

Short abstract
We tested the hypothesis that BRCA1 may play a role in the regulation of ovarian tumor cell death as well as the inhibition of ovarian cell proliferation. Introduction of BRCA1 antisense retroviral constructs into BG-1 estrogen-dependent ovarian adenocarcinoma cells resulted in reduced BRCA1 expression. BRCA1 antisense pooled populations and derived subclones were able to proliferate in monolayer culture without estrogen, whereas control cells began to die after 10 days of estrogen deprivation. In addition, both populations and subclones of BRCA1 antisense infected cells demonstrated a growth advantage in monolayer culture in the presence of estrogen and were able to proliferate in monolayer culture without estrogen, while control cells did not. Furthermore, clonal studies demonstrated that reduced levels of BRCA1 protein correlated with growth in soft agar and greater tumor formation in nude mice in the absence of estrogen. These data suggest that reduction of BRCA1 protein in BG-1 ovarian adenocarcinoma cells may have an effect on cell survival during estrogen deprivation both in vitro and in vivo.
Keywords: antisense, BRCA1, cell death, estrogen, ovarian cancer, proliferation
Abstract
Introduction:
Germline mutations in the breast and ovarian cancer susceptibility gene BRCA1, which is located on chromosome 17q21, are associated with a predisposition to the development of cancer in these organs [1,2]. No mutations in the BRCA1 gene have been detected in sporadic breast cancer cases, but mutations have been detected in sporadic cases of ovarian cancer [3,4]. Although there is debate regarding the level of cancer risk associated with mutations in BRCA1 and the significance of the lack of mutations in sporadic tumors, it is possible that alterations in the function of BRCA1 may occur by mechanisms other than mutation, leading to an underestimation of risk when it is calculated solely on the basis of mutational analysis. Such alterations cannot be identified until the function and regulation of BRCA1 are better understood.
The BRCA1 gene encodes a 220-kDa nuclear phosphoprotein that is regulated in response to DNA damaging agents [5,6,7] and in response to estrogen-induced growth [8,9,10,11]. Germline mutations that cause breast and ovarian cancer predisposition frequently result in truncated and presumably inactive BRCA1 protein [12].
BG-1 cells were derived from a patient with stage III, poorly differentiated ovarian adenocarcinoma [13]. This cell line, which expresses wild-type BRCA1, is estrogen responsive and withdrawal of estrogen results in eventual cell death. Previous studies suggest that BRCA1 is stimulated as a result of estrogen treatment [8,9,10,11], and also that BRCA1 may be involved in the cell death process [14]. Therefore, we examined the effect of reduction of BRCA1 levels in BG-1 cells on the cellular response to hormone depletion as well as estrogen stimulation. The results suggest that reduced levels of BRCA1 correlates with a survival advantage when BG-1 cells are placed under growth-restrictive and hormone-depleted conditions. In optimum growth conditions, significantly reduced levels of BRCA1 correlates with enhanced growth both in vitro and in vivo.
Aims:
To test the hypothesis that BRCA1 may play a role in the regulation of ovarian tumor cell death as well as in the inhibition of ovarian cell proliferation.
Materials and methods:
The estrogen receptor-positive, BG-1 cell line [13], which contains an abundant amount of estrogen receptors (600 fmoles/100 μg DNA), was infected using a pLXSN retroviral vector (provided by AD Miller) containing an inverted partial human cDNA 900-base-pair sequence of BRCA1 (from nucleotide 121 in exon 1 to nucleotide 1025 in exon 11, accession #U14680). After 2 weeks of selection in 800 μg/ml of geneticin-G418 (Gibco/Life Technologies, Gaithersburg, MD, USA), BG-1 G418-resistant colonies were pooled, or individually isolated, and assayed for growth in the presence or absence of supplemented estrogen. Virally infected pooled populations of BG-1 cells were examined for BRCA1 message levels by ribonuclease protection assay (Fig. 1a). BRCA1 ribonuclease protection probe was made using an in vitro transcription kit (Ambion, Inc, Austin, TX, USA) as previously described [10] and derived clones were tested for protein levels by Western blot analysis using an anti-BRCA1 (Oncogene Research, Ab-1, Cambridge, MA, USA) antibody. Growth curve analysis of Infected populations and were pretreated for 5 days in phenol red-free, Dulbecco's modified eagle medium (DMEM)/F-12 medium (Gibco/Life Technologies) supplemented with 10% charcoal/dextran treated serum (Hyclone, Logan, UT, USA), then plated at 2.5 × 106 cells per 100mm dish in triplicate in the absence or presence of estrogen (10-8 mol/l; 17β-Estradiol; 1,3,5 (10) - Estratriene 3,17β-diol; Sigma, St Louis, MO, USA). For soft agar assay, clones were plated into 10 60-mm dishes at 1 × 105 cells/dish containing 0.3% bactopeptone agar with or without added estrogen (10-8 mol/l) in phenol red-free medium with 10% stripped serum in order to test for anchorage independent growth. BG-1 infected clones were tested for tumorigenicity by injection of cells (106 cells in 0.1cm2 50% matrigel; Collaborative Biomedical Products, Bedford, MA, USA) into subcutaneous sites in 6-week-old athymic Ncr-nude mice (NCI Animal Program, Bethesda, MD, USA) that were ovariectomized at approximately 4 weeks of age. Half of the ovariectomized mice received an implanted 0.18mg estrogen 60-day pellet (Innovative Research of America, Sarasota, FL, USA).
Results:
Antisense technology was effective in decreasing both RNA and protein levels of BRCA1 in the BG-1 human ovarian adenocarcinoma cells. BRCA1 antisense-infected populations contained significantly less BRCA1 message than control LXSN-infected pools and selected clones contained varying reduced levels of BRCA1 protein compared with control clones (Figs 1a and 1b).
Three independent BRCA1 antisense-infected cultures demonstrated a resistance to cell death induced by withdrawal from estrogen over a 6- to 20-day period (Fig. 2a). The BRCA1 antisense population also exhibited a threefold to sixfold increase in cell growth compared with control cells in the presence of estrogen treatment. BG-1 BRCA1 antisense clones demonstrated a similar response to pooled population studies, enhanced growth with estrogen, and failure to die upon estrogen depletion (Fig. 2b).
