

Abstract
The cell cycle is an intricate process of DNA replication and cell division that concludes with the formation of two genetically equivalent daughter cells. In this progression, the centrosome is duplicated once and only once during the G1/S transition to produce the bipolar spindle and ensure proper chromosome segregation. The presence of multiple centrosomes in cancer cells suggests that this process is mis-regulated during carcinogenesis. This suggests that certain factors exist that license the progression of centrosome duplication and serve to inhibit further duplications during a single cell cycle. Recent studies suggest that the Ran/Crm1 complex not only regulates nucleocytoplasmic transport but is also independently involved in mitotic spindle assembly. Factors that are capable of interacting with Ran/Crm1 through their nuclear export sequences, such as cyclins/cdks, p53 and Brca1/2, may potentially function as centrosome checkpoints akin to the G1/S and G2/M checkpoints of the cell cycle. Our recent findings indicate that nucleophosmin, a protein whose trafficking is mediated by the Ran/Crm1 network, is one of such checkpoint factors for maintaining proper centrosome duplication. We propose that Ran/Crm1 may act as a ‘loading dock’ to coordinate various checkpoint factors in regulating the fidelity of centrosome duplication during cell cycle progression, and the disruption of these processes may lead to genomic instability and an acceleration of oncogenesis.
Keywords: nucleophosmin, Crm1, Ran, mitosis, centrosome, hepatocellular carcinoma, liver cancer
CELL CYCLE, CENTROSOME DUPLICATION AND GENOMIC INSTABILITY
The cell cycle is a highly coordinated process involved in the growth and proliferation of cells, organizational development and regulation of DNA damage repair. This multi-stage process culminates in mitosis during which a bipolar mother cell partitions into two genetically equivalent daughter cells.1,2 To ensure the fidelity of this process, major pathways referred to as cell cycle checkpoints occur at the G1/S and G2/M transitions.3 At these checkpoints a variety of factors including cell size, nutrition and DNA status are ascertained and cell cycle arrest, delay or exit are induced if these factors are deemed unfavorable for proper duplication. Numerous regulatory complexes containing cyclins and cyclin dependent kinases direct this sequence of events and are in turn regulated by signaling, sensor and adapter/mediator proteins to produce hierarchical amplified signal transduction pathways that connect the checkpoints with the core cell cycle machinery.
In mammalian cells, two critical events must be completed before mitosis begins: replication of DNA, such that each new daughter cell inherits an identical copy of the genome, and duplication of the centrosome. Centrosomes are the major microtubule organizing centers of mammalian cells and direct the assembly of a bipolar spindle and balance chromosome segregation during mitosis, controlling the number, polarity, orientation and nucleation of microtubules.4 Centrosome duplication begins at G1/S and is completed at S phase, coinciding with DNA replication.5 During the process of bipolar mother cell division to two daughter cells, a complex regulatory network ensures that duplication of the centrosome occurs only once per cell cycle.6 Thus, centrosome duplication and cell cycle progression must be tightly regulated by a fine control network.
The mechanisms underlying the regulation of centrosome duplication is poorly understood. However, many cellular factors have been implicated in this process. These findings imply that there are intricate mechanisms to coordinate and control unwanted centrosome synthesis throughout the cell cycle. Thus, conditions that favor disruption of cell cycle transitions may also contribute to centrosome amplification. This in turn, may result in the initiation of chromosome imbalance, through the formation of multipolar spindles and aberrant mitosis, leading to a random loss or gain of chromosomes.7,8 Therefore disruption of the centrosome synthesis cycle may lead to aneuploidy, an initiation event that facilitates carcinogenesis.
