

Abstract
This work assesses relationships for 30 complete prokaryotic genomes between the presence of the Shine-Dalgarno (SD) sequence and other gene features, including expression levels, type of start codon, and distance between successive genes. A significant positive correlation of the presence of an SD sequence and the predicted expression level of a gene based on codon usage biases was ascertained, such that predicted highly expressed genes are more likely to possess a strong SD sequence than average genes. Genes with AUG start codons are more likely than genes with other start codons, GUG or UUG, to possess an SD sequence. Genes in close proximity to upstream genes on the same coding strand in most genomes are significantly higher in SD presence. In light of these results, we discuss the role of the SD sequence in translation initiation and its relationship with predicted gene expression levels and with operon structure in both bacterial and archaeal genomes.
In bacteria, translation initiation is commonly considered the rate-limiting step of translation and a major determinant of the overall expression level of a gene (11, 16). The components of the initiation complex have been identified as the ribosome, an mRNA sequence, fMet-tRNAfMet, and three initiation factors. RNase protection experiments and sequence alignment studies indicate that the ribosome may contact a short region at the 5′ end of the mRNA called the translation initiation region, which extends from about 20 bp 5′ to the initiation codon to 13 bp 3′ to the initiator codon. Sequence elements in the translation initiation region important for translation initiation include the initiation codon and nucleotides in its vicinity, a short motif 5′ to the initiator, called the Shine-Dalgarno (SD) sequence, and the mRNA secondary structure (11).
The SD sequence plays an important role in formation of the initiation complex by base-pairing with the anti-SD sequence found at the 3′ end of 16S rRNA. This has been demonstrated by extensive experiments with Escherichia coli (9, 19, 46), other bacteria, and even archaea (8, 32, 35, 37). The SD sequences could be different subsequences of the complementary sequence of the anti-SD sequence (see Table 1); however, most SD sequences are slight variations of the GGAGG core (43). The effectiveness of an SD sequence is determined by both its base-pairing potential with the anti-SD sequence and its spacing from the start codon (10, 34). The aligned spacing of the SD sequences (see the legend to Fig. 1A for definition) generally varies from 5 to 13 bases, with optimal spacings of about 8 to 10 bases for E. coli genes (7, 34). Although it is not mandatory in translation initiation, a strong SD sequence may compensate for a weak start codon and counteract mRNA secondary structures that hinder access to the start (10, 55).
TABLE 1.
Features of the prokaryotic genomes studied
| Genomea | G+C% | Anti-SDb | Genesc | SD%d | OASe |
|---|---|---|---|---|---|
| Bacteria | |||||
| Proteobacteria | |||||
| γ-Type | |||||
| ESCCO | 50.8 | AUCACCUCCUUA | 3,908 | 57.1 | 7, 8, 9 |
| HAEIN | 38.2 | AUCACCUCCUUA | 1,533 | 53.7 | 7, 8, 9 |
| VIBCH | 47.5 | AUCACCUCCUUA | 3,260 | 48.3 | 7, 8, 9 |
| PSEAE | 66.6 | AUCACCUCCUUA | 5,246 | 69.2 | 7, 8, 9 |
| ɛ-Type | |||||
| CAMJE | 30.5 | AUCACCUCCUUU | 1,502 | 58.7 | 6, 7, 8 |
| HELPY | 38.9 | AUCACCUCCUUU | 1,391 | 59.4 | 6, 7, 8 |
| α-Type | |||||
| RICPR | 29.0 | AUCACCUCCUUA | 773 | 18.6 | 7, 8, 9 |
| β-Type | |||||
| NEIME | 51.5 | AUCACCUCCUUU | 1,674 | 49.3 | 6, 7, 8 |
| Chlamydiae | |||||
| CHLPN | 40.6 | AUCACCUCCUUU | 965 | 42.8 | 7, 8, 9 |
| CHLTR | 41.3 | AUCACCUCCUUU | 831 | 46.8 | 7, 8, 9 |
| Spirochetes | |||||
| BORBU | 28.6 | AUCACCUCCUUU | 771 | 52.3 | 6, 7, 8 |
| TREPA | 52.8 | AUCACCUCCUUU | 916 | 60.0 | 6, 7, 8 |
| Fermicutes (gram-positive) | |||||
| Bacilli | |||||
| BACSU | 43.5 | AUCACCUCCUUU | 3,624 | 89.4 | 9, 10, 11 |
| Mollicutes | |||||
| MYCGE | 31.7 | AUCACCUCCUUU | 448 | 10.8 | 6, 7, 8* |
| MYCPN | 40.0 | AUCACCUCCUUU | 658 | 17.5 | 11, 12, 13 |
| UREUR | 25.5 | AUCACCUCCUUU | 556 | 57.4 | 7, 8, 9 |
| Actinobacteria | |||||
| MYCTU | 65.6 | AUCACCUCCUUU | 3,677 | 47.7 | 7, 8, 9 |
| Cyanobacteria | |||||
| SYNSQ | 47.7 | AUCACCUCCUUU | 2,906 | 26.0 | 9, 10, 11 |
| Thermus/Deinococcus | |||||
| DEIRA | 66.6 | AUCACCUCCUUU | 2,923 | 42.0 | 7, 8, 9 |
| Gram-negative thermophiles | |||||
| AQUAE | 43.5 | AUCACCUCCUUU | 1,487 | 48.1 | 8, 9, 10 |
| THEMA | 46.2 | AUCACCUCCUUU | 1,685 | 90.1 | 9, 10, 11 |
| Archaea | |||||
| Euryarchaea | |||||
| Methanogens | |||||
| METJA | 31.4 | AUCACCUCCU | 1,471 | 48.8 | 9, 10, 11 |
| METTH | 49.5 | AUCACCUCCUUA | 1,641 | 60.7 | 8, 9, 10 |
| ARCFU | 48.6 | AUCACCUCCUAA | 2,083 | 46.2 | 9, 10, 11 |
| Hyperthermophiles | |||||
| PYRAB | 44.7 | AUCACCUCCUAU | 1,686 | 71.3 | 9, 10, 11 |
| PYRHO | 41.9 | AUCACCUCCUAU | 1,999 | 54.9 | 9, 10, 11 |
| THEAC | 46.0 | AUCACCUCC | 1,386 | 23.5 | 9, 10, 11 |
| Halobacteriales | |||||
| HALSP | 67.9 | AUCACCUCCUAA | 1,755 | 26.6 | 9, 10, 11 |
| Crenarchaea | |||||
| SULSO | 35.8 | AUCACCUCAU | 2,741 | 23.0 | 10, 11, 12 |
| PYRAE | 51.4 | AUCACCUCC | 2,137 | 22.9 | 4, 5, 6* |
For abbreviations, see Fig. 2 legend.
3′ end of the 16S rRNA sequence; the core anti-SD sequence CCUCC is in bold.
Number of genes with at least 100 codons.
Fraction of genes having an SD sequence.
OAS for the SD sequences (see the text for a formal definition of OAS). *, OAS that may not reflect real optimal spacings as in other genomes (see text for details).
FIG. 1.

Distribution of aligned spacings of SD sequences for RP, PHX, and PMX gene classes. (A) Simplified diagram of the translation initiation complex formed between an E. coli mRNA and the 30S ribosomal subunit. The aligned spacing is designated as the distance between the center of the SD sequence GGAGG and the start codon AUG. (B and C) Histograms of the SD aligned spacings in the genomes of Escherichia coli K-12 (ESCCO) and Pyrococcus abyssi (PYRAB), respectively.
Previous studies on the SD sequence have centered on clarifying its role in translation initiation in bacteria, especially E. coli. Comparative analysis of this motif in different classes of genes and across different genomes may provide insights into the function and evolution of the SD interaction. This kind of analysis became possible only recently with the availability of many complete genome sequences (3).
The main objective of this paper is to investigate, in 30 complete prokaryotic genomes (available as of June 2001), the correlation between the presence of an SD sequence and predicted expression levels of genes based on codon usage biases, functional gene classes, type of start codon, and distance between successive genes.
MATERIALS AND METHODS
Genome sequences.
All genome sequences were extracted from the National Center for Biotechnology Information (NCBI) GenBank database (http://www.ncbi.nlm.nih.gov). The species names as well as the abbreviations and GenBank accession numbers are given as follows. Bacteria included Aquifex aeolicus (NC_000918), Bacillus subtilis (NC_000964), Borrelia burgdorferi (NC_001318), Campylobacter jejuni (NC_002163), Chlamydophila pneumoniae (NC_000922), Chlamydia trachomatis (NC_000117), Deinococcus radiodurans (NC_001263), Escherichia coli K-12 (NC_000913), Haemophilus influenzae (NC_000907), Helicobacter pylori 26695 (NC_000915), Mycobacterium tuberculosis (NC_000962), Mycoplasma genitalium (NC_000908), Mycoplasma pneumoniae (NC_000912), Neisseria meningitidis (NC_002183), Pseudomonas aeruginosa (NC_002516), Rickettsia prowazekii (NC_000963), Synechocystis sp. strain PCC6803 (NC_000911), Thermotoga maritima (NC_000853), Treponema pallidum (NC_000919), Ureaplasma urealyticum (NC_002162), and Vibrio cholerae (NC_002505 and NC_002506). Archaea included Archaeoglobus fulgidus (NC_000917), Halobacterium sp. strain NRC-1 (NC_002607), Methanobacterium thermoautotrophicum (NC_000916), Methanococcus jannaschii (NC_000909), Pyrobaculum aerophilum (NC_003364), Pyrococcus abyssi NC_000868), Pyrococcus horikoshii (NC_000961), Sulfolobus solfataricus (NC_002754), and Thermoplasma acidophilum (NC_002578).