The BRCA1 antisense clones were further examined for other associated tumorigenic properties. All of the antisense clones were able to form colonies in soft agar (2-23 colonies per 104 cells plated; data not shown), whereas control clones were deficient in their ability to form colonies (0-0.8 colonies per 104 cells plated). Table 1 shows, in the presence of estrogen, the clone with the lowest levels of BRCA1 (AS-4) produced significantly more colonies (133 ± 17.9 colonies per 104 cells plated) than the control clone (NEO; 6 ± 3.1 colonies per 104 cells plated). Clones AS-4 and NEO were also injected with matrigel subcutaneously into ovariectomized athymic mice. Almost twice as many sites were positive for the AS-4 clone (14 out of 14) as for the NEO clone (eight out of 14) 42 days after injection. In addition, BRCA1 antisense tumors averaged twice the size of control tumors. The BRCA1 reduced cells also formed tumors with half the latency of control cells in the presence of implanted estrogen (11 days versus 21 days until tumor formation).
Discussion:
The present studies show that reduction in BRCA1 levels, using an antisense retroviral vector in the estrogen dependent BG-1 ovarian carcinoma cell line, contributes to confirmation of the hypothesis that BRCA1 plays a pivotal role in the balance between cell death and cell proliferation. BRCA1 RNA and protein levels were successfully reduced in populations and isolated clones of antisense infected BG-1 cells. Decreased BRCA1 levels rescued the BG-1 cells from growth arrest or cell death in adverse growth conditions in monolayer or soft agar conditions. Furthermore, a BRCA1 antisense clone that had significantly low levels of BRCA1 protein was able to form twice as many tumors in ovariectomized nude mice with a decreased latency compared with a control clone.
In multicellular mammalian organisms, a balance between cell proliferation and cell death is extremely important for the maintenance of normal healthy tissues. In support of this hypothesis, it has been shown that p53 and BRCA1 can form stable complexes, and can coactivate p21 and bax genes, which may lead to the activation of the apoptosis pathway [15]. The present data, which show that cells with a reduction of BRCA1 have a survival advantage in conditions where control cells fail to thrive, also supports this hypothesis. BRCA1 levels appear to affect the ability of cells to arrest growth or die in the absence of estrogenic growth-inducing conditions. Although mutations in this gene are uncommon in sporadic breast and ovarian tumors, BRCA1 expression levels and protein levels have been found to be reduced in sporadic human breast carcinomas [16,17,18,19]. In addition it has been demonstrated [20] that hormone-dependent tumors such as breast and ovarian cancers have a decreased ability to undergo apoptosis. Other mechanisms involving gene regulation may allow for decreased expression of BRCA1 in sporadic tumors. The response of BRCA1 mRNA and protein levels to mitogens and hormones in vitro suggests that BRCA1 may play a role in regulation of cell growth or maintenance [21]. The BRCA1 gene product may be involved in the regulation of hormone response pathways, and the present results demonstrate that loss of BRCA1 may result in loss of inhibitory control of these mitogenic pathways. These studies show that reduction in BRCA1 mRNA and protein can result in increased proliferation of BG-1 ovarian cancer cells in both in vitro and in vivo conditions, suggesting that BRCA1 may normally be acting as a growth inhibitor. Low BRCA1 levels found in sporadic cancers may be an important factor in tumorigenesis. The present data suggest that diminished levels of BRCA1 not only accelerate proliferation in the BG-1 ovarian carcinoma cell line, but also appear to promote tumorigenesis. We propose that the loss or reduction of BRCA1 may predispose a cell population to neoplastic transformation by altering the balance between cell death and proliferation/survival, rendering it more sensitive to secondary genetic changes.
Introduction
Germline mutations in the breast and ovarian cancer susceptibility gene BRCA1, which is located on chromosome 17q21, are associated with a predisposition to the development of cancer in these organs [1,2]. Initial analyses [22] suggested that women with germline mutations in the BRCA1 gene and a strong family history of breast or ovarian cancer have 85 and 44% lifetime risks of developing breast and ovarian cancer, respectively. Recent studies [23], however, have suggested that analyses based on women who were not selected for a familial history of cancer indicate that the risk for cancer associated with mutations in these genes is 50 and 16% for breast and ovarian cancers, respectively. No mutations in the BRCA1 gene have been detected in sporadic breast cancer cases; however, mutations have been detected in sporadic cases of ovarian cancer [3,4]. Although there is debate regarding the level of cancer risk associated with mutations in BRCA1 and the significance of the lack of mutations in sporadic tumors, it is possible that alterations in the function of BRCA1 may occur by mechanisms other than mutation. This would lead to an underestimation of risk when it is calculated solely on the basis of mutational analysis. Such alterations cannot be identified until the function and regulation of BRCA1 are better understood.
The BRCA1 gene encodes a 220-kDa nuclear protein that may be regulated by phosphorylation through the cell cycle and in response to DNA damaging agents [5,6,7]. The level of BRCA1 is also regulated in response to estrogen or estrogen-induced growth in breast [8,9,10,11] and ovarian cell lines. BRCA1 has been shown to colocalize in nuclear dots with other cellular proteins, including BARD-1 [24], Rad51, PCNA, and BRCA2 [7,25]. In addition, BRCA1 can act as a transcriptional transactivator in yeast reporter assays [26,27] and binds the RNA polymerase II holoenzyme, a component of the basal transcription machinery [25]. The precise mechanism of action and the specific signaling pathway affected by BRCA1 remain unknown, however.
Studies of BRCA1 expression patterns in mouse tissue reveal that BRCA1 is most highly expressed in tissues undergoing rapid proliferation and differentiation, and that expression in vivo is also hormone responsive. For example, analyses of mammary gland growth and development show high levels of BRCA1 expression in terminal end buds during puberty and in budding alveoli during pregnancy. In addition, hormonal stimulation in ovariectomized mice results in induction of BRCA1 expression in the breast [28]. Attempts to develop homozygous, BRCA1-deleted mouse models have resulted in embryonic lethality [29,30]. For example, when the BRCA1 gene deletion was targeted in exons 5 and 6, mutant mice died before day 7.5 of embryogenesis. Analysis of DNA synthesis in the mutant embryos indicated that cell proliferation was impaired, suggesting that BRCA1 may paradoxically play a positive role in the regulation of embryonic cell growth [29].
Most of the mechanistic BRCA1 studies to date have been conducted in breast carcinoma cell lines; therefore, we decided to conduct a study to determine the effect of BRCA1 expression on the cellular phenotype of an ovarian carcinoma cell line, BG-1. BG-1 cells were derived from a patient with stage III, poorly differentiated ovarian adenocarcinoma [13]. This cell line, which expresses wild-type BRCA1, is estrogen responsive, and withdrawal of estrogen results in eventual cell death. Previous studies suggested that BRCA1 is stimulated as a result of estrogen treatment [8,9,10,11], and that BRCA1 may be involved in the cell death process [14]. Therefore, we examined the effect of reduction of BRCA1 levels in BG-1 cells on the cellular response to estrogen stimulation as well as hormone depletion. Our results suggest that when BG-1 cells are subjected to growth restrictive and hormone-depleted conditions, cells that have even moderately reduced levels of BRCA1 protein have a distinct advantage for survival. In addition, significant reduction in BRCA1 protein level correlates with enhanced estrogen proliferation when compared with cells that express moderate to wild-type BRCA1 levels, grown under optimal growth conditions both in vitro and in vivo.