Genomic instability denotes the propensity for genomic changes, often including aneuploidy. The hallmarks of genomic instability include gene amplification and chromosome aberrations.9 Centrosome amplification is associated with aneuploidy,10–12 suggesting that abnormalities in chromosome segregation are due to the presence of abnormal spindles. Since centrosome duplication and DNA replication are tightly coupled, the various features of genomic instability likely result from failure of coordination of S phase events and/or failure of S phase and mitotic checkpoints. The presence of multiple centrosomes in tumor cells suggests that this regulatory network goes awry during carcinogenesis. Consistently, unscheduled activation of certain cell cycle-dependent kinases (cyclin E-cdk2 or cyclin A-cdk2) can result in premature centriole splitting and centrosome over-duplication. Disruption of cell cycle checkpoint proteins with tumor suppressive function, such as p53, APC, Brca1, Brca2 or Gadd45a, or abnormal expression of oncoproteins, such as Mdm2, aurora kinases, viral oncoproteins such as HBx of HBV and HPV E7 are associated with centrosome amplification.13–17 Furthermore, disruptions of genes, such as Mad2 and Bub3 that regulate mitotic spindle checkpoint, can also lead to abnormal mitosis and genomic instability, which facilitate tumorigenesis.18,19 It is plausible that different inhibitors may be responsible for preventing unscheduled centriole splitting in duplicated centrosomes during the cell cycle.
THE ROLE OF NUCLEOCYTOPLASMIC TRANSPORT MACHINARY IN CENTROSOME DUPLICATION AND SPINDLE ASSEMBLY
Recent studies indicate that cellular components that regulate nucleocytoplasmic transport are also independently involved in spindle assembly.20 In principle, these processes are accomplished by specific receptors of the β-importin family, e.g., the importin receptors (importins α and β) that bind to nuclear localization signals (NLS) and the export receptor Crm1 that binds to nuclear export signals (NES). These processes require a small GTPase, Ran, which controls the interaction of these receptors with their substrates. The guanine nucleotide exchange factor RCC1 facilitates Ran binding to Crm1, while RanBP1, a major regulator of Ran, promotes Crm1 dissociation from Ran. Our recent findings indicate that Crm1 may regulate the fidelity of centrosome duplication by acting as a licensing factor to prevent unscheduled duplication.21 A fraction of Ran, Crm1 and RanBP1 is found on centrosomes.21–23 Inactivation of Crm1 either by a Crm1-specific inhibitor, leptomycin B (LMB), or through hepatitis B virus (HBV) HBx protein interaction with its NES motif results in supernumerary centrosomes and multipolar spindles.21,24 Aneuploidy can be detected in livers chronically infected with HBV, a preneoplastic condition that predisposes individuals to develop HCC. Similar multipolar spindles and mitotic abnormalities are also a consequence of Ran mutations or overexpression of RanBP1.23 Thus, the Ran/Crm1 complex may negatively regulate the initiation of centrosome duplication, possibly through its association with NES-containing proteins.21 Our data indicate that inactivation of Crm1 can lead to unscheduled centriole splitting resulting in multipolar spindles, implying that Crm1 may be involved in preventing unscheduled centriole splitting during mitosis, thereby ensuring the formation of bipolar spindles (Fig. 1). Since Ran/Crm1 acts as a receptor to shuttle cellular proteins and many of the known cell cycle regulators can bind to this receptor, it is likely that components of the Ran/Crm1 pathway may function as licensing factors to ensure appropriate centrosome duplication during different stages of the cell cycle (Fig. 2). Thus, one attractive model is that the Ran/Crm1 complex may serve as a loading dock through its NES binding activity to ensure that proteins that regulate centrosome duplication are present at the correct location and time to ensure the fidelity of this process.
Figure 1.
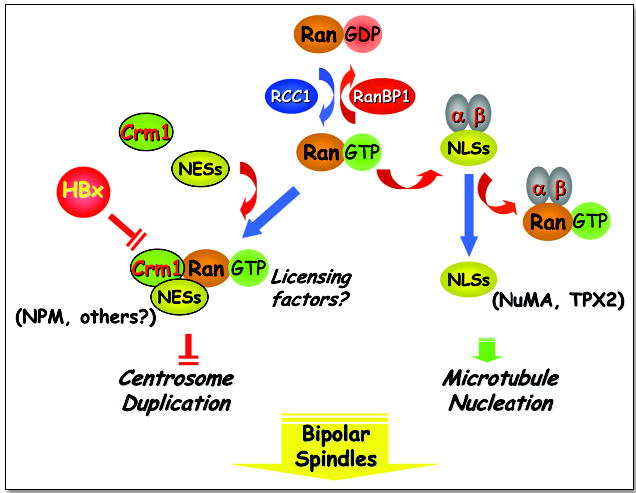
The Ran/Crm1 network: Nucleocytoplasmic transport and mitotic spindle assembly. The small GTPase, Ran, shuttles between an inactive GDP and an active GTP-bound state through interaction with RanBP1 and RCC1 respectively. In its GTP-bound state, Ran can interact with importin receptors (a and b) to promote the cytoplasmic to nuclear transport of proteins containing nuclear localization signals (NLS). The transport of certain NLS-containing proteins such as NuMA and TPX2 can promote microtubule nucleation. Ran-GTP can also interact with the nuclear export receptor, Crm1 that binds to proteins containing nuclear export signals (NES). The hepatitis B viral oncoprotein HBx, interacts with and inactivates Crm1 through its NES, leading to centrosome over-duplication and multipolar spindles. Other NES-containing substrates that bind Crm1, such as nucleophosmin (NPM), may have tumor suppressive effects and function as licensing factors to regulate centrosome duplication during the cell cycle.