Detection of SD sequences.
To detect putative SD sequences, we calculated the free energy (designated ΔGSD, in kilocalories per mole) for all possible duplexes between the anti-SD sequence and the 20 bases upstream of the start codon of a gene. Dynamic programming was implemented to find the duplex that gave the lowest free energy. This method has been described in several publications and is well accepted in SD detection (12, 32, 37, 43). The stacking energy was calculated based on the rules developed by Freier et al. (13). Only canonical Watson-Crick base pairs and G-U pairings flanked by Watson-Crick base pairs were allowed, and the free energy loss by duplex initiation, 3.4 kcal/mol (13), was subtracted. To reduce ambiguity, a cutoff value of ΔGSD = −4.4 kcal/mol was used, which is the ΔGSD for the core SD motifs GGAG, GAGG, and AGGA in bacteria (43). A specific anti-SD sequence was used for each genome (Table 1).
There are several rationales in favor of a threshold of −4.4 kcal/mol. (i) An effective SD sequence usually binds to the core CCUCC of the anti-SD sequence, which is conserved in all but one of the genomes (Table 1). It seems unlikely that the basic mechanism of the SD interaction will change from genome to genome, given the conservation of the core anti-SD motif. Thus, we define the SD sequences GGAG, GAGG, and AGGA as core SD motifs, all of which have a free energy of binding of −4.4 kcal/mol. (ii) Constrained by the base composition of the anti-SD sequence, its complementary motifs with a free energy of binding greater than −4.4 kcal/mol most often bind parts other than the core CCUCC and are likely to be random motifs. Thus, we have designated this threshold to exclude these random motifs. (iii) We analyzed several genomes with SD sequences defined by the core SD motifs GAGG, GGAG, and AGGA (SD sequences were defined as sequences harboring any of these motifs) and obtained highly concordant results (data not shown). Also, we relaxed the stringency and accepted the 3-bp motifs GGA, GAG, and GGA as SD sequences. The final results are consistent (see Supplementary Data Fig. S-1 and Table S-1; all supplementary data can be accessed at http://gnomic.stanford.edu/jiongm/SD/).
For most genes, this cutoff value effectively leaves only one or no SD sequence in the 5′ region (20 nucleotides). In rare cases there might be two or more competing motifs that qualify as SD sequences. When this happens, we chose the one with the lowest free energy of binding.
The aligned spacing of an SD sequence is defined as the number of bases between the first base of the start codon and the U in the core anti-SD motif CCUCC (Table 1) in the duplex formed (Fig. 1A). The aligned spacing of the SD sequence GGAGG in Fig. 1A is 7 bases. There are 22 possible spacings (0 to 21 bases). However, generally more than 80% of all the SD sequences occur at spacings of 5 to 13 bases to the start codon (see below).
Theoretical measures of gene expression.
We used a method introduced by Karlin and Mrázek (22) to assess codon biases of a class of genes (or a single gene) relative to a second class of genes. Let G be a group of genes with average codon frequencies g(x,y,z) for the codon triplet (x,y,z) such that Σg(x,y,z) = 1 for each amino acid family. Similarly, let {f(x,y,z)} indicate the average codon frequencies for the gene group F, normalized to 1 in each amino acid codon family. The codon usage difference of F relative to G is calculated by the formula
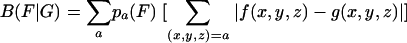 |
(1) |
where {pa(F)} is the average amino acid frequency of the genes of F. When no ambiguity is likely, we refer to B(F|G) as the codon bias of F with respect to G. The assessments of equation 1 can be made for any two gene groups from the same genome or from different genomes. In particular, there are four classes of genes as standards: C, all genes; ribosomal proteins (RP); transcription, translation processing factors (TF); and major chaperon/degradation (CH) genes functioning in protein folding, trafficking, and secretion. Qualitatively, gene g is predicted to be highly expressed (PHX) if B(g|C) is high while B(g|RP), B(g|CH), and B(g|TF) are low (i.e., the codon usage of g is very different from that of the average genes but rather similar to that of the gene classes RP, CH, and TF). Similarly, g is putatively alien (PA) if its codon biases relative to the four classes are all large. Predicted expression levels of g with respect to these standards are calculated by
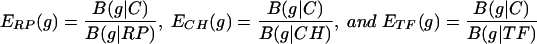 |
(2) |
We propose a general expression measure for g as follows:
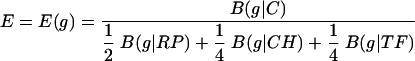 |
(3) |
Other weights can also be used and give similar results.
Definition of PHX and putative alien (PA) gene classes.
We defined a gene as PHX if the following two conditions were satisfied: (i) at least two among the three expression values ERP(g), ECH(g), and ETF(g) exceeded 1.05 and (ii) the overall expression level E(g) was ≥ 1.00. A gene was defined as PA if it fulfilled the following criteria: B(g|RP) ≥ M + 0.10, B(g|CH) ≥ M + 0.10, B(g|TF) ≥ M + 0.10, and B(g|C) ≥ M + 0.10, where M is the median value among B(g|C) for all g (22, 23, 27). Predicted moderately expressed (PMX) genes are genes that are neither PHX nor PA. PMX genes constitute roughly more than 90% of a genome and thus represent average genes. We often use PMX as a standard to compare to PHX and PA genes.
Logistic regression analysis.
To study the correlation between SD presence and E(g) values, we used a logistic regression model (18). Considering a genome with n genes (each ≥100 codons), we observe n pairs (xi, yi), i = 1, 2, . . . , n, where xi = E(gi) is the predicted expression level of gene gi calculated by equation 3 and where yi designates the presence or absence of the SD sequence in gi (1 if present and 0 if not). We attempted to fit the data pairs to the logistic regression model
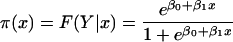 |
(4) |
where π(x) = F(Y|x) represents the conditional mean of Y (SD presence), given x = E(g). The logit transformation is defined as
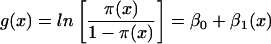 |
(5) |
where β1 is the regression coefficient and the measure of correlation. The SPSS statistics software (version 6.1.4; SPSS Inc., Chicago, Ill.) was used for the model fitting. The estimated β1 (as β) and the estimated standard error are given for each genome in Table 2; other estimated parameters not shown include β0 and the P value for a likelihood ratio test of the regression.
TABLE 2.