Materials and methods
Cells and cell culture
The estrogen receptor-positive, BG-1 line [13], which contains an abundant amount of estrogen receptors (600 fmol/100 μg DNA), was provided by J Boyd (Sloan-Kettering Cancer Center, New York, NY, USA). GPE86 and PA317 viral packaging cell lines were provided by AD Miller (Fred Hutchinson Cancer Center, Seattle, WA, USA). BG-1 cells were maintained in Dulbecco's modified eagle medium (DMEM)/F12 medium supplemented with 10% fetal calf serum (Summit, Fort Collins, CO, USA), and 50 units/ml penicillin/streptomycin. BG-1 cells arrest to gamma radiation consistent with a wildtype p53 phenotype. These cells were tested negative for mycoplasmas.
Retroviral vector preparation and infection of cells
A partial human cDNA sequence of BRCA1 (from nucleotide 121 in exon 1 to nucleotide 1025 in exon 11, accession #U14680) was inserted in the antisense orientation into the EcoR1 site of the pLXSN retroviral vector (provided by AD Miller). The vector alone, or the antisense BRCA1 vector, was transfected using the calcium-phosphate precipitation method into the ecotropic packaging cell line GPE86 [31]. Supernatant, generated from transfected GPE86 cells [31], was then used to infect the amphotropic packaging cell line PA317 [31] in the presence of 4 μg/ml polybrene (Abbott Laboratories, Abbott Park, IL, USA). PA317-infected cells were grown in selection media for 2 weeks and pooled for supernatant collection. Supernatants were filtered (0.20 μm filter) and tested for virus-producing cells. Titer efficiencies of the LXSN virus ranged from 104 to 105 colony-forming units/ml on mouse cells (A9). Log phase BG-1 cells were exposed to supernatant containing either the LXSN control retrovirus or retroviruses containing the antisense BRCA1 cDNA sequence. After 2 weeks of selection in 800 μg/ml of geneticin-G418 (Gibco/Life Technologies, Gaithersburg, MD, USA), BG-1 G418-resistant colonies were pooled or individually isolated and assayed for growth in the presence or absence of supplemented estrogen. Only isolated clones were used in anchorage dependence and tumorigenicity studies.
BRCA1 ribonuclease protection assay and protein analysis
A BRCA1 ribonuclease protection probe was made using an In Vitro Transcription Kit (Ambion, Inc, Austin, TX, USA). The DNA template spanned part of exon 22, all of exon 23, and part of exon 24 of the BRCA1 gene. Template DNA was incubated for 45min at 37°C with (α-32P)-uridine triphosphate and T7 polymerase in the presence of buffer and nucleotides. DNA template was removed by ribonuclease-free deoxyribonuclease incubation at 37°C for 30min. The reaction was stopped by the addition of 0.5 mol/l ethylenediaminetetra-acetic acid, and the labeled probe was purified on a 5% polyacrylamide gel. Sample RNA (20μg total RNA) was coprecipitated with the BRCA1 probe and the cyclophilin control probe [32], resuspended in Hyb-speed RPA (Ambion) hybridization buffer at 95°C, and then incubated at 68°C for 10min. Ribonuclease was added and the sample was incubated for 45min at 37°C. Protected fragments were precipitated, and resuspended in loading buffer, followed by separation on a 5% polyacrylamide-urea gel, and exposed to X-ray film.
BRCA1 protein was analyzed by Western blot analysis. Whole cell lysate (50 μg) was loaded onto a 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, transferred to nitrocellulose, and hybridized with an anti-BRCA1 (Ab-1; Oncogene Research, Cambridge, MA, USA) antibody as previously described [10].
Estrogen treatment and growth curve analysis
G418-resistant colonies from BRCA1 antisense infected BG-1 cells and control vector LXSN-infected BG-1 cells were pooled, pretreated for 5 days in phenol red-free, DMEM/F-12 medium (Gibco/Life Technologies) supplemented with 10% charcoal/dextran treated serum (Hyclone, Logan, UT, USA), then plated at 2.5 × 106 cells per 100mm dish in triplicate for growth curve analysis in the absence or presence of estrogen (10-8mol/l; 17β-Estradiol; 1,3,5 (10) - Estratriene 3, 17β-diol; Sigma, St Louis, MO, USA). Extended growth curve analysis was plated at 2.5 × 106 in 100mm dishes for an extended treatment of 20 days without estrogen. Clones were isolated from the BRCA1 antisense and LXSN BG-1 pooled populations and grown in phenol red-free, DMEM/F-12 medium (Gibco/Life Technologies) supplemented with charcoal/dextran treated serum (Hyclone) for 5 days before plating for growth curve analysis. Cells were then plated in triplicate at 1 × 106 cells per 60mm dish in either the absence or presence of estrogen (10-8 mol/l) and grown for 8-10 days. Cell number was calculated on indicated days using a Coulter counter. The number of dead cells for the extended growth curve experiment was calculated by counting trypan blue incorporated cells using a hemocytometer. Statistical analyses of P values were calculated based on the fold differences between growth of the clones by computing a mean ratio and the corresponding standard deviation [33].
Anchorage independence analysis and tumorigenicity
Selected BG-1 clones were tested for anchorage independent growth in 0.3% bacto-peptone agar with a 0.6% bacto-peptone agar base plus or minus added estrogen (10-8 mol/l) in phenol red-free medium with 10% stripped serum. Each BG-1, neomycin-resistant clone was plated into 10 60-mm dishes containing the agar at 1 × 105 cells per dish. Colonies (greater than 30 cells) were scored after 14 days. Pairwise comparisons were made by a two-sided Mann-Whitney U test to calculate P values [34].
BG-1-infected clones were tested for tumorigenicity in 6-week-old athymic Ncr-nude mice (NCI Animal Program, Bethesda, MD, USA) that were ovariectomized at approximately 4 weeks of age. BRCA1 antisense clone (AS-4) was injected (106 cells in 0.1cm2 50% matrigel; Collaborative Biomedical Products, Bedford, MA, USA) into two subcutaneous sites on one side of 16 mice (32 injection sites), whereas a LXSN control clone was injected on the opposite side of the same mice. Nine of the 16 ovariectomized mice also received an implanted 0.18mg estrogen 60-day pellet (Innovative Research of America, Sarasota, FL, USA). Mice were periodically examined and tumor size was measured during the 3-month period after injection.