Figure 2.
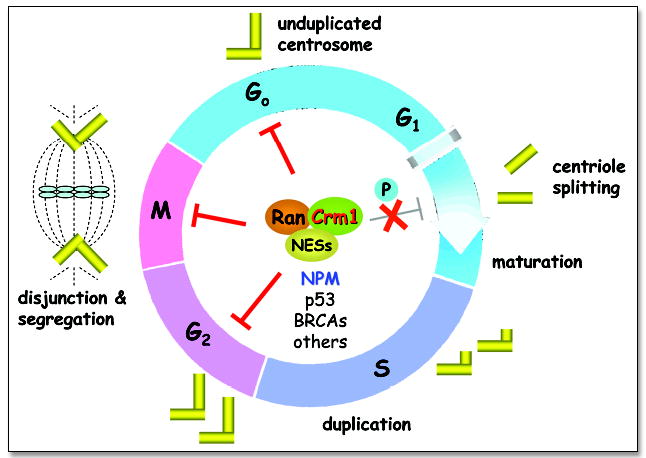
Ran/Crm1 functions as a loading dock to coordinate cell cycle checkpoints and centrosome duplication. The centrosome duplication cycle is shown through the various stages of the cell cycle. The Ran/Crm1 network functions as a loading dock to spatially and temporally coordinate NES-containing factors that ensure the fidelity of the centrosome duplication process. The Ran/Crm1 complex interacts with various NES-containing proteins, such as NPM, p53, BRCAs, etc., in a cell cycle dependent manner to inhibit centriole splitting. At the G1/S transition of a normal cell, such a process may be inactivated by phosphorylation of the substrates, such as NPM, thereby preventing their bindings to Ran/Crm1. Disruption of this coordination may lead to genomic instability and carcinogenesis.
NUCLEOPHOSMIN AS A SUBSTRATE OF RAN/CRM1 IN CENTROSOME DUPLICATION
Early studies demonstrated that nucleophosmin (NPM, or B23) is a multifunctional nucleolar phospho-protein that is localized at high levels in the granular region of the nucleolus.25 NPM shuttles between the nucleus and the cytoplasm during the cell cycle and plays a role in the sub-cellular localization of human T-cell leukemia and human immunodeficiency viral proteins.25 NPM can modulate the activity of p53 and is upregulated in response to stress stimuli, including DNA damage and hypoxia.26
Since NPM has been implicated as a regulator of centrosome synthesis,27 it is plausible that NPM may be a substrate for Ran/Crm1 to regulate centrosome duplication. Consistent with this model, we have recently demonstrated that local trafficking of NPM is mediated by a Ran/Crm1-dependent process, whereby NPM acts as a Crm1 substrate through its NES domain.28 In this study, the association of NPM and Crm1 was shown by immunofluorescence-based colocalization on centrosomes and cofractionation from sucrose gradients to isolate centrosomes. NES mutation of NPM or disruption of Crm1 function by LMB, RanBP1 or HBx led to dissociation of NPM from centrosomes and initiation of premature centrosome duplication. Therefore, this process was mediated by the Ran/Crm1 pathway and required a functional NES motif. To examine the role of nucleophosmin in regulating centrosome duplication, NPM siRNA was employed to knock-down the expression of endogenous NPM. Consistently, NPM siRNA resulted in supernumerary centrosomes that could be nucleated in mitotic cells to form mitotic spindles, an activity that could be effectively inhibited by coexpression of NPM. Additionally, site-directed mutagenesis revealed a potential regulatory role of a novel NPM phosphorylation site at threonine-95. A mutant mimicking phosphorylation at this site displayed decreased centrosome binding and supernumerary centrosomes. Since this phosphorylation site is within the NPM NES, it may regulate the binding of NPM to Crm1. Taken together, these results suggest that proper centrosome duplication is mediated by NPM binding to centrosomes through the interaction of its NES motif with Crm1.