SD% for RP, PHX, PMX, and PA genesa
| Genomeb | RP
|
PHX
|
PMX
|
PA
|
βc | SEd | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of genes | SD% | No. of genes | SD% | No. of genes | SD% | No. of genes | SD% | |||
| ESCCO | 57 | 75.4 | 306 | 71.6 | 3,321 | 57.1 | 269 | 40.9 | 0.71 | 0.12 |
| HAEIN | 57 | 82.5 | 140 | 75.7 | 1,328 | 51.6 | 60 | 48.3 | 1.82 | 0.26 |
| VIBCH | 56 | 80.4 | 172 | 64.0 | 2,907 | 47.5 | 174 | 44.8 | 0.51 | 0.14 |
| PSEAE | 57 | 87.7 | 128 | 82.8 | 4,836 | 69.1 | 274 | 65.3 | 0.77 | 0.19 |
| CAMJE | 54 | 92.6 | 119 | 79.0 | 1,351 | 57.1 | 11 | 45.5 | 2.09 | 0.42 |
| HELPY | 54 | 81.5 | 73 | 83.6 | 1,287 | 58.1 | 27 | 55.6 | 3.54 | 0.79 |
| RICPR | 54 | 50.0 | 41 | 31.7 | 728 | 17.9 | 0 | 2.97 | 1.17 | |
| NEIME | 57 | 84.2 | 91 | 83.5 | 1,368 | 48.4 | 209 | 40.2 | 1.90 | 0.28 |
| CHLPN | 53 | 75.5 | 85 | 62.4 | 864 | 40.7 | 14 | 50.0 | 3.20 | 0.63 |
| CHLTR | 51 | 70.6 | 52 | 69.2 | 766 | 45.4 | 9 | 33.3 | 2.66 | 0.83 |
| BORBU* | 54 | 79.6 | 71 | 67.6 | 700 | 50.7 | 0 | 0.98 | 0.59 | |
| TREPA | 51 | 98.0 | 99 | 73.7 | 791 | 59.2 | 26 | 34.6 | 2.33 | 0.60 |
| BACSU** | 62 | 100.0 | 147 | 93.2 | 3,292 | 89.4 | 172 | 84.9 | 0.33 | 0.26 |
| MYCGE** | 51 | 22.4 | 27 | 14.8 | 419 | 10.5 | 0 | 0.09 | 1.31 | |
| MYCPN | 53 | 34.0 | 57 | 28.1 | 546 | 17.0 | 50 | 10.0 | 2.81 | 1.00 |
| UREUR | 52 | 84.6 | 72 | 73.6 | 469 | 55.0 | 11 | 54.5 | 2.31 | 0.64 |
| MYCTU | 55 | 74.5 | 560 | 55.4 | 2,939 | 46.4 | 158 | 46.2 | 0.96 | 0.27 |
| SYNSQ | 57 | 49.1 | 380 | 37.6 | 2,329 | 24.5 | 186 | 21.5 | 1.54 | 0.29 |
| DEIRA | 55 | 85.5 | 337 | 58.5 | 2,329 | 39.5 | 254 | 42.5 | 1.16 | 0.14 |
| AQUAE | 56 | 92.9 | 233 | 69.5 | 1,174 | 43.5 | 74 | 54.1 | 3.18 | 0.39 |
| THEMA | 54 | 98.1 | 174 | 93.7 | 1,443 | 89.9 | 64 | 84.4 | 2.35 | 0.78 |
| METJA* | 62 | 87.1 | 113 | 54.9 | 1,325 | 48.5 | 28 | 35.7 | 0.53 | 0.29 |
| METTH | 60 | 85.0 | 160 | 71.3 | 1,393 | 59.6 | 76 | 59.2 | 1.66 | 0.46 |
| ARCFU | 59 | 78.0 | 343 | 59.8 | 1,601 | 43.5 | 133 | 43.6 | 2.35 | 0.36 |
| PYRAB | 60 | 96.7 | 241 | 80.5 | 1,351 | 70.5 | 89 | 59.6 | 1.87 | 0.36 |
| PYRHO | 54 | 81.2 | 173 | 69.9 | 1,680 | 55.7 | 134 | 29.9 | 1.52 | 0.38 |
| THEAC | 49 | 85.7 | 149 | 33.6 | 1,140 | 22.9 | 94 | 14.9 | 2.24 | 0.83 |
| HALSP* | 58 | 53.4 | 328 | 24.1 | 1,283 | 27.4 | 140 | 25.7 | −0.82 | 0.45 |
| SULSO | 65 | 58.5 | 654 | 27.1 | 1,760 | 21.7 | 322 | 21.4 | 1.29 | 0.42 |
| PYRAE** | 66 | 33.3 | 59 | 25.4 | 1,985 | 23.3 | 92 | 14.1 | 0.45 | 1.01 |
See text for definitions of PHX, PMX, and PA gene classes. For the gene classes RP, PHX, and PA, SD% in bold indicates a significant increase over the group PMX, while underlining signifies a significant decrease (P < 0.05 for a χ2 test using the Yates correction). The P value of the logistic regression (for a likelihood ratio test of the regression) was <0.01 for all genomes except those marked with * (0.05 < P < 0.1) and ** (P > 0.1).
See Fig. 2 legend for abbreviations.
Estimated coefficient for the logistic regression model (β1 in equation 5).
Standard error of the estimated β.
RESULTS AND DISCUSSION
SD sequences in bacterial and archaeal genomes.
We defined SD% as the fraction of genes in a given group that possesses an SD sequence. For each genome, Table 1 reports the G+C content, the count of genes encoding products of at least 100 amino acids in length, the anti-SD sequence at the 3′ end of the 16S rRNA sequence, the SD% of all the genes in the genome (≥100 amino acids), and the optimal aligned spacings (OAS) for the SD sequences (discussed below). In bacterial genomes, the anti-SD sequence is AUCACCUCCUUU, although the archaeal genomes show some variation in their anti-SD sequences around the conserved core CCUCC (Table 1).
Using the free-energy method and a cutoff value of −4.4 kcal/mol, all the SD sequences detected were at least 4 bases in length, and most harbored the motif GGAG, GAGG, or AGGA (e.g., 88% of the SD sequences in Escherichia coli K-12). In some natural mRNAs an SD sequence can consist of a weaker motif, e.g., AAGG, with a ΔGSD of −2.9 kcal/mol (57). For our purposes we prefer to find only unambiguous SD sequences. In terms of base-pairing potential with the anti-SD sequence, the SD sequences defined by our method may be considered strong SD sequences. Most of them are present at an aligned spacing of between 5 and 13 bases, as verified by histograms of spacings of all the SD sequences in a genome (Fig. 1B and C; also see Supplementary Data Fig. S-2). An SD sequence at this range of spacings has been established to be effective (7, 16, 34).
Of the 30 genomes, 22 had an SD% exceeding 40% for all genes. Bacillus subtilis and Thermotoga maritima registered the highest SD%, 89.4% and 90.1%, respectively. The lowest genome SD% occurred for Rickettsia prowazekii, Mycoplasma genitalium, Mycoplasma pneumoniae, Halobacterium sp. strain NRC-1, Thermoplasma acidophilum, Sulfolobus solfataricus, and Pseudomonas aeruginosa, each at around 20%. In general, fast-growing bacteria, gram-negative thermophiles, spirochetes, methanogens, and hyperthermophilic archaea achieved relatively high SD%, while obligate intracellular parasites, surface parasites, pathogens, and cyanobacteria had diminished genome SD%.
We carried out a simulation study to determine whether these SD% values represent real DNA elements or just random motifs. For each genome, we generated 100 (1,000 for Escherichia coli K-12) data sets of random sequences 20 nucleotides long according to the base composition of the original 20-nucleotide 5′ end sequence data set, each with the same number of sequences as in the given genome. SD sequences were detected and SD% was calculated for each set of these random sequences. The SD% values shown in Table 1 were found to represent real motifs in all the genomes except for Mycoplasma genitalium and Halobacterium sp. strain NRC-1, as assessed by distributions of the SD% for these simulated data sets (the probability of these SD% values coming from random sequences was <0.01).
Correlation between SD presence and predicted gene expression levels.
It is known that not all genes contain an SD sequence. In some genomes, the majority of genes do not have such a motif (Table 1). Although an SD sequence is not compulsory for the translation of many genes (21), it may still be effective for genes that contain such a motif. This raises the question of how the SD sequences are distributed in different gene classes.
First we examined SD sequences for the RP genes. Primarily highly expressed during fast growth, the RP gene class showed a very high SD%, around 80% in most genomes (Table 2). Even for genomes with a low overall SD%, the RP SD% was significantly high. For example, the SD% was 85.7% for RP genes in Thermoplasma acidophilum (23.5% for the genome) and 58.5% for RP in Sulfolobus solfataricus (23.0% for the genome). This is consistent with a greater SD presence for highly expressed genes.
We then divided the genes of a genome (≥100 codons) into three classes, PHX, PA, and PMX, based on codon usage biases (22). The percentage of PHX genes in different genomes ranged from 2% to 19%, whereas PA genes ranged from 0 to 13% (Table 2). PMX genes constitute the bulk of a genome and consisted mostly of average genes. The major PHX genes were RP, TF, and CH genes. Other PHX genes included those encoding enzymes of essential energy metabolism pathways and the principal genes of amino acid and nucleotide biosyntheses (22, 23, 27). Our results on PHX agree well with two-dimensional gel experimental assessments in several prokaryotes (1, 22, 23, 42, 53, 54). The PHX genes in most of the 30 genomes carried a significantly higher SD% than PMX genes. PA genes generally showed an SD% about the same as or less than that of the PMX genes (Table 2). Since PA genes are largely composed of putative lateral transfer genes, they tend to have low expression levels (28).
To verify the positive correlation of SD presence and gene expression levels, we applied logistic regression analysis. The regression coefficient β and its estimated standard error for each genome are given in Table 2. All but six genomes (Borrelia burgdorferi, Bacillus subtilis, Mycoplasma genitalium, Methanococcus jannaschii, Halobacterium sp. strain NRC-1, and Pyrobaculum aerophilum) recorded a significant positive correlation between SD presence and E(g) values (P < 0.01 for a likelihood ratio test of the regression). For the genomes of Borrelia burgdorferi, Methanococcus jannaschii, and Halobacterium sp. strain NRC-1, the P value for the likelihood test was between 0.05 and 0.1, indicating a relatively strong correlation. Of the three genomes that did not record a significant correlation (P > 0.1), Mycoplasma genitalium had the lowest SD% (10.8%); Bacillus subtilis was among the highest in SD%; and Pyrobaculum aerophilum was low at about 23% (Tables 1 and 2).
Since all the data sets used were original genome annotations, a reasonable concern was that incorrect annotations of the gene start sites may have affected the accuracy of our SD analysis. To better determine how the genome data would compare to more reliable data sets, we analyzed the SD% for genes from several human-curated Escherichia coli K-12 data sets and achieved very similar results, as shown in Table 3. The data sets on essentiality were from the Profiling of E. coli Chromosome (PEC) database (http://www.shigen.nig.ac.jp/ecoli/pec/). The PEC data set classifies all E. coli genes into three groups: genes essential for cell growth (“essential”; total of 191 genes), those dispensable for cell growth (“nonessential”), and those unknown to be essential or nonessential (“unknown”), mainly using information from the literature. The “verified” (total, 656 genes) data set was extracted from EcoMap12 (http://bmb.med.miami.edu/EcoGene/EcoWeb/), which consists of genes whose starts have been confirmed by N-terminal protein sequencing (41). There are 65 genes in the verified set whose start sites were incorrectly annotated in the NCBI genome (4), giving an accuracy of about 90% for start site annotation, which is consistent with the average accuracy estimated for various gene-finding programs (25).