Results
Effective decrease in BRCA1 expression using antisense technology
Antisense technology was effective in decreasing both RNA and protein levels of BRCA1 in the BG-1 human ovarian adenocarcinoma cells. BG-1 human ovarian adenocarcinoma cells were infected with a retroviral construct composed of an antisense 900 base-pair cDNA sequence of the amino-terminal region of BRCA1. Three experiments (two of which were from independently made supernatants) showed that infection of pLXSN (vector alone) and BRCA1 antisense retroviral constructs into BG-1 cells yielded G418-resistant colonies at similar rates (titers ranged from 0.78 to 4.2 × 104 colony-forming units/ml). The same vectors were also directly transfected into BG-1 cells at an efficiency of 6.3×10-5 for the anti-sense BRCA1 or 9.9 × 10-5 for the control plasmid (pLXSN). Neomycin-resistant colonies were pooled and examined for BRCA1 message levels by ribonuclease protection assay. BRCA1 antisense infected cells contained significantly less BRCA1 message than control LXSN infected cells, whether cultured in the presence or absence of estrogen (Fig. 1a). Although there appears to be no detectable amounts of BRCA1 RNA present after estrogen withdrawal, low levels of protein can be detected by western blot analysis [10].
Figure 1.
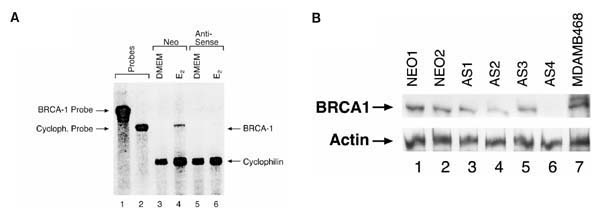
Expression of BRCA1 is reduced in BG-1 cells following infection with antisense BRCA1. (A) Ribonuclease protection analysis of BRCA1 mRNA. Lanes 1 and 2 show undigested probe for BRCA1 and loading control, cyclophilin. Lanes 3 and 4 are pooled BG-1 neo-infected control cells grown minus [Dulbecco's modified eagle medium (DMEM)] and plus (E2) 10-8 mol/l estrogen 24 h. Lanes 5 and 6 are pooled BG-1 BRCA1 antisense-infected cells minus and plus 10-8 mol/l estrogen, respectively, for 24 h. The observed doublet is the result of incomplete digestion. (B) Western blot analysis of individual control (NEO) and BRCA1 antisense (AS) clones. MDAMB468 is a BRCA1-positive breast cancer cell line.
Subclones were also isolated from BRCA1 antisense infected cells, or LXSN infected cells (NEO). Western blot analysis demonstrated that all of the antisense BRCA1 clones had reduced levels of BRCA1 protein compared with the NEO clones, and one antisense clone (AS-4) had very low levels of BRCA1 protein, although it was not totally absent (Fig. 1b).
Effects of reduced BRCA1 expression on in vitro growth
Pooled populations of antisense BRCA1 BG-1 colonies were examined for growth in the absence or presence of supplemented estrogen. Three independently infected cultures of BRCA1 antisense cells demonstrated a resistance to cell death induced by withdrawal from estrogen over a 6-day period, as well as a threefold to sixfold increase in cell growth compared with control cells in the presence of estrogen treatment (Fig. 3). In order to investigate further whether reduction of BRCA1 protein had an effect on hormone-dependent cell growth, BG-1 antisense and control cells were grown in estrogen-deprived conditions for an extended period of time. During the first 5 days, both groups continued to proliferate in the absence of estrogen, but the BRCA1 antisense group continued to grow for the next 10 days, whereas control cells decreased in number (Fig. 2a).
Figure 3.
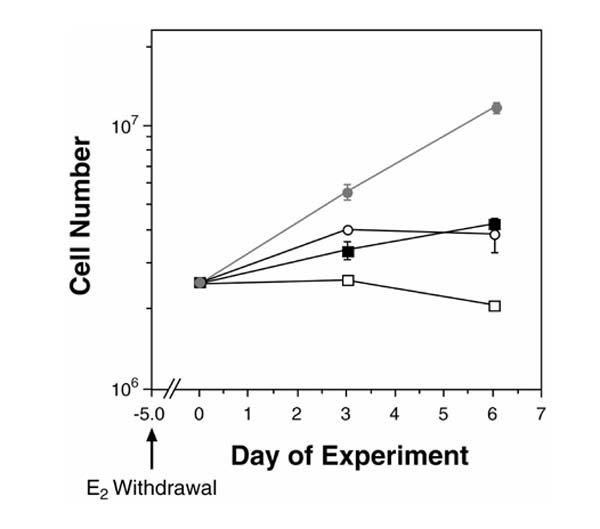
The growth curve of BG-1 populations infected with BRCA1 antisense treated with estrogen (10-8 mol/l; grey circle) or without estrogen (white circle); and the growth curve of BG-1 pooled population infected with pLXSN control vector treated with estrogen (black square) or without estrogen (white square) over a 6-day period. Cells were deprived of estrogen for 5 days before plating 2.5×106 cells per 100-mm dish and treatment with estrogen. The results shown indicate the mean cell number of three pooled populations after replating.
Figure 2.
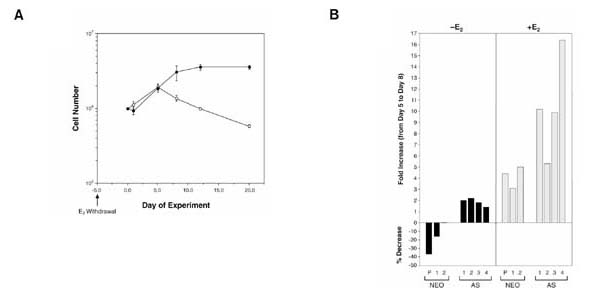
The effect of estrogen deprivation and estrogen treatment on the growth of BRCA1 antisense and control cells. (A) The mean cell number of BRCA1 pooled population antisense infected cells (filled circle) and vector-only infected cells (white circle) deprived of estrogen for up to 20 days after plating 2.5×106 cells per 100-mm dish. Cells received a pretreatment of 5 days of estrogen deprivation before plating. (B) The percentage decrease or fold increase of BG-1 antisense (AS) or NEO clones and parental cells plated at 106 cells per 60-mm dish after deprivation of estrogen (solid bars) or treatment with 10-8 mol/l estrogen (spotted bars) for 8 days. Percentage decrease or fold increase was calculated on the basis of the change in cell number between days 5 and 8 after treatment or nontreatment with 10-8 mol/l estrogen. Cells were pretreated with 5 days of estrogen deprivation before plating.