The association of NPM and Ran/Crm1 provides novel mechanistic insight regarding the regulation of centrosome duplication, a process that may be disrupted in viral hepatitis-mediated liver carcinogenesis.21,24 NPM thus acts as a licensing factor to prevent any unscheduled centrosome duplication during the cell cycle. In this manner, NPM may serve as a tumor suppressor since it provides a centrosome duplication checkpoint during cell division and abrogation of its functions may lead to tumorigenic phenotypes. Consistent with this view, alterations of NPM are associated with genomic instability and human malignancies. For example, translocations that produce N-terminal NPM fusions with various partner genes, including ALK, RARα or MLF1, lead to hematological disorders such as anaplastic large cell lymphoma, acute promyelocytic leukemia (APL) or myelodysplastic syndrome (MDS), respectively.29–31 Cytoplasmic dislocation of NPM due to frameshift mutations at the C-terminus has also been reported to be associated with a subgroup of acute myelogenous leukemia (AML) patients who have a normal karyotype.32 The physiological function of NPM in carcinogenesis has been confirmed by the generation of Npm1-deficient mice.33 Npm was found to be an essential gene for embryonic development and the maintenance of genomic stability. Cells with Npm inactivation had unrestricted centrosome duplication and multi-polar spindles. Moreover, mice with haploinsufficiency of NPM developed MDS with an acceleration of oncogenesis. Thus, NPM may have a tumor suppressive function by regulating centrosome duplication and maintaining genomic stability.
It is interesting to note that the recent article by Falini et al.32 and the letter by Nakagawa et al.34 put forward a working model on the mechanism underlying the abnormal cytoplasmic localization of NPM in AML patients. While still hypothesizing, both groups suggested that mutated NPM may utilize a Crm1-mediated nuclear export mechanism leading to cytoplasmic accumulation either through a frameshift mutation resulting in the creation of a functional NES motif or by amino acid substitutions at residues 288 and 299 to alter nucleolar localization. The authors suggest that such NPM mutants that are sequestered in the cytoplasm might interfere with normal NPM functions including interactions with p53 and ARF. However, we recently found that NPM mutated at residues 288 and 299 still largely accumulated in the nucleoplasm due to an NLS motif.28 In contrast, we identified a functional NES motif in residues 94–102. Of note, NPM translocation sites always occur downstream of this NES, resulting in fusion products with a functional export motif. This suggests that cytoplasmic localization of NPM mutants found in AML may be regulated by Crm1. In addition, the AML patients with cytoplasmic NPM have a normal karyotype. It remains intriguing whether NPM cytoplasmic accumulation is associated with carcinogenesis, as we and others demonstrated its key role in the maintenance of centrosome duplication.
OTHER CELL CYCLE REGULATORS AS CANDIDATE SUBSTRATES OF RAN/CRM1 IN CENTROSOME DUPLICATION
A family of serine/threonine kinases known as cyclin-dependent-kinases (cdks) control the onset of major cell cycle events such as DNA synthesis and mitosis.3,35 The activity of cdks is regulated by their association with different cyclins and cdk inhibitors which are temporally expressed at specific stages of the cell cycle. Cyclins A and E complex with cdk2 and regulate both nuclear and cytoplasmic events through their ability to shuttle between these cellular compartments. Cdk2/cyclin A has been shown to play a predominant role in the duplication of the centrosome in mammalian cells.36 Studies have also shown that Cdk2/cyclin E regulates the initiation of centrosome duplication and its overexpression is strongly correlated with a higher degree of centrosome abnormality in tumors.37 Although the export of cyclins A and E are not affected by treatment with LMB, a Crm1-specific inhibitor, Cyclin B does bind to and is exported by Crm1.38,39 Cyclin B export is sensitive to LMB and it is thought to play a possible role in the DNA-damage-induced G2 checkpoint. Cdk1 (cdc2)/Cyclin B is physically associated with centrosomes and activation in early mitosis is associated with centrosome separation. Gadd45a has been shown to associate with Cyclin B and to dissociate it from Cdk1 thereby inhibiting Cdk1/cyclin B activity while p21 binds to and inactivates Cdk1/Cyclin E and A.40 Thus controlling the presence and duration of cdk/cyclins is a key mechanism to prevent improper amplification of centrosomes.