TABLE 3.
E. coli data setsa
| Data set | Type | PHX
|
PHX(RP−)
|
PMX
|
PA
|
||||
|---|---|---|---|---|---|---|---|---|---|
| No. of genesb | SD% | No. of genes | SD% | No. of genes | SD% | No. of genes | SD% | ||
| EcoMap12 | Verified | 178 | 81.5 | 157 | 81.5 | 468 | 73.3 | 10 | 70.0 |
| PEC database | Essential | 86 | 69.8 | 51 | 62.7 | 105 | 49.5 | 0 | |
| Nonessential | 183 | 73.8 | 183 | 73.8 | 1,798 | 62.6 | 117 | 38.5 | |
| Unknown | 37 | 64.9 | 37 | 64.9 | 1,408 | 50.6 | 151 | 43.0 | |
| NCBI genome | Annotations | 306 | 71.6 | 271 | 70.5 | 3,320 | 57.1 | 269 | 40.9 |
See text for definitions of PHX, PMX, and PA gene classes. For the gene classes PHX, PHX(RP−), and PA, SD% in bold indicates a significant increase over the group PMX, while underlining signifies a significant decrease (P < 0.05 for a χ2 test using the Yates correction). PHX(RP−), PHX excluding RP genes.
Number of genes comprising at least 100 codons.
Naturally, both the essential and the verified data sets have much higher fractions of PHX genes, and thus higher overall SD%, than do the other data sets (Table 3). However, the PHX genes in all these data sets registered an even higher SD% than the PMX or PA genes (Table 3). Furthermore, the collection of 591 correctly annotated genes in the verified set displayed a significant positive correlation between SD presence and predicted expression levels by logistic regression analysis (β = 0.62, standard error = 0.22; P < 0.005).
To further reduce potential errors caused by annotation inaccuracies, we compiled a “single-start genes” data set for each genome, which consists of genes with a single start codon (AUG, GUG, or UUG as the first codon) within 90 nucleotides of their annotations. Of the 65 wrongly annotated genes in the E. coli “verified” data set, the correct start was found within 30 codons of the annotations for 54 (83%). Therefore, the single-start genes may have a chance of <0.02 of being wrongly annotated if the error rate for the genome annotations is 10% or about only 0.04 if the error rate reaches as high as 25% in certain genomes, as estimated by some authors (3). In general, these genes constitute about 26% of a genome (29% PHX genes, 25% PMX, and 26% PA; see Supplementary Data Table S-2). Compared to the whole-genome data, they registered highly comparable SD% for the three gene classes PHX, PMX, and PA, indicating that the inaccuracies in start site prediction could only slightly affect the validity of our results obtained from genome annotations (see Supplementary Data Table S-2).
There was also evidence suggesting that wrong starts are likely to be distributed evenly among the different classes of genes (PHX, PMX, and PA) that we used and thus would not significantly affect our comparisons of SD presence between PHX and PMX or PA gene classes. Of the 65 E. coli genes with incorrect starts mentioned above, 20% were PHX, 77% were PMX, and 3% were PA, indicating that incorrect annotations do not tend to bias strongly toward PMX or PA genes.
Taken together, our results on the correlation of SD presence and predicted expression levels have been verified by both human-curated E. coli data sets and the high-quality single-start gene data sets. The validity of the results holds despite the existence of a few incorrectly predicted gene start sites in the genome data.
It is also evident that the increased SD% for PHX genes is not due solely to the presence of RP genes, as shown in Table 3 for Escherichia coli K-12. The collection of PHX genes, excluding RP genes, achieved an SD% similar to that of the complete PHX class for the verified, essential, and whole-genome data sets (Table 3).
The results corroborate our assignment of genes as PHX based on codon usage, even in the many prokaryotes for which little direct information on protein abundances is available. Although many factors affect protein abundances, a high rate of translational initiation is essential to achieve a high level of expression and is the factor most simply observed by genome analysis.
SD sequences for PHX and PMX genes.
We also tried to determine whether the SD sequences of RP and PHX genes are stronger than those of PMX genes in terms of base-pairing potential with the anti-SD sequence and with respect to their aligned spacings, which reflect the two major determinants of the strength of an SD sequence (17). Ringquist et al. (34) showed experimentally that the SD sequence UAAGGAGG is about fourfold more effective than AAGGA. The former SD has a ΔGSD of −12 kcal/mol, while the latter has a ΔGSD of −5.3 kcal/mol. Spacing has a substantial effect only when the SD sequence is short (17). Experimental evidence demonstrated that an aligned spacing of 8 to 10 bases is optimal for E. coli genes (7, 34).
We first determined the OAS for each genome based on the distribution of SD spacings for all the genes in general and the PHX and RP gene classes in particular. The genomes of Escherichia coli K-12 and Pyrococcus abyssi are shown as two examples in Fig. 1. The OAS are 7, 8, and 9 bases for Escherichia coli K-12 and 9, 10, and 11 bases for Pyrococcus abyssi (Fig. 1B and C). Notably, 6, 7, and 8 bases are the most occupied SD spacings for PMX genes from Escherichia coli K-12, whereas 7, 8, and 9 bases are preferred by PHX and RP genes (Fig. 1B). In fact, no SD sequence for the Escherichia coli K-12 RP genes occurs at an aligned spacing of 6 bases.
Assuming that the SD sequences for RP genes are the most optimal, the three aligned spacings of 7, 8, and 9 bases were chosen as the OAS for SD sequences in Escherichia coli K-12. These OAS agree excellently with experimental evidence that 8 to 10 bases are optimal for SD sequences in Escherichia coli K-12 genes (7, 34). These also indicate that SD sequences for PHX genes may have a distribution closer to the actual optimal spacings than PMX genes.
For the genomes of Haemophilus influenzae, Vibrio cholerae, Campylobacter jejuni, Helicobacter pylori 26695, Chlamydophila pneumoniae, and Chlamydia trachomatis, the OAS were determined in a way similar to that used for Escherichia coli K-12. In other genomes, the OAS were aligned spacings occupied by the largest fraction of SD sequences for both PHX and PMX genes, e.g., for Pyrococcus abyssi (Fig. 1C; see also Supplementary Data Fig. S-2). However, the SD sequences in the genomes of Mycoplasma genitalium and Pyrobaculum aerophilum were spread to all positions. Their OAS were chosen in the same way but may not represent optimal spacings (see Supplementary Data Fig. S-3).
Table 1 displays the OAS for each genome. In general, bacterial genomes attain similar OAS, with position 8 being the most common optimal spacing. Archaeal genomes show a preference for OAS about 2 bases longer than that of most bacterial genomes, usually at positions of 9 to 11 bases (Table 1, Fig. 1B and C).
We display in Fig. 2 for each genome the mean ΔGSD of the SD sequences and the frequencies of the SD sequences at the OAS (designated OAS%) in RP, PHX, and PMX genes. The mean ΔGSD indicates the average affinity of the SD sequences for a given gene class. The 30 genomes are divided into three groups (Fig. 2). The first group consists of the proteobacteria. Their SD sequences were among the weakest, with a mean ΔGSD of −6.5 kcal/mol, and about 50% to 70% occurred at the OAS. The most common SD sequence for these genomes was AGGAG (ΔGSD = −6.5 kcal/mol). In comparison, AGGAGG had a ΔGSD of −9.8 kcal/mol. It is also noteworthy that these genomes were highly similar in SD sequences for all three classes of genes (Fig. 2).
FIG. 2.

SD sequences for RP, PHX, and PMX gene classes. (A) The y axis, OAS%, is the fraction of SD sequences present at the three OAS (given in Table 1) for each gene class. * indicates genomes where the OAS% for RP is significantly higher than for PMX genes (P < 0.05 for a χ2 test using the Yates correction). ** indicates that the OAS% for both the RP and PHX genes are significantly higher than for the PMX genes. (B) The y axis shows mean ΔGSD, the mean free energy of binding of the SD sequences in a gene group. * indicates genomes where the mean ΔGSD for the RP genes is significantly less than that for the PMX genes (the difference is at least 20% of the bacterial mean ΔGSD, or 1.3 kcal/mol); ** indicates that the mean ΔGSD for both the RP and PHX genes is significantly less than for the PMX genes. Abbreviations: ESCCO, Escherichia coli; HAEIN, Haemophilus influenzae; VIBCH, Vibrio cholerae; PSEAE, Pseudomonas aeruginosa; CAMJE, Campylobacter jejuni; HELPY, Helicobacter pylori; RICPR, Rickettsia prowazekii; NEIME, Neisseria meningitidis; CHLPN, Chlamydophila pneumoniae; CHLTR, Chlamydia trachomatis; BORBU, Borrelia burgdorferi; TREPA, Treponema pallidum; BACSU, Bacillus subtilis; MYCGE, Mycobacterium genitalium; MYCPN, Mycobacterium pneumoniae; UREUR, Ureaplasma urealyticum; MYCTU, Mycobacterium tuberculosis; SYNSQ, Synechocystis sp. strain PCC6803; DEIRA, Deinococcus radiodurans; AQUAE, Aquifex aeolicus; THEMA, Thermotoga maritima; METJA, Methanococcus jannaschii; METTH, Methanobacterium thermoautotrophicum; ARCFU, Archaeoglobus fulgidus; PYRAB, Pyrococcus abyssi; PYRHO, Pyrococcus horikoshii; THEAC, Thermoplasma acidophilum; HALSP, Halobacterium sp. strain NRC-1; SULSO, Sulfolobus solfataricus; PYRAE, Pyrococcus aerophilum.