In order to avoid the problem of a mixed population of cells expressing various levels of BRCA1, subclones were isolated from infected populations of BRCA1 antisense infected BG-1 cells and control LXSN infected BG-1 cells (NEO). Figure 2b shows BG-1 parental and NEO clones exhibited up to a 37% decrease in cell number during a 3-day period of estrogen withdrawal, whereas antisense BRCA1 clones showed as much as a twofold increase in cell number during the same time period. In an attempt to determine if increased survival of the antisense cells was do to increased proliferation of the antisense cells or decreased death rate, the number of trypan blue positive, non-viable were examined after 14 days without estrogen. There were 5-10-fold more dead cells present in the media of control cells (BG-1 parental and NEO clone) then in the BRCA1 antisense clone AS-4 (data not shown). It appeared that resistance to cell death plays a significant role in the survival of BRCA1 antisense cells to estrogen withdrawal. Figure 2b again demonstrates the ability of the BRCA1 antisense sub-clones to survive estrogen deprivation. In the presence of estrogen, three out of the four antisense BRCA1 clones exhibited a growth advantage over NEO clones or the BG-1 parental population (Fig. 2b). Antisense BRCA1 clones 1, 3 and 4 showed a 10-fold to 16-fold increase in cell number between days 5 and 8 after estrogen treatment compared with only a threefold to fivefold increase of cell number in NEO clones and BG-1 parental cells (Fig. 2b). The AS-4 clone, which had the lowest levels of BRCA1 protein, showed a highly significant (16-fold; P<0.01) stimulation of growth between days 5 and 8 of estrogen induction (Figs 2b and 4). In summary, although three out of four of the antisense BRCA1 clones had a growth advantage in the presence of estrogen, all four antisense BRCA1 clones showed enhanced survival in estrogen-depleted media.
Figure 4.
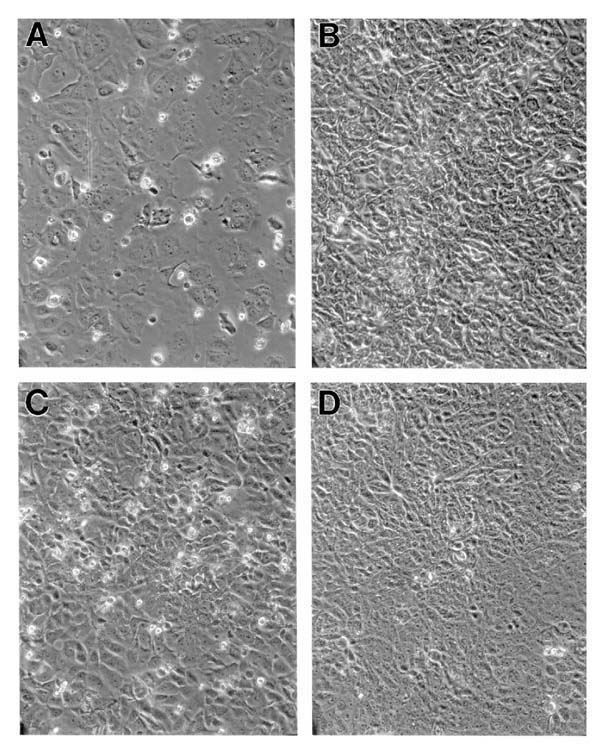
Estrogen dependent BG-1 cells infected with BRCA1 antisense continue to proliferate in the absence of estrogen. (A) BG-1 NEO control subclone on day 14 of estrogen deprivation. (B) BG-1 NEO at day 9 of 10-8 mol/l estrogen treatment. (C) BG-1 BRCA1 antisense AS-4 subclone on day 14 of estrogen deprivation. (D) BG-1 BRCA1 antisense infected cells at day 9 of 10-8 mol/l estrogen treatment.
Anchorage independent growth of BG-1 clones
Anchorage independent growth is a common property of many transformed cells. Therefore, the BRCA1 antisense subclones were also studied for anchorage independent growth in a semisoft agar medium with and without supplemented estrogen. Table1 shows that colony formation efficiencies on plastic of control (NEO) and BRCA1 anti-sense (AS-4) cells were similar in estrogen-depleted and estrogen-containing media. However, the BG-1 control clone (NEO) was unable to form colonies (fewer than one colony per 104 cells plated) in agar without the addition of estrogen, whereas the BG-1 antisense BRCA1 clone was able to form soft agar colonies in estrogen depleted conditions (10 ± 2.9 colonies per 104 cells plated). In the presence of estrogen, both NEO and AS-4 were able to form colonies; however, there was a significant difference (P <0.01) in the ability to form colonies in agar between AS-4 (133 colonies) and the control clone (six colonies). These data suggest a correlation between the loss of BRCA1 protein and an increased survival/growth advantage in anchorage-independent conditions.
Effects of reduced BRCA1 protein on in vivo tumor cell growth
Because the AS-4 clone showed a growth advantage in soft agar, a phenotype that may be correlated with the ability to form tumors in vivo, the BRCA1 antisense sub-clone AS-4 was evaluated for its ability to form subcutaneous tumors in ovariectomized athymic mice in the presence or absence of an estrogen pellet. Mice were injected subcutaneously with AS-4 cells in matrigel on one side of each mouse and NEO cells in matrigel on the other side. Of the mice injected, 50% received an implanted estrogen pellet (0.18 mg estrogen) that was designed to release estrogen for 60 days. In the absence of estrogen, a significant difference was detected in tumorigenic growth between AS-4 and NEO cells (Fig. 5b). Almost twice as many sites were tumor positive for the AS-4 clones than for NEO injected sites. 100% (14/14) Tumor formation was reached for all AS-4 clones 42 days after injection, compared with 57% (eight out of 14) positive tumor formation of the NEO sites (Fig. 5b, upper panel). AS-4 cells also formed tumors that averaged twice the size of NEO control tumors (Fig. 5b, lower panel).
Figure 5.