The first gene identified to have a role in genomic instability was p53, a transcription factor that is either lost or mutated in a majority of human tumors. The regulation of p53 is tightly controlled through several mechanisms including cellular localization. Its nuclear export is mediated by Crm1 and is enabled by two nuclear export signals.41,42 p53 has been implicated in the regulation of centrosome duplication and loss of p53 has multiple effects, including centrosome amplification and cell cycle checkpoint disturbances. Mouse embryonic fibroblasts (MEFs) lacking p53 result in multiple copies of functionally competent centrosomes that are generated in a single cell cycle.13 These profoundly affect mitotic fidelity resulting in unequal segregation of chromosomes. p53 localizes to centrosomes and can regulate centrosome duplication independent of its function as a transcription factor.43 Inactivation of p53 can be achieved by cellular oncoproteins that associate with p53, including mdm2. Mdm2 binds to p53 and inhibits its transactivation function. Mdm2 amplification is observed in human cancers, preferentially in tumors without p53 mutation. Centrosome hyper-amplification has been observed in carcinoma and in cultured cells overexpressing Mdm2.44 p53 is a major activator of Gadd45a, a protein that has been implicated in the regulation of centrosome duplication.16,45 Gadd45a expression, which is stress inducible in all examined mammalian cells, has been frequently associated with growth arrest and in some cases, apoptosis. Recent findings indicate a role for Gadd45a in the control of G1 and G2 cell cycle progression.16,46 Targeted disruption of Gadd45a has revealed a clear role for this protein in the prevention of genomic instability and maintenance of normal centrosome numbers.16 Cells lacking Gadd45a exhibit genomic instability with similar characteristics to that of p53-deficient mice. Gadd45a−/− MEFs exhibit a high degree of tetraploidy and at later passages, aneuploidy. These cells are often found to contain more than two centrosomes and mitosis with multiple spindles.
Brca1 and Brca2 are recognized as tumor suppressors that shuttle between the nucleus and cytoplasm implicating that control of their subcellular localization serves as a potential switch for regulating their function.47 A nuclear export signal was identified at the amino-terminus of Brca1 and loss of endogenous nuclear Brca1 in T47D cells results from overexpression of Crm1. Brca1 and Brca2 mutations are found in breast tumors of women with familial breast cancer and these tumors show a greater degree of genomic instability than breast tumors without Brca1 or Brca2 mutations.48 The absence of Brca1 in cultured cells prevents p53-mediated induction of Gadd45a.49 Brca1 has been shown to localize to centrosomes and cells from embryos lacking Brca1 exhibit genomic instability, including aneuploidy and chromosome structural abnormalities similar to those seen in Gadd45−/− cells.14 Brca1 binds to and activates p53 which then transcriptionally downregulates Brca1. Brca1 is normally phosphorylated during S phase and in response to DNA damage although the relevance of Brca1 phosphorylation to its function as a tumor suppressor is unclear. Mutation in the Brca1 gene results in centrosome amplification and Brca1−/− tumors display genetic instability and cells from Brca1 mutant mice display centrosome amplification and genetic instability.14 It is interesting to note that the absence of Brca2, a structurally unrelated tumor suppressor that forms a complex with Brca1 leads to centrosome amplification15 although Brca2 has not been localized to centrosomes. It will be interesting to see whether Brca2 has similar effects as Brca1.