The second group included the other bacteria except Aquifex aeolicus and Thermotoga maritima. The SD sequences in these genomes were more variable in the mean ΔGSD and with an OAS% of around 40%. Bacillus subtilis was the only genome in this cluster to have very strong SD sequences (lower mean ΔGSD).
The third group consisted of Aquifex aeolicus, Thermotoga maritima, and all the archaea. The SD sequences in this cluster were the strongest, except for the genomes with a very low genome SD% (Halobacterium sp. strain NRC-1, Sulfolobus solfataricus, and Pyrobaculum aerophilum). In Bacillus subtilis, Aquifex aeolicus, Thermotoga maritima, and the euryarchaea, the SD sequences for RP genes were significantly higher in OAS% and significantly lower in mean ΔGSD than the PMX genes. This was mostly valid also for the PHX gene classes in these genomes (Fig. 2). In particular, Bacillus subtilis did not show a significant correlation between SD presence and predicted expression levels (Table 2), but the SD sequences for its PHX genes did tend to be stronger than those of its PMX genes in both ΔGSD and OAS% (Fig. 2). In contrast, the genomes of Mycoplasma genitalium and Pyrobaculum aerophilum appeared to have SD sequences that were weak and not at optimal spacings, even for the PHX and RP genes (Fig. 2). The SD sequences in these genomes may not play any significant role in translation initiation as in other genomes, which is also implied by the logistic regression analysis (Table 2; see below).
It was previously suggested that there is no direct correlation between the affinity of the SD sequence for the anti-SD sequence and the efficiency of initiation complex formation under certain experimental conditions (10). An SD interaction that involves the center of the anti-SD sequence, CCUCC, may be more efficient in facilitating translation initiation than when it involves off-center sequences (24). This could explain the results of Ringquist et al. (34) and also the twofold-higher yields for GAGGU (ΔGSD = −6.6 kcal/mol) than for UAAGG (−4.2 kcal/mol) found by Chen et al. (7). Not coincidentally, the core anti-SD sequence CCUCC provides the greatest contribution to ΔGSD, as a G:C pair is more stable than an A:U pair.
Since a majority of the SD sequences that we detected involved interaction with the core anti-SD sequence, it might be reasonable to speculate that a lower mean ΔGSD indeed signifies a higher efficiency for SD sequences of PHX genes. We also found that, in Escherichia coli K-12, SD sequences for PHX genes had a higher frequency of GGAG and GAGG (24.7%) and a lower frequency of AGGA (5.0%) than the PMX genes (16.7% and 7.8%, respectively). These three SD sequences had the same ΔGSD of −4.4 kcal/mol, but AGGA was apparently a weaker SD sequence than the other two. In fact, 72% of all the SD sequences for PHX genes in Escherichia coli K-12 harbored the core SD motif GGAG or GAGG, compared to 62% for PMX genes. This trend appears to be valid for most genomes, even those for which no significant decreases in the mean ΔGSD were found for PHX genes versus PMX genes, e.g., proteobacterial genomes (see Supplementary Data Fig. S-4). Therefore, it appears that PHX genes tend to have an SD sequence that has higher affinity to the anti-SD sequence, occurs at a more optimal spacing, and involves interaction with the core anti-SD region. Such an SD sequence is very likely to have a higher efficiency in translation initiation.
Variation of SD% for different functional gene classes.
We also tried to find out whether SD presence is correlated with certain gene classes by assessing the SD% for different functional classes defined in the Cluster of Orthologous Groups (COG) database (50, 51). The two COG categories that are persistently highest in SD% are J (translation, ribosome structure, and biogenesis) and C (energy production and conversion) (see Supplementary Data Table S-3), consistent with the recognition that most genes in these groups are PHX (22). In contrast, the COG categories with low SD% include L (DNA replication, recombination and repair), M (cell envelope biogenesis, outer membrane), and I (lipid metabolism) (see Supplementary Data Table S-3). Genes in these classes usually attain the expression levels of PMX genes (22). Thus, variations in SD% for different COG classes seem to reflect an association with the expression levels of the genes in the class.
Relationship between SD presence and start codon.
Most genes rely on AUG as a start codon, while GUG and UUG are used sparsely (Table 4). Moreover, genes with an AUG start codon tend to have a higher SD% than genes with either GUG or UUG. The increase was significant in 12 genomes and most pronounced in the five euryarchaeal genomes with SD% exceeding 40% (Table 4).
TABLE 4.
SD% for genes with different start codonsa
| Genome | AUG
|
GUG
|
UUG
|
|||
|---|---|---|---|---|---|---|
| No. of genes | SD% | No. of genes | SD% | No. of genes | SD% | |
| ESCCO | 3,544 | 60.5 | 612 | 40.8 | 130 | 33.8 |
| HAEIN | 1,523 | 57.0 | 192 | 21.9 | 0 | |
| VIBCH | 3,248 | 49.8 | 161 | 40.4 | 136 | 23.5 |
| PSEAE | 4,938 | 70.0 | 548 | 66.1 | 78 | 73.1 |
| CAMJE | 1,426 | 60.0 | 92 | 59.8 | 128 | 50.8 |
| HELPY | 1,285 | 61.2 | 152 | 47.4 | 126 | 64.3 |
| RICPR | 774 | 18.7 | 54 | 22.2 | 0 | |
| NEIME | 1,880 | 48.5 | 66 | 50.0 | 43 | 30.2 |
| CHLPN | 865 | 42.1 | 110 | 50.9 | 76 | 35.5 |
| CHLTR | 777 | 46.1 | 67 | 62.7 | 31 | 48.4 |
| BORBU | 588 | 54.1 | 79 | 51.9 | 183 | 43.7 |
| TREPA | 600 | 65.3 | 346 | 53.8 | 85 | 37.6 |
| BACSU | 3,185 | 89.4 | 387 | 87.6 | 512 | 90.4 |
| MYCGE | 430 | 11.2 | 36 | 8.3 | 0 | |
| MYCPN | 626 | 18.2 | 30 | 10.0 | 18 | 11.1 |
| UREUR | 565 | 59.3 | 28 | 39.3 | 18 | 44.4 |
| MYCTU | 2,387 | 50.7 | 1,312 | 43.2 | 189 | 47.6 |
| SYNSQ | 2,617 | 26.7 | 548 | 29.9 | 0 | |
| DEIRA | 1,887 | 47.4 | 839 | 34.4 | 377 | 34.2 |
| AQUAE | 1,251 | 49.0 | 159 | 46.5 | 111 | 47.7 |
| THEMA | 1,230 | 89.9 | 421 | 86.2 | 195 | 85.6 |
| METJA | 1,526 | 52.9 | 152 | 22.4 | 2 | |
| METTH | 1,172 | 66.6 | 420 | 41.9 | 275 | 62.5 |
| ARCFU | 1,828 | 48.5 | 523 | 39.0 | 56 | 71.4 |
| PYRAB | 1,315 | 77.4 | 286 | 59.8 | 160 | 48.8 |
| PYRHO | 1,470 | 60.4 | 588 | 41.7 | 0 | |
| THEAC | 979 | 26.0 | 267 | 18.7 | 232 | 25.4 |
| HALSP | 2,055 | 26.3 | 3 | 33.3 | 0 | |
| SULSO | 1,881 | 23.3 | 543 | 18.6 | 552 | 25.7 |
| PYRAE | 1,617 | 24.7 | 883 | 21.1 | 103 | 32.0 |
For genes with AUG, SD% in bold indicates a significant increase over the collection of genes using other start codons, while underlining signifies a significant decrease (P < 0.05 for a χ2 test using the Yates correction). For genes with GUG or UUG, SD% in bold indicates a significant increase over the collection of genes using AUG, while underlining signifies a significant decrease (P < 0.05 for a χ2 test using the Yates correction). See Fig. 2 legend for abbreviations.
Considering that AUG is a more potent initiator than GUG and UUG (34), the weak start codons GUG and UUG, in conjunction with lack of an SD sequence, might substantially reduce the expression level of a given gene. For example, of the 449 genes in Escherichia coli K-12 that start with either GUG or UUG and do not have an SD sequence, 228 are annotated as “orf, hypothetical protein” and many others encode “putative” proteins. Only 2 of these 228 open reading frames (ORFs) are PHX, whereas 21 are PA. Many of these ORFs may code for low-expression proteins or may be wrongly annotated. For these genes, a strong SD sequence could compensate for their weak start codons, especially UUG, as shown in laboratory manipulations (10, 55). Our results suggest that the SD sequence might work in concert with the start codons as part of an elaborate regulatory system for gene expression to maintain different expression levels for different genes.