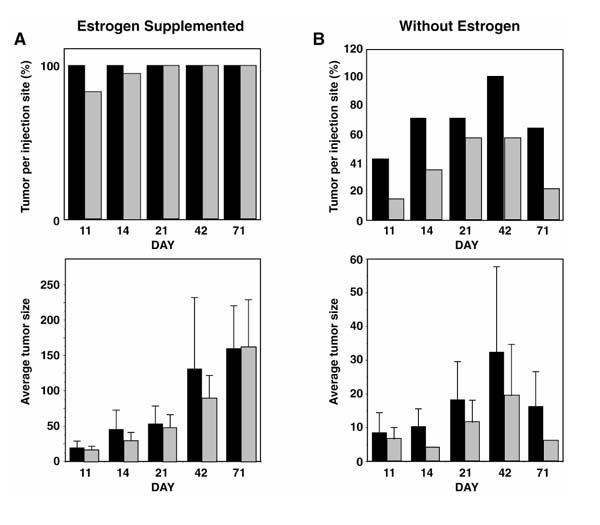
The effect of reduced BRCA1 protein in BG-1 cells on incidence and size of tumor formation in ovariectomized nude mice. (A) BG-1 BRCA1 antisense clone AS-4 (solid bars) and control clone NEO (gray bars) injected at 105 cells per site in matrigel in the presence of an 0.18mg estrogen implanted pellet. (B) BG-1 BRCA1 antisense clone AS-4 (solid bars) and control clone NEO (gray bars) injected at 105 cells per site in matrigel without supplemental estrogen. The upper panels indicate the percentage of injected sites that had tumor development. The lower panels provide the average tumor size in millimeters (length × width) for each clone on indicated days.
BG-1 cells without matrigel were nontumorigenic in athymic male or female mice (0 positive sites/20 sites injected at 5×106 cells per site), but these cells formed large, progressively growing tumors when injected with matrigel in the presence of estrogen (Fig. 5a). These tumors were very large (>1cm diameter) and did not regress even though the estrogen pellet was effective for only 60 days (Fig. 5a, lower panel). Similar to the agar experiments, both the BRCA1 antisense clone and LXSN control clone were positive for tumor formation in the presence of estrogen. AS-4 cells formed tumors with half of the latency of control cells in the presence of implanted estrogen (Fig. 5a, upper panel; 11 days versus 21 days until tumor formation). Neither AS-4 nor NEO cells formed progressively growing tumors in the absence of estrogen, however. All tumors in the mice without estrogen pellets had started to regress by 71 days after injection. The observed tumor regression was not surprising, because matrigel is not stable for longer than 14 days in culture, and probably not in vivo either (personal communication; Collaborative Biomedical Products, Bedford, MA, USA). By day 71, the matrigel would no longer confer an optimal growth environment for BG-1 cells.
Discussion
The present studies show that reduction of BRCA1 levels, using an antisense retroviral vector in the estrogen dependent BG-1 ovarian carcinoma cell line, may aid in confirmation of the hypothesis that BRCA1 functions as a tumor suppressor gene by playing a pivotal role in the balance between cell death and cell proliferation. BRCA1 RNA and protein levels were successfully reduced in pooled and isolated subclones of antisense-infected populations of BG-1 cells. Decreased BRCA1 levels appeared to affect the ability of BG-1 cells to arrest growth or die in the absence of estrogenic growth-inducing conditions. We found that BRCA1 antisense cells, both as pooled populations and individual subclones, also exhibited enhanced growth in monolayer culture on plastic in the presence of estrogen compared with control vector-infected colonies. All BRCA1 antisense subclones were able to proliferate as well as exhibit a decreased death rate in estrogen-deprived media, whereas parental and control subclones failed to grow. Death after estrogen withdrawal has been shown in previous studies using BG-1 cells [13,35]. BRCA1 antisense subclones demonstrated other traits associated with a tumorigenic phenotype, such as the ability to grow in soft agar independent of estrogen, whereas control clones could only form colonies with the addition of estrogen. In ovariectomized nude mice, a BRCA1 antisense clone (AS-4) was examined for tumorigenicity compared with a control clone (NEO). The AS-4 clone formed a greater number of and larger tumors than NEO in the absence of estrogen, and in general formed tumors faster in the presence of estrogen. The main conclusion from these studies is that BG-1 clones with reduced levels of BRCA1 protein have a survival advantage over controls in the absence of estrogen both in vitro and in vivo.
The response of BRCA1 mRNA and protein levels to mitogens and hormones in vitro suggests that BRCA1 may play a role in regulation of cell growth or maintenance [21]. During estrous, many hormones and growth factors interact in a complex manner as survival factors and inducers of cell proliferation, which are then balanced with growth inhibitors [36,37,38,39,40]. The mechanism by which BRCA1 can regulate or influence these processes has not yet been identified. It has been shown that BRCA1 is induced as a result of the mitogenic activity of the estrogen receptor in estrogen receptor-positive cells [9,10]. Direct estrogen stimulation is not required for BRCA1 transcription, however [9,41]. In support of this, BRCA1 expression has been shown to occur in the mouse ovary (granulosa and thecal cells of small and medium follicles) independent of hormonal status, and even in ovaries from estrogen receptor -/- deficient mice [41,42]. In contrast, the tumors from patients with BRCA1 mutations appear to have downregulation of estrogen receptors [43,44,45]. Previous experiments in our laboratory showed that another hormone, progesterone, could also cause a modest increase of BRCA1 mRNA in BG-1 cells after 24 h exposure without an increase in growth (unpublished data). Progesterone has been found [46] to inhibit cell proliferation and induce apoptosis significantly in two ovarian carcinoma cell lines. Thus, although BRCA1 may not be regulated directly by hormones, the BRCA1 gene product may be involved in the regulation of hormone response pathways, and the present results may demonstrate that loss of BRCA1 may result in loss of inhibitory control of these mitogenic pathways.
BRCA1 transcription is regulated with the cell cycle, and highest levels correlate with the G1/S-phase boundary [5,9,41,47,48,49]. The present studies show that reduction of BRCA1 mRNA and protein can result in increased proliferation of BG-1 ovarian cancer cells in vitro and in vivo, suggesting that BRCA1 may normally be acting as a growth inhibitor. Similar to our findings with ovarian carcinoma cells, accelerated growth, anchorage independence and tumorigenicity is associated with BRCA1 antisense introduction into mouse NIH3T3 cells [50]. In addition, increased proliferation of mammary cells is induced with antisense oligonucleotides to BRCA1 [51]. Conversely, introduction of full-length BRCA1 by retrovirus-mediated gene transfer inhibited growth of breast and ovarian cancer cell lines in both in vitro and in vivo experiments [51], and transfection of BRCA1 into colon cancer cells inhibited new DNA synthesis by 50% in addition to inhibition of S-phase progression, possibly through direct transactivation of the cell cycle inhibitor p21 WAF1/CIP1 [49].
