determined at 2.6 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 1993;49:403–417. doi: 10.1107/S0907444993002628. [DOI] [PubMed] [Google Scholar]
- 45.Waley S. G. Refolding of triosephosphate isomerase. Biochem. J. 1973;135:165–172. doi: 10.1042/bj1350165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Téllez-Valencia A., Olivares-Illana V., Hernández-Santoyo A., Pérez-Montfort R., Costas M., Rodríguez-Romero A., López-Calahorra F., de Gómez-Puyou M. T., Gómez-Puyou A. Inactivation of triosephosphate isomerase from Trypanosoma cruzi by an agent that perturbs its dimer interface. J. Mol. Biol. 2004;341:1355–1365. doi: 10.1016/j.jmb.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 47.Espinoza-Fonseca L. M., Trujillo-Ferrara J. G. Structural considerations for the rational design of selective anti-trypanosomal agents: the role of the aromatic clusters at the interface of triosephosphate isomerase dimer. Biochem. Biophys. Res. Commun. 2005;328:922–928. doi: 10.1016/j.bbrc.2005.01.043. [DOI] [PubMed] [Google Scholar]
- 48.Singh S. K., Maithal K., Balaram H., Balaram P. Synthetic peptides as inactivators of multimeric enzymes: inhibition of Plasmodium falciparum triosephosphate isomerase by interface peptides. FEBS Lett. 2001;501:19–23. doi: 10.1016/s0014-5793(01)02606-0. [DOI] [PubMed] [Google Scholar]
- 49.Maithal K., Ravindra G., Nagaraj G., Singh S. K., Balaram H., Balaram P. Subunit interface mutation disrupting an aromatic cluster in Plasmodium falciparum triosephosphate isomerase: effect on dimer stability. Protein Eng. 2002;15:575–584. doi: 10.1093/protein/15.7.575. [DOI] [PubMed] [Google Scholar]
- 50.Salminen A., Parfenyev A. N., Salli K., Efimova I. S., Magretova N. N., Goldman A., Baykov A. A., Lahti R. Modulation of dimer stability in yeast pyrophosphatase by mutations at the subunit interface and ligand binding to the active site. J. Biol. Chem. 2002;277:15465–15471. doi: 10.1074/jbc.M200101200. [DOI] [PubMed] [Google Scholar]