We have shown that SD presence is significantly correlated with predicted gene expression levels in most prokaryotic genomes. In particular, the RP genes and more generally the PHX genes display a higher SD% than the PMX genes (i.e., the average genes). Also, in some genomes the SD sequences of RP and PHX genes are closer to optimal in both base-pairing potential with the anti-SD sequence and spacing to the start codon (Fig. 2). This provides further evidence that the SD sequence is important in translation of these genes. A strong SD sequence may also work together with other features of the highly expressed genes, e.g., the stronger start codon AUG and favorable secondary structure around the translation initiation region (16), that ameliorate the translation initiation efficiency.
Relationship between SD presence and distance between successive genes.
The intergenic distance (Dg) is another important feature of prokaryotic genes that might correlate with the SD presence. For ease of discussion, we refer to the Dg of gene g as the distance (in base pairs) from g's start codon to the end of its immediate upstream gene in the same orientation. Negative values of Dg signify genes that overlap their immediate upstream genes. In most genomes, the most prevalent value of Dg is −4 bp (the junction is always AUGA; also see reference 38), which is observed for on average 7.8% and as much as 18% for Thermotoga maritima.
The median Dg in a genome varies from 9 bp for Campylobacter jejuni and 11 bp for both Thermotoga maritima and Mycoplasma genitalium to 187 bp for Methanococcus jannaschii and 201 bp for Halobacterium sp. strain NRC-1 (see Supplementary Data Table S-4). In most archaeal genomes, the SD% for genes with a Dg of −4 bp is marked higher than the SD% for all the other genes, at a level comparable to the SD% of the RP genes. In contrast, many genomes recorded a reduced SD% for the collection of genes with a Dg of >20 bp, compared to genes with a Dg of <20 bp. This is especially valid for all the archaeal genomes (see Supplementary Data Table S-4).
We then assessed SD% for genes with different Dg ranges. Since the SD% does not show much variation among the groups with a Dg of greater than 30 bp, we focused on genes with a Dg of below 30 bp, which on average constitute 35% of a genome. We divided all the genes in a genome into seven Dg groups: genes with a Dg below −20 bp; five groups with a Dg of from −20 to 30 bp, with 10-bp intervals; and genes with a Dg exceeding 30 bp (see Supplementary Data Table S-5). In most genomes, each group contained more than 30 genes. The gene group with a Dg of −10 to 0 bp was the largest among the five groups of 10-bp intervals. Figure 3 shows the SD% for these Dg groups.
FIG. 3.

Relationship between SD% and distances between successive genes (Dg). The y axis represents SD%. The symbols for the lines and points for each plot are shown. In each plot, the seven data points represent seven Dg groups (from left to right): genes with a Dg of less than −20 bp; five groups of genes with a Dg from −20 to 30 bp, at 10-bp intervals; and genes with a Dg of more than 30 bp (see Supplementary Data Table S-5 for details of the groups). For abbreviations, see the legend to Fig. 2.
In bacterial genomes, the first group (Dg of below −20 bp) persistently carried a much reduced SD%, except for Pseudomonas aeruginosa, Aquifex aeolicus, and Mycoplasma genitalium (Fig. 3A to E). One possible explanation for the low SD% is that many of these genes might be incorrectly annotated. At the other end, the last group (genes with a Dg in excess of 30 bp) contained about 60% to 80% of the genome and had an SD% at about the genome level. In 16 genomes, the group with a Dg of −10 to 0 bp was significantly higher in SD% over the genome level. The groups with a Dg of between 10 and 20 bp were significantly higher in SD% for 10 bacterial and three archaeal genomes (see Supplementary Data Table S-5). The increased SD% in these Dg groups were not due to higher expression levels (data not shown). Of particular interest was the genome of Mycoplasma genitalium, which contained 75 genes with a Dg of between −10 and 0 bp, of which 25% had an SD sequence (Fig. 3E). Whether these SD sequences are functional remains unclear.
Genes with a Dg of 0 to 20 bp may have strong biases in base composition in their translation initiation region because their 5′ end is located in the regions around the stop codon of the upstream gene (49). Rocha et al. (35) found that the 6 bases following the stop codon in Bacillus subtilis genes are AU rich. Such biases could discount the occurrence of an SD sequence, which might be the reason for the somewhat reduced SD% for the group with a Dg of 0 to 10 bp in bacterial genomes (Fig. 3). On the other hand, Eyre-Walker (12) showed that Escherichia coli K-12 genes overlapping a downstream gene tend to have low codon preferences at the 3′ end, which would more easily enable the presence of an SD for the downstream gene (e.g., with a Dg of −20 to 0 bp).
The archaeal genomes revealed a common trend distinctive from the bacteria. The genes with a Dg of less than 20 bp (Fig. 3F) or less than 10 bp (Fig. 3G) were strongly biased with an extant SD compared to genes with a larger Dg. This was even more emphatic for genomes with less than 30% overall SD%, especially for gene groups with a Dg of between −20 and 10 bp (Fig. 3G). These increased SD% were again not correlated with higher expression levels (data not shown). It is interesting that Bacillus subtilis, Aquifex aeolicus, and Thermotoga maritima were distinctively like bacteria in their relationship between Dg and SD presence (Fig. 3D), even though they were very similar to the archaea in the SD sequences with respect to ΔGSD and OAS (Table 1; Fig. 2). Thus, the parameters of translation initiation do not sort along simple phylogenetic lines.
Relationship between SD presence and operon structure.
The greatly increased SD presence in genes in close proximity to their upstream genes led us to investigate the connection between the SD sequence and operon structure. Apparently many genes in the groups with a Dg of −20 to 20 bp are genes within operons (38). It has been suggested that operon structure might have arisen during the evolution of both bacteria and archaea by thermoreduction from a common thermophilic ancestor (14). The operon structures in the two kingdoms thus might have some common features, such as the SD sequence. The high SD presence suggests that the SD sequences may play an essential role in translation of these genes.
We analyzed SD sequences for 391 documented operons from Escherichia coli K-12 (each with at least two genes) extracted from the RegulonDB database (39). Of the 601 internal genes within these operons, 69.2% had a Dg of between −20 and 30 bp, compared to only 6.6% of the 391 initial operon genes. The SD% was 71.0% for genes within operons and 67.3% for initial genes.
We then conducted a more general analysis over the 30 genomes. Based on the Dg, we partitioned the genes in a genome into three classes, types I to III, as illustrated in Fig. 4A. Type I consists of genes at least 100 bp in distance from both the upstream and downstream genes; type I genes are presumably single genes. Type II consists of genes with a Dg larger than 50 bp and followed by at least two consecutive downstream genes with a Dg below 20 bp; type II genes are likely initial genes of operons. Type III comprises all genes with a Dg below 20 bp following a type II gene; type III genes are likely genes within operons. The three classes encompass about half of a genome. We found that more than one third of the type II and type III genes in Escherichia coli K-12 were present in the 391 known operons, and most of them were also predicted to be operons by Salgado's method (38). On average, there were three type III genes following each type II gene (see Supplementary Data Table S-6). Figure 4B presents the SD% for these three gene classes.
FIG. 4.

SD sequences for genes with different internal positions. (A) How the three types of genes were classified (see text for details). (B) Asterisks indicate genomes where the SD% for type III genes is significantly higher than that for type I genes. Boldface indicates that the SD% for the type II genes was significantly higher than for the type I genes (P < 0.05 for a χ2 test using the Yates correction). For abbreviations, see the legend to Fig. 2.
Type II genes always attain an SD% about the same as or lower than that of type I genes in most genomes. In fact, only Chlamydia trachomatis, Halobacterium sp. strain NRC-1, and Sulfolobus solfataricus recorded significantly higher SD% for type II genes than for type I genes (Fig. 4B). It appears that initial genes of operons are similar to single genes in SD presence. This may be expected because these genes are both at the start of a transcript. In contrast, type III genes recorded significantly higher SD% than type I genes in all the thermophiles, from Deinococcus radiodurans to Pyrobaculum aerophilum, and four other bacteria: Escherichia coli K-12, Haemophilus influenzae, Vibrio cholerae, and Mycoplasma genitalium (Fig. 4B). These results imply that the presence of an SD sequence is especially conserved for genes within operons in bacterial and archaeal genomes, prominently for thermophiles.
This conservation was even more significant in the genomes where the overall SD% was very low and/or no correlation between the SD presence and predicted expression levels was observed. Such genomes included those of Mycoplasma genitalium, Mycoplasma pneumoniae, Synechocystis sp. strain PCC6803, Halobacterium sp. strain NRC-1, Sulfolobus solfataricus, and Pyrobaculum aerophilum (Fig. 2, 3, and 4B). Thus, it is tempting to speculate that the SD sequence may have coevolved with the operon gene structure in both bacteria and archaea (14). The correlation of SD presence with gene expression levels might have been established later. This would explain the observation that, in all archaeal genomes and Aquifex aeolicus, PHX genes with a Dg of below 50 bp recorded a significantly higher SD% than other PHX genes (data not shown). The RP genes are both highly expressed and profusely expressed in operons, and not surprisingly, they always attained the highest SD% (Table 2).