In multicellular mammalian organisms, a balance between cell proliferation and cell death is extremely important for the maintenance of normal healthy tissues. This is especially important during early embryonic development as well as in the development and function of adult tissues such as the gonadal cells (ie ovarian and testes) [41,48]. For example, BRCA1 expression is critical during development, as evidenced by the embryonic lethality in transgenic knockout mice [29,30,52]. Alternatively, overexpression of BRCA1 may activate apoptosis or cell death [14]. Human prostate cells with an introduced wild-type BRCA1 cDNA demonstrated a threefold to sixfold increase in chemosensitivity, as well as an increased susceptibility to drug-induced apoptosis [53]. We found that clones with even moderately reduced levels of BRCA1 protein appeared to be relatively resistant to death due to estrogen deprivation. Previous studies in our laboratory showed that response of parental BG-1 cells and antisense clones to gamma radiation were consistent with a p53 wildtype phenotype, indicating that loss of estrogen dependence is probably not due to a p53 mutation (unpublished data). Shao et al [14] demonstrated that BRCA1 transfected into mouse 3T3 fibroblasts resulted in increased programmed cell death. In support of this hypothesis, it has been shown that p53 and BRCA1 can form stable complexes, and can coactivate p21 and bax genes, which may lead to the activation of the apoptosis pathway [15]. The present data, which show that cells with a reduction in BRCA1 have a survival advantage in conditions where control cells fail to thrive, also supports this hypothesis.
Like p53, BRCA1 has also been implicated in DNA damage and repair pathways [7,48,54]. According to this model, cells without normal BRCA1 activity may accumulate genetic alterations as a result of failure to arrest and repair DNA damage or self-destruct, thereby leading to genomic instability and neoplastic progression. It may not be coincidental that BRCA1-mutant breast cancers are preferentially linked to a 'specific' histopathologic pattern that includes a high S-phase fraction of cells, aneuploidy, and hormone receptor-negative status [45]. In addition, it has been demonstrated [20] that hormone-dependent tumors such as breast and ovarian cancers have a decreased ability to undergo apoptosis. Although mutations in this gene are uncommon in sporadic breast and ovarian tumors, BRCA1 expression levels and protein levels have been found to be reduced in sporadic human breast carcinomas [16,17,18,19]. Other mechanisms that involve gene regulation may allow for decreased expression of BRCA1 in sporadic tumors. Hypermethylation has been observed in some sporadic breast tumors in the promoter region of BRCA1, which may account for decreased BRCA1 transcription [55]. Low BRCA1 levels found in sporadic cancers may play an important role in tumorigenesis. The present data suggest that diminished levels of BRCA1 not only accelerate proliferation in the BG-1 ovarian carcinoma cell line, but appear to alter tumorigenesis. The exact mechanism may be unknown, but decreased BRCA1 levels appear to affect the ability to arrest growth or die in the absence of estrogenic growth-inducing conditions. We propose that the loss or reduction of BRCA1 may predispose a cell population to neoplastic transformation by altering the balance between cell death and proliferation/survival, rendering it more sensitive to secondary genetic changes.
Table 1.
Number of colonies per 104 cells in agar
| - Estrogen | + Estrogen | |||
|
|
|
|||
| Clone | Colonies in Agara | CFE on Plasticb | Colonies in Agara | CFE on Plasticb |
| NEO | 0.0± 0.0 | 21 | 6.0 ± 3.1 | 19 |
| AS-4 | 10.0 ± 2.9* | 38 | 133.0 ± 17.9* | 39 |
a Mean agar colonies ± STD; b% colony forming efficiency on plastic. * P <0.01 versus neo 1; n = 5.
Acknowledgments
Acknowledgements
We gratefully acknowledge the expertise and contribution to this work by Drs William Baldwin, Donato Romagnoto, Minoru Koi, and Joseph Haseman. We also thank Drs Roger Wiseman and Barbara Davis for critical review of the manuscript.
References
- Futreal PA, Liu Q, Shattuck-Eidens D, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. . Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Merajver SD, Pham TM, Caduff RF, et al. Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nature Genet. 1995;9:439–443. doi: 10.1038/ng0495-439. [DOI] [PubMed] [Google Scholar]
- Hosking L, Trowsdale J, Nicolai H, et al. A somatic BRCA1 mutation in an ovarian tumour [letter]. Nature Genet. 1995;9:343–344. doi: 10.1038/ng0495-343. [DOI] [PubMed] [Google Scholar]
- Thomas JE, Smith M, Tonkinson JL, Rubinfeld B, Polakis P. Induction of phosphorylation on BRCA1 during the cell cycle and after DNA damage. Cell Growth Differ. 1997;8:801–809. [PubMed] [Google Scholar]
- Ruffner H, Verma IM. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc Natl Acad Sci USA. 1997;94:7138–7143. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, et al. Dynamic changes of BRCA1 sub-nuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Gudas JM, Nguyen H, Li T, Cowan KH. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995;55:4561–4565. [PubMed] [Google Scholar]
- Marks JR, Huper G, Vaughn JP, et al. BRCA1 expression is not directly responsive to estrogen. Oncogene. 1997;14:115–121. doi: 10.1038/sj.onc.1200808. [DOI] [PubMed] [Google Scholar]
- Romagnolo D, Annab LA, Thompson TE, et al. Estrogen upregulation of BRCA1 expression with no effect on localization. . Mol Carcinogenesis. 1998;22:102–109. doi: 10.1002/(sici)1098-2744(199806)22:2<102::aid-mc5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Xu CF, Chambers JA, Solomon E. Complex regulation of the BRCA1 gene. J Biol Chem. 1997;272:20994–20997. doi: 10.1074/jbc.272.34.20994. [DOI] [PubMed] [Google Scholar]
- Gayther SA, Warren W, Mazoyer S, et al. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nature Genet. 1995;11:428–433. doi: 10.1038/ng1295-428. [DOI] [PubMed] [Google Scholar]
- Geisinger KR, Kute TE, Pettenati MJ, et al. Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors. Cancer. 1989;63:280–288. doi: 10.1002/1097-0142(19890115)63:2<280::aid-cncr2820630213>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Shao N, Chai YL, Shyam E, Reddy P, Rao VN. Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene. 1996;13:1–7. [PubMed] [Google Scholar]
- Zhang H, Somasundaram K, Peng Y, et al. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene. 1998;16:1713–1721. doi: 10.1038/sj.onc.1201932. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Ramos L, Villasenor MR, et al. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. . Nature Genet. 1999;21:236–240. doi: 10.1038/6029. [DOI] [PubMed] [Google Scholar]
- Taylor J, Lymboura M, Pace PE, et al. An important role for BRCA1 in breast cancer progression is indicated by its loss in a large proportion of non-familial breast cancers. Int J Cancer. 1998;79:334–342. doi: 10.1002/(sici)1097-0215(19980821)79:4<334::aid-ijc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Ozcelik H, To MD, Couture J, Bull SB, Anrulis IL. Preferential allelic expression can lead to reduced expression of BRCA1 in sporadic breast cancers. Int J Cancer. 1998;77:1–6. doi: 10.1002/(sici)1097-0215(19980703)77:1<1::aid-ijc1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nature Genet. 1995;9:444–450. doi: 10.1038/ng0495-444. [DOI] [PubMed] [Google Scholar]
- Thompson C. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Gudas JM, Li T, Nguyen H, et al. Cell cycle regulation of BRCA1 messenger RNA in human breast epithelial cells. Cell Growth Differ. 1996;7:717–723. [PubMed] [Google Scholar]
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. . Lancet. 1994;343:692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. . Nature Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- Scully R, Anderson SF, Chao DM, et al. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci USA. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MS, Verma IM. Transcriptional activation by BRCA1 [letter; comment]. Nature. 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. . Proc Natl Acad Sci USA. 1996;93:13595–13599. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis ST, Rajan JV, Wynshaw-Boris A, et al. The developmental pattern of Brca1 expression implies a role in differentiation of the breast and other tissues. Nature Genet. 1995;11:17–26. doi: 10.1038/ng0995-17. [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neu-roepithelial abnormalities. Nature Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980-990, 984–986, 989-990. [PMC free article] [PubMed] [Google Scholar]
- Haendler B, Hofer E. Characterization of the human cyclophilin gene and of related processed pseudogenes. Eur J Biochem. 1990;190:477–482. doi: 10.1111/j.1432-1033.1990.tb15598.x. [DOI] [PubMed] [Google Scholar]
- Finney DJ. Statistical Method in Biological Assay New York: Hafner Publishing Co, 1964.