CONCLUDING REMARKS
The functional details of the Ran/Crm1 complex and its regulation are emerging. Current studies indicate that the Ran-GTP gradient model that has been used to describe Ran-complex function in various processes, including mitosis, is oversimplified. Our understanding thus far has detailed a complex interaction and localization of various players of the Ran pathway that are not only involved in nucleotide turnover but recruitment and stabilization of factors to specific sites of action. In particular, NPM, and perhaps other NES-containing proteins such as p53, Brca1 and cyclin B that can interact with components of the Ran/Crm1 complex, may serve as crucial safeguards to ensure the fidelity of the mitotic process. Current findings suggest that the Ran/Crm1 complex may act as a loading dock to allow other factors to properly regulate their cell-cycle related targets, such as the centrosome, both spatially and temporally. Future studies of such licensing factors are warranted to promote our knowledge of how these processes are regulated in normal cells and how they are disrupted during carcinogenesis.
References
- 1.Jacks T, Weinberg RA. Cell-cycle control and its watchman. Nature. 1996;381:643–4. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- 2.Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000;100:71–8. doi: 10.1016/s0092-8674(00)81684-0. [DOI] [PubMed] [Google Scholar]
- 3.Elledge SJ. Cell cycle checkpoints: Preventing an identity crisis. Science. 1996;274:1664–72. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 4.Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- 5.Lange BM. Integration of the centrosome in cell cycle control, stress response and signal transduction pathways. Curr Opin Cell Biol. 2002;14:35–43. doi: 10.1016/s0955-0674(01)00291-5. [DOI] [PubMed] [Google Scholar]
- 6.Sluder G, Hinchcliffe EH. The coordination of centrosome reproduction with nuclear events during the cell cycle. Curr Top Dev Biol. 2000;49:267–89. doi: 10.1016/s0070-2153(99)49013-1. [DOI] [PubMed] [Google Scholar]
- 7.Doxsey S. Duplicating dangerously: Linking centrosome duplication and aneuploidy. Mol Cell. 2002;10:439–40. doi: 10.1016/s1097-2765(02)00654-8. [DOI] [PubMed] [Google Scholar]
- 8.Nigg EA. Centrosome aberrations: Cause or consequence of cancer progression? Nat Rev Cancer. 2002;2:815–25. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 10.Brinkley BR. Managing the centrosome numbers game: From chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- 11.Salisbury JL, Whitehead CM, Lingle WL, Barrett SL. Centrosomes and cancer. Biol Cell. 1999;91:451–60. [PubMed] [Google Scholar]
- 12.Sluder G, Hinchcliffe EH. Control of centrosome reproduction: The right number at the right time. Biol Cell. 1999;91:413–27. [PubMed] [Google Scholar]
- 13.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–7. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- 14.Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389–95. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 15.Tutt A, Gabriel A, Bertwistle D, Connor F, Paterson H, Peacock J, Ross G, Ashworth A. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol. 1999;9:1107–10. doi: 10.1016/s0960-9822(99)80479-5. [DOI] [PubMed] [Google Scholar]
- 16.Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan Q, Fernandez-Salguero PM, Morgan WF, Deng CX, Fornace AJ., Jr Genomic instability in Gadd45a-deficient mice. Nat Genet. 1999;23:176–84. doi: 10.1038/13802. [DOI] [PubMed] [Google Scholar]
- 17.Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–8. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 18.Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–9. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- 19.Kalitsis P, Earle E, Fowler KJ, Choo KH. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 2000;14:2277–82. doi: 10.1101/gad.827500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weis K. Regulating access to the genome: Nucleocytoplasmic transport throughout the cell cycle. Cell. 2003;112:441–51. doi: 10.1016/s0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 21.Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, Valerie K, Fukasawa K, Wang XW. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23:5282–92. doi: 10.1128/MCB.23.15.5282-5292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keryer G, Di Fiore B, Celati C, Lechtreck KF, Mogensen M, Delouvee A, Lavia P, Bornens M, Tassin AM. Part of Ran is associated with AKAP450 at the centrosome: Involvement in microtubule-organizing activity. Mol Biol Cell. 2003;14:4260–71. doi: 10.1091/mbc.E02-11-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Fiore B, Ciciarello M, Mangiacasale R, Palena A, Tassin AM, Cundari E, Lavia P. Mammalian RanBP1 regulates centrosome cohesion during mitosis. J Cell Sci. 2003;116:3399–411. doi: 10.1242/jcs.00624. [DOI] [PubMed] [Google Scholar]