The archaeal genomes provide an excellent system with which to analyze the evolution of both the SD sequence and the bacterial translation mechanism utilizing the SD-anti-SD interaction. Some euryarchaea (Thermoplasma acidophilum and Halobacterium sp. strain NRC-1), and especially crenarchaea (Sulfolobus solfataricus and Pyrobaculum aerophilum), seem to have gradually lost conservation of both the anti-SD and the SD sequences (Table 1; Fig. 2). Accumulating evidence suggests that many single genes, or initial genes of operons, in these genomes are translated through leaderless mRNA by mechanisms that do not involve the SD-anti-SD interaction (45, 47, 52). The SD sequence may thus become dispensable for these genes. However, for genes within operons, the SD sequence appears to be particularly important, evidenced by the prevalence of the SD motifs in those genes (Fig. 3F and G). Experimental evidence supporting this hypothesis has been reported for Sulfolobus solfataricus (8).
SD presence and other gene features.
It has been suggested that the SD sequence is especially important in a genome where an S1 ribosomal protein is missing, e.g., Bacillus subtilis, which has only a reduced S1 homologue and achieves the second highest SD% of all the genomes (Table 1) (35). However, we did not find such a correlation for other genomes. Three bacteria (Ureaplasma urealyticum, Mycoplasma genitalium, and Mycoplasma pneumoniae) and all archaeal genomes did not have an S1 or any S1 homologues. But, unlike Bacillus subtilis, the genomes of Ureaplasma urealyticum, Mycoplasma genitalium, and Mycoplasma pneumoniae recorded a very low SD% (Table 1). On the other hand, genomes with an S1 gene can achieve very high SD%, e.g., Thermotoga maritima, which had the highest SD% (Table 1). Thus, SD presence is not correlated with the presence or absence of an S1 RP gene. Also, the SD sequence seems to be uncorrelated with factors such as copy number of the 16S rRNA, G+C content, total number of genes, gene length, or lifestyle (data not shown).
Further comments.
Given the correlation between the SD sequence and other gene features, especially expression levels and distances between successive genes, it is suggested that the SD sequence should be incorporated in algorithms for gene start determination, expression level prediction, and operon prediction to improve accuracy. Most of the genomes studied in this report were annotated with the programs GeneMark (20, 26) and GLIMMER (40) or a combination of automatic gene-finding methods and similarity searches in protein databases. Now SD information has been incorporated in recent programs, such as GeneMark.hmm and GeneMarkS (3, 25). It appears to work well for genomes with high SD%, such as low-G+C gram-positive bacteria (e.g., Listeria monocytogenes [15]). However, for many genomes, the SD% is around 30 to 50% and thus would provide only marginal improvements (36).
On the other hand, the relationship between SD presence and intergenic distances may contribute greatly to operon predictions, an important part of prokaryotic genomics. No highly reliable method to date has been developed for operon prediction (38). Also, little is known about operons in archaeal genomes. Our findings that archaeal genes that are presumably within operons have remarkably increased SD presence should help in developing an effective method for operon characterization in these genomes.
Recently, the crystal structures of both the 50S and 30S complexes of the bacterial ribosome have been determined at high resolution (2, 41, 56). A structure of the 80S ribosome from Saccharomyces cerevisiae was also reported (48). These accomplishments greatly augment our understanding of the mechanisms of protein synthesis at the atomic level (5, 6, 29-31, 33, 44). Furthermore, Yusupova et al. (58) directly observed the path of mRNA in the 70S ribosome from Thermus thermophilus at 7 Å resolution. The model mRNA was based on the phage T4 gene 32 mRNA except that the SD sequence was expanded to AAGGAGGU. They found that about 30 nucleotides are bound to the 30S subunit (15 bp upstream of the initiator to 15 bp downstream), which is roughly the whole translation initiation region. The SD interaction was clearly observed to form a helix, which was accommodated in a cleft formed by 16S rRNA elements and the ribosomal proteins S11 and S18 (58). These results provide additional proof that the SD interaction can be an important part of translation initiation.
The SD sequence in the mRNA, AAGGAGGU, had an aligned spacing of 7 bases. It is interesting that of the 67 AAGGAGGU SD sequences in the 21 bacterial genomes (Table 1), only 4 occurred at an aligned spacing of 7 bases, while 10, 19, and 12 conferred 8, 9, and 10 bases of spacing, respectively. A total of 55 (82.1%) were present at a spacing larger than 7 bases. Thus, most likely an aligned spacing of 9 bases should be more preferable for the mRNA in the structure. There are apparently structural constraints that require such an optimal spacing, and three-dimensional simulation studies based on the structure using different SD sequences and spacings could provide insights into these structural constraints and a better understanding of the SD interaction.
Acknowledgments
This work was supported in part by National Institutes of Health grants 5R01GM10452-38 and 5R01HG00335-13.
REFERENCES
- 1.Antelmann, H., J. Bernhardt, R. Schmid, H. Mach, U. Volker, and M. Hecker. 1997. First steps from a two-dimensional protein index towards a response-regulation map for Bacillus subtilis. Electrophoresis 18:1451-1463. [DOI] [PubMed] [Google Scholar]
- 2.Ban, N., P. Nissen, J. Hansen, P. B. Moore, and T. A. Steitz. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 289:905-920. [DOI] [PubMed] [Google Scholar]
- 3.Besemer, J., A. Lomsadze, and M. Borodovsky. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29:2607-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 5.Brodersen, D. E., W. M. Clemons, Jr., A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly, and V. Ramakrishnan. 2000. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103:1143-1154. [DOI] [PubMed] [Google Scholar]
- 6.Carter, A. P., W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly, and V. Ramakrishnan. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407:340-348. [DOI] [PubMed] [Google Scholar]
- 7.Chen, H., M. Bjerknes, R. Kumar, and E. Jay. 1994. Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res. 22:4953-4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Condo, I., A. Ciammaruconi, D. Benelli, D. Ruggero, and P. Londei. 1999. Cis-acting signals controlling translational initiation in the thermophilic archaeon Sulfolobus solfataricus. Mol. Microbiol. 34:377-384. [DOI] [PubMed] [Google Scholar]
- 9.de Smit, M. H., and J. van Duin. 1993. Translational initiation at the coat-protein gene of phage MS2: native upstream RNA relieves inhibition by local secondary structure. Mol. Microbiol. 9:1079-1088. [DOI] [PubMed] [Google Scholar]
- 10.de Smit, M. H., and J. van Duin. 1994. Translational initiation on structured messengers. Another role for the Shine-Dalgarno interaction. J. Mol. Biol. 235:173-184. [DOI] [PubMed] [Google Scholar]
- 11.Draper, D. E. 1996. Translational initiation, p. 902-908. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 12.Eyre-Walker, A. 1996. The close proximity of Escherichia coli genes: consequences for stop codon and synonymous codon use. J. Mol. Evol. 42:73-78. [DOI] [PubMed] [Google Scholar]
- 13.Freier, S. M., R. Kierzek, J. A. Jaeger, N. Sugimoto, M. H. Caruthers, T. Neilson, and D. H. Turner. 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA 83:9373-9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glansdorff, N. 1999. On the origin of operons and their possible role in evolution toward thermophily. J. Mol. Evol. 49:432-438. [DOI] [PubMed] [Google Scholar]
- 15.Glaser, P., L. Frangeul, C. Buchrieser, C. Rusniok, A. Amend, F. Baquero, P. Berche, H. Bloecker, P. Brandt, T. Chakraborty, A. Charbit, F. Chetouani, E. Couve, A. de Daruvar, P. Dehoux, E. Domann, G. Dominguez-Bernal, E. Duchaud, L. Durant, O. Dussurget, K. D. Entian, H. Fsihi, F. G. Portillo, P. Garrido, L. Gautier, W. Goebel, N. Gomez-Lopez, T. Hain, J. Hauf, D. Jackson, L. M. Jones, U. Kaerst, J. Kreft, M. Kuhn, F. Kunst, G. Kurapkat, E. Madueno, A. Maitournam, J. M. Vicente, E. Ng, H. Nedjari, G. Nordsiek, S. Novella, B. de Pablos, J. C. Perez-Diaz, R. Purcell, B. Remmel, M. Rose, T. Schlueter, N. Simoes, A. Tierrez, J. A. Vazquez-Boland, H. Voss, J. Wehland, and P. Cossart. 2001. Comparative genomics of Listeria species. Science 294:849-852. [DOI] [PubMed] [Google Scholar]
- 16.Gold, L. 1988. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu. Rev. Biochem. 57:199-233. [DOI] [PubMed] [Google Scholar]
- 17.Gualerzi, C. O., and C. L. Pon. 1990. Initiation of mRNA translation in prokaryotes. Biochemistry 29:5881-5889. [DOI] [PubMed] [Google Scholar]
- 18.Hosmer, D., and S. Lemeshow. 2000. Applied logistic regression, 2nd ed. John Wiley & Sons, New York, N.Y.