- Siegel S. Nonparametric Statistics New York: McGraw-Hill. 1956.
- Baldwin WS, Curtis SW, Cauthen CA, et al. BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro. . In Vitro Cell Dev Biol . 1998;19:1895–1900. doi: 10.1007/s11626-996-0015-9. [DOI] [PubMed] [Google Scholar]
- Foghi A, Teerds KJ, van der Donk H, Dorrington J. Induction of apop-tosis in rat thecal/interstitial cells by transforming growth factor alpha plus transforming growth factor beta in vitro. J Endocrinol. 1997;153:169–178. doi: 10.1677/joe.0.1530169. [DOI] [PubMed] [Google Scholar]
- Kenny N, Williams RE, Kelm LB. Spontaneous apoptosis of cells prepared from the nonregressing corpus luteum. Biochem Cell Biol . 1994;72:531–536. doi: 10.1139/o94-071. [DOI] [PubMed] [Google Scholar]
- Einspanier R, Lauer B, Gabler C, Kamhuber M, Schams D. Egg-cumulus-oviduct interactions and fertilization. Adv Exp Med Biol. 1997;424:279–289. doi: 10.1007/978-1-4615-5913-9_50. [DOI] [PubMed] [Google Scholar]
- Rueda BR, Tilly KI, Botros IW, et al. Increased bax and interleukin-1beta-converting enzyme messenger ribonucleic acid levels coincide with apoptosis in the bovine corpus luteum during structural regression. Biol Reprod. 1997;56:186–193. doi: 10.1095/biolreprod56.1.186. [DOI] [PubMed] [Google Scholar]
- Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. . Annu Rev Physiol. 1997;59:349–363. doi: 10.1146/annurev.physiol.59.1.349. [DOI] [PubMed] [Google Scholar]
- Phillips KW, Goldsworthy SM, Bennett LM, et al. Brca1 is expressed independently of hormonal stimulation in the mouse ovary. . Lab Invest. 1997;76:419–425. [PubMed] [Google Scholar]
- Blackshear PE, Goldsworthy SM, Foley JF, et al. Brca1 and Brca2 expression patterns in mitotic and meiotic cells of mice. . Oncogene. 1998;16:61–68. doi: 10.1038/sj.onc.1201506. [DOI] [PubMed] [Google Scholar]
- Schmutzler RK, Bierhoff E, Werkhausen T, et al. Genomic deletions in the BRCA1, BRCA2 and TP53 regions associate with low expression of the estrogen receptor in sporadic breast carcinoma. Int J Cancer. 1997;74:322–325. doi: 10.1002/(sici)1097-0215(19970620)74:3<322::aid-ijc15>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Karp SE, Tonin PN, Begin LR, et al. Influence of BRCA1 mutations on nuclear grade and estrogen receptor status of breast carcinoma in Ashkenazi Jewish women. Cancer. 1997;80:435–441. doi: 10.1002/(sici)1097-0142(19970801)80:3<435::aid-cncr11>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Johannsson OT, Idvall I, Anderson C, et al. Tumour biological features of BRCA1-induced breast and ovarian cancer. Eur J Cancer. 1997;33:362–371. doi: 10.1016/s0959-8049(97)89007-7. [DOI] [PubMed] [Google Scholar]
- Bu SZ, Yin DL, Ren XH, et al. Progesterone induces apoptosis and up-regulation of p53 expression in human ovarian carcinoma cell lines. Cancer. 1997;79:1944–1950. doi: 10.1002/(sici)1097-0142(19970515)79:10<1944::aid-cncr15>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Rajan JV, Wang M, Marquis ST, Chodosh LA. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc Natl Acad Sci USA. 1996;93:13078–13083. doi: 10.1073/pnas.93.23.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Somasundaram K, Zhang H, Zeng YX, et al. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- Rao VN, Shao N, Ahmad M, Reddy ES. Antisense RNA to the putative tumor suppressor gene BRCA1 transforms mouse fibroblasts. Oncogene. 1996;12:523–528. [PubMed] [Google Scholar]
- Holt JT, Thompson ME, Szabo C, et al. Growth retardation and tumour inhibition by BRCA1. Nature Genet. 1996;12:298–302. doi: 10.1038/ng0396-298. [DOI] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- Fan S, Wang J-A, Yuan R-q, et al. BRCA1 as a potential human prostate tumor suppressor: modulation of proliferation, damage responses and expression of cell regulatory proteins. . Oncogene. 1998;16:3069–3082. doi: 10.1038/sj.onc.1202116. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Jacks T. Double indemnity: p53, BRCA and cancer. p53 mutation partially rescues developmental arrest in Brca1 and Brca2 null mice, suggesting a role for familial breast cancer genes in DNA damage repair [news]. Nature Med. 1997;3:721–722. doi: 10.1038/nm0797-721. [DOI] [PubMed] [Google Scholar]
- Dobrovic A, Simpfendorfer D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997;57:3347–3350. [PubMed] [Google Scholar]