- 24.Forgues M, Marrogi AJ, Spillare EA, Wu CG, Yoshida M, Wang XW. Interaction of the hepatitis b virus x protein with the Crm1-dependent nuclear export pathway. J Biol Chem. 2001;276:22797–803. doi: 10.1074/jbc.M101259200. [DOI] [PubMed] [Google Scholar]
- 25.Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379–90. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 26.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol. 2002;4:529–33. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 27.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–40. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Budhu A, Forgues M, Wang XW. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat Cell Biol. 2005;7:823–830. doi: 10.1038/ncb1282. [DOI] [PubMed] [Google Scholar]
- 29.Morris SW, Kirstein MN, Valentine MB, Dittmer K, Shapiro DN, Look AT, Saltman DL. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in nonHodgkin’s lymphoma. Science. 1995;267:316–7. doi: 10.1126/science.267.5196.316-b. [DOI] [PubMed] [Google Scholar]
- 30.Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ. The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood. 1996;87:882–6. [PubMed] [Google Scholar]
- 31.Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, Carroll AJ, Morris SW. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12:265–75. [PubMed] [Google Scholar]
- 32.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–66. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 33.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–53. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa M, Kameoka Y, Suzuki R. Nucleophosmin in acute myelogenous leukemia. N Engl J Med. 2005;352:1819–20. doi: 10.1056/NEJM200504283521719. [DOI] [PubMed] [Google Scholar]
- 35.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: A perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 36.Meraldi P, Nigg EA. The centrosome cycle. FEBS Lett. 2002;521:9–13. doi: 10.1016/s0014-5793(02)02865-x. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol. 1999;9:429–32. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Bardes ES, Moore JD, Brennan J, Powers MA, Kornbluth S. Control of cyclin B1 localization through regulated binding of the nuclear export factor CRM1. Genes Dev. 1998;12:2131–43. doi: 10.1101/gad.12.14.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toyoshima F, Moriguchi T, Wada A, Fukuda M, Nishida E. Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J. 1998;17:2728–35. doi: 10.1093/emboj/17.10.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ., Jr Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999;18:2892–900. doi: 10.1038/sj.onc.1202667. [DOI] [PubMed] [Google Scholar]
- 41.Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999;18:1660–72. doi: 10.1093/emboj/18.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Xiong Y. A p53 Amino-Terminal Nuclear Export Signal Inhibited by DNA Damage-Induced Phosphorylation. Science. 2001;292:1910–5. doi: 10.1126/science.1058637. [DOI] [PubMed] [Google Scholar]
- 43.Tarapore P, Okuda M, Fukasawa K. A mammalian in vitro centriole duplication system: Evidence for involvement of CDK2/cyclin E and nucleophosmin/B23 in centrosome duplication. Cell Cycle. 2002;1:75–81. [PubMed] [Google Scholar]
- 44.Carroll PE, Okuda M, Horn HF, Biddinger P, Stambrook PJ, Gleich LL, Li YQ, Tarapore P, Fukasawa K. Centrosome hyperamplification in human cancer: Chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene. 1999;18:1935–44. doi: 10.1038/sj.onc.1202515. [DOI] [PubMed] [Google Scholar]
- 45.Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in Ataxia-Telangiectasia. Cell. 1992;71:587–97. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 46.Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O’Connor PM, Fornace AJ, Jr, Harris CC. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA. 1999;96:3706–11. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson BR. Regulation of BRCA1, BRCA2 and BARD1 intracellular trafficking. Bioessays. 2005;27:884–93. doi: 10.1002/bies.20277. [DOI] [PubMed] [Google Scholar]
- 48.Bertwistle D, Ashworth A. The pathology of familial breast cancer: How do the functions of BRCA1 and BRCA2 relate to breast tumour pathology? Breast Cancer Res. 1999;1:41–7. doi: 10.1186/bcr12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harkin DP, Bean JM, Miklos D, Song YH, Truong VB, Englert C, Christians FC, Ellisen LW, Maheswaran S, Oliner JD, Haber DA. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 1999;97:575–86. doi: 10.1016/s0092-8674(00)80769-2. [DOI] [PubMed] [Google Scholar]