- 19.Hui, A., and H. A. de Boer. 1987. Specialized ribosome system: preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA 84:4762-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isono, K., J. D. McIninch, and M. Borodovsky. 1994. Characteristic features of the nucleotide sequences of yeast mitochondrial ribosomal protein genes as analyzed by computer program GeneMark. DNA Res. 1:263-269. [DOI] [PubMed] [Google Scholar]
- 21.Jacques, N., and M. Dreyfus. 1990. Translation initiation in Escherichia coli: old and new questions. Mol. Microbiol. 4:1063-1067. [DOI] [PubMed] [Google Scholar]
- 22.Karlin, S., and J. Mrázek. 2000. Predicted highly expressed genes of diverse prokaryotic genomes. J. Bacteriol. 182:5238-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlin, S., J. Mrázek, A. Campbell, and D. Kaiser. 2001. Characterizations of highly expressed genes of four fast-growing bacteria. J. Bacteriol. 183:5025-5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozak, M. 1983. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 47:1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukashin, A. V., and M. Borodovsky. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26:1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McIninch, J. D., W. S. Hayes, and M. Borodovsky. 1996. Applications of GeneMark in multispecies environments. Proc. Int. Conf. Intell. Syst. Mol. Biol. 4:165-175. [PubMed] [Google Scholar]
- 27.Mrazek, J., D. Bhaya, A. R. Grossman, and S. Karlin. 2001. Highly expressed and alien genes of the Synechocystis genome. Nucleic Acids Res. 29:1590-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mrazek, J., and S. Karlin. 1999. Detecting alien genes in bacterial genomes. Ann. N. Y. Acad. Sci. 870:314-329. [DOI] [PubMed] [Google Scholar]
- 29.Muth, G. W., L. Ortoleva-Donnelly, and S. A. Strobel. 2000. A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science 289:947-950. [DOI] [PubMed] [Google Scholar]
- 30.Nissen, P., J. Hansen, N. Ban, P. B. Moore, and T. A. Steitz. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science 289:920-930. [DOI] [PubMed] [Google Scholar]
- 31.Ogle, J. M., D. E. Brodersen, W. M. Clemons, Jr., M. J. Tarry, A. P. Carter, and V. Ramakrishnan. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292:897-902. [DOI] [PubMed] [Google Scholar]
- 32.Osada, Y., R. Saito, and M. Tomita. 1999. Analysis of base-pairing potentials between 16S rRNA and 5′ UTR for translation initiation in various prokaryotes. Bioinformatics 15:578-581. [DOI] [PubMed] [Google Scholar]
- 33.Ramakrishnan, V., and P. B. Moore. 2001. Atomic structures at last: the ribosome in 2000. Curr. Opin. Struct. Biol. 11:144-154. [DOI] [PubMed] [Google Scholar]
- 34.Ringquist, S., S. Shinedling, D. Barrick, L. Green, J. Binkley, G. D. Stormo, and L. Gold. 1992. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol. Microbiol. 6:1219-1229. [DOI] [PubMed] [Google Scholar]
- 35.Rocha, E. P., A. Danchin, and A. Viari. 1999. Translation in Bacillus subtilis: roles and trends of initiation and termination, insights from a genome analysis. Nucleic Acids Res. 27:3567-3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruepp, A., W. Graml, M. L. Santos-Martinez, K. K. Koretke, C. Volker, H. W. Mewes, D. Frishman, S. Stocker, A. N. Lupas, and W. Baumeister. 2000. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 407:508-513. [DOI] [PubMed] [Google Scholar]
- 37.Saito, R., and M. Tomita. 1999. Computer analyses of complete genomes suggest that some archaebacteria employ both eukaryotic and eubacterial mechanisms in translation initiation. Gene 238:79-83. [DOI] [PubMed] [Google Scholar]
- 38.Salgado, H., G. Moreno-Hagelsieb, T. F. Smith, and J. Collado-Vides. 2000. Operons in Escherichia coli: genomic analyses and predictions. Proc. Natl. Acad. Sci. USA 97:6652-6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salgado, H., A. Santos-Zavaleta, S. Gama-Castro, D. Millan-Zarate, E. Diaz-Peredo, F. Sanchez-Solano, E. Perez-Rueda, C. Bonavides-Martinez, and J. Collado-Vides. 2001. RegulonDB (version 3.2): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res. 29:72-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salzberg, S. L., A. L. Delcher, S. Kasif, and O. White. 1998. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 26:544-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schluenzen, F., A. Tocilj, R. Zarivach, J. Harms, M. Gluehmann, D. Janell, A. Bashan, H. Bartels, I. Agmon, F. Franceschi, and A. Yonath. 2000. Structure of functionally activated small ribosomal subunit at 3.3 angstrom resolution. Cell 102:615-623. [DOI] [PubMed] [Google Scholar]
- 42.Schmid, R., J. Bernhardt, H. Antelmann, A. Volker, H. Mach, U. Volker, and M. Hecker. 1997. Identification of vegetative proteins for a two-dimensional protein index of Bacillus subtilis. Microbiology 143:991-998. [DOI] [PubMed] [Google Scholar]
- 43.Schurr, T., E. Nadir, and H. Margalit. 1993. Identification and characterization of E. coli ribosomal binding sites by free energy computation. Nucleic Acids Res. 21:4019-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sengupta, J., R. K. Agrawal, and J. Frank. 2001. Visualization of protein S1 within the 30S ribosomal subunit and its interaction with messenger RNA. Proc. Natl. Acad. Sci. USA 98:11991-11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sensen, C. W., H. P. Klenk, R. K. Singh, G. Allard, C. C. Chan, Q. Y. Liu, S. L. Penny, F. Young, M. E. Schenk, T. Gaasterland, W. F. Doolittle, M. A. Ragan, and R. L. Charlebois. 1996. Organizational characteristics and information content of an archaeal genome: 156 kb of sequence from Sulfolobus solfataricus P2. Mol. Microbiol. 22:175-191. [DOI] [PubMed] [Google Scholar]
- 46.Shine, J., and L. Dalgarno. 1974. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. USA 71:1342-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slupska, M. M., A. G. King, S. Fitz-Gibbon, J. Besemer, M. Borodovsky, and J. H. Miller. 2001. Leaderless transcripts of the crenarchaeal hyperthermophile Pyrobaculum aerophilum. J. Mol. Biol. 309:347-360. [DOI] [PubMed] [Google Scholar]
- 48.Spahn, C. M., R. Beckmann, N. Eswar, P. A. Penczek, A. Sali, G. Blobel, and J. Frank. 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae tRNA-ribosome and subunit-subunit interactions. Cell 107:373-386. [DOI] [PubMed] [Google Scholar]
- 49.Tate, W. P., and S. A. Mannering. 1996. Three, four or more: the translational stop signal at length. Mol. Microbiol. 21:213-219. [DOI] [PubMed] [Google Scholar]
- 50.Tatusov, R. L., E. V. Koonin, and D. J. Lipman. 1997. A genomic perspective on protein families. Science 278:631-637. [DOI] [PubMed] [Google Scholar]
- 51.Tatusov, R. L., D. A. Natale, I. V. Garkavtsev, T. A. Tatusova, U. T. Shankavaram, B. S. Rao, B. Kiryutin, M. Y. Galperin, N. D. Fedorova, and E. V. Koonin. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29:22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolstrup, N., C. W. Sensen, R. A. Garrett, and I. G. Clausen. 2000. Two different and highly organized mechanisms of translation initiation in the archaeon Sulfolobus solfataricus. Extremophiles 4:175-179. [DOI] [PubMed] [Google Scholar]
- 53.VanBogelen, R. A., K. Z. Abshire, A. Pertsemlidis, R. L. Clark, and F. C. Neidhardt. 1996. Gene-protein database of Escherichia coli K-12, edition 6, p. 2067-2117. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 54.VanBogelen, R. A., E. E. Schiller, J. D. Thomas, and F. C. Neidhardt. 1999. Diagnosis of cellular states of microbial organisms using proteomics. Electrophoresis 20:2149-2159. [DOI] [PubMed] [Google Scholar]
- 55.Weyens, G., D. Charlier, M. Roovers, A. Pierard, and N. Glansdorff. 1988. On the role of the Shine-Dalgarno sequence in determining the efficiency of translation initiation at a weak start codon in the car operon of Escherichia coli K12. J. Mol. Biol. 204:1045-1048. [DOI] [PubMed] [Google Scholar]
- 56.Wimberly, B. T., D. E. Brodersen, W. M. Clemons, Jr., R. J. Morgan-Warren, A. P. Carter, C. Vonrhein, T. Hartsch, and V. Ramakrishnan. 2000. Structure of the 30S ribosomal subunit. Nature 407:327-339. [DOI] [PubMed] [Google Scholar]
- 57.Wood, C. R., M. A. Boss, T. P. Patel, and J. S. Emtage. 1984. The influence of messenger RNA secondary structure on expression of an immunoglobulin heavy chain in Escherichia coli. Nucleic Acids Res. 12:3937-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yusupova, G. Z., M. M. Yusupov, J. H. Cate, and H. F. Noller. 2001. The path of messenger RNA through the ribosome. Cell 106:233-241. [DOI] [PubMed] [Google Scholar]