


Abstract
Human TAFII55 (hTAFII55), a component of the general transcription factor TFIID, is the only general transcription factor encoded by an intronless gene identified thus far. Analysis of the TATA-less hTAFII55 promoter-proximal sequence reveals putative binding sites for STAT-1, MEF2, E2F, Sp1, AP2, AREB6 and E47. Using chromatin immunoprecipitation, DNase I footprinting and electrophoretic mobility shift assays, we demonstrate that Sp1 and AP2 can bind simultaneously to juxtaposed Sp1- and AP2-binding sites in the hTAFII55 promoter-proximal region and functionally modulate hTAFII55 promoter activity, as evidenced by reporter gene assays performed in transiently transfected human C-33A and insect SL2 cell lines. Interestingly, removal of all the promoter-proximal Sp1-binding sites does not impair the function of the hTAFII55 core promoter. Moreover, a 52-bp DNA fragment containing only the hTAFII55 initiator (Inr) and downstream promoter element (DPE) is able to support Gal4–VP16-mediated activation in vivo and in vitro. Our data suggest that Sp1, although it plays an enhancing role in hTAFII55 gene expression, is not essential for hTAFII55 core promoter activity. Interestingly, mutations introduced at the Inr and DPE differentially affect the selection of transcription start sites, suggesting that these two core promoter elements play a non-redundant role in the function of TATA-less promoters.
INTRODUCTION
Human TAFII55 (hTAFII55) was initially identified as an RNA polymerase II-specific TBP-associated factor (TAFII) in TFIID (1,2) and was also found in the TBP-free-TAFII-containing complex (3). It can interact with the hTAFII250, hTAFII100, hTAFII28, hTAFII20 and hTAFII18 components of TFIID (1,2) and blocks the acetyltransferase activity of TAFII250 (4). Moreover, hTAFII55 has been shown to interact with many transcription factors and appears to function as a transcriptional coactivator (1,5). The hTAFII55 homolog has been identified in many species (6–8) and is an essential gene in yeast (6,7). In addition to its role in transcription, TAFII55 has also been implicated in mRNA transport in fission yeast (7) and can interact with the human cleavage and polyadenylation specificity factor (9). These observations support the notion that mRNA synthesis and processing reactions are intimately linked (10).
To shed light on the regulation of general transcription factor-encoding genes, we set out to dissect the regulatory elements important for hTAFII55 promoter function. Our previous study demonstrated that hTAFII55 is encoded by an intronless gene (11) which has been localized at chromosome 5q31 (12). The hTAFII55 gene is driven by a TATA-less promoter with a functional initiator (Inr) and a downstream promoter element (DPE), and is one of the few human DPE-containing TATA-less promoters reported (11). Using promoter constructs containing deletions and point mutations, we found that a GC-rich sequence, spanning –99 to –26 relative to the transcription start site of the hTAFII55 gene, is critical for a high level of hTAFII55 gene expression in a human cervical carcinoma-derived C-33A cell line (11). This GC-rich sequence contains putative binding sites for specificity protein 1 (Sp1) and activator protein 2 (AP2). However, it remains unclear whether the hTAFII55 gene is indeed regulated by Sp1 and AP2.
Sp1 is ubiquitously expressed, and its GC-rich DNA-binding core sequence (GGGCGG) (13) has been found in many, especially TATA-less, promoters. It is commonly believed that Sp1 may function as a tethering moiety to recruit the general transcription machinery to a TATA-less promoter (14). Sp1 is a member of a related protein family implicated in a wide range of cellular processes (15), including cell cycle regulation (16–18), maintenance of gene activation during embryonic development by preventing de novo methylation of the CpG islands at house-keeping gene promoters (19–21), and preservation of chromatin structures at gene loci (22). Sp1 may work cooperatively with other tissue-specific transcription factors to modulate the activities of a variety of promoters (18,23–25). Sp1 protein undergoes post-translational modifications, including phosphorylation and glycosylation (26,27). The mechanisms by which Sp1 mediates transcriptional activation have been under intensive studies. Sp1 has been shown to interact with dTAFII110 (28,29) and hTAFII130 (30) through its activation domain and with hTAFII55 through its DNA-binding domain (1), suggesting that Sp1 may facilitate the assembly of the general transcription machinery via multiple protein–protein interactions with components of TFIID. Sp1 also requires an additional cofactor complex, CRSP, to mediate synergistic activation with SREBP-1a on chromatin templates (31,32). In addition to its role as a transcriptional activator, Sp1 has also been reported to repress transcription by recruiting histone deacetylase activity (33).
AP2 belongs to a family of transcription factors encoded by structurally related genes (34–37) and is evolutionally conserved among different species (38–40). AP2 recognizes a core sequence of 5′-CC(C/G)C(A/G)GGC-3′ (41), and is a cell type-specific transcription factor important in retinoid-controlled morphogenesis and differentiation, especially in neural crest-derived cell lineages and epithelial cells (42). AP2 responds to signals from secondary messengers of both the phorbol ester/PKC and the PKA signaling pathways (43,44) and many of its target genes are involved in cellular signaling important for the development and progression of cancers (36,45–48). Interestingly, positive cofactor 4 (PC4), poly(ADP-ribose) polymerase (PARP), as well as CREB-binding protein/p300-interacting transactivator with ED-rich tail 2 and 4 (CITED2/4) have been suggested to function as coactivators for AP2-mediated transcriptional activation (49–52).
To define the roles of Sp1 and AP2 in modulating hTAFII55 promoter activity, we examined the in vivo association of the hTAFII55 promoter with Sp1 and AP2 proteins, using the chromatin immunoprecipitation (ChIP) approach. We report here that Sp1 and AP2 can bind simultaneously to the hTAFII55 promoter and activate hTAFII55 transcription in a combinatorial manner. Given the prevalence of Sp1-binding sites in TATA-less promoters, Sp1 is commonly thought to be essential for transcription initiation from TATA-less promoters. We investigated the requirement of Sp1-binding sites for TATA-less transcription, and conclude that Sp1 is important in maintaining a high level of hTAFII55 promoter activity, but it is not essential for transcription from the TATA-less hTAFII55 core promoter. Importantly, our studies also uncover a non-redundant role of Inr and DPE in the selection of transcription start sites driven by TATA-less promoters.
MATERIALS AND METHODS
Plasmid constructions
The FLAG-tagged human AP2α expression plasmid, pF:AP2α-11d, was constructed by first cloning the human AP2α open reading frame, amplified by PCR from plasmid SPRSV-AP2 (53) with an NdeI site-containing upstream primer (5′-AAGTCGACATATGAAAATGCTTTGGAAA-3′) and a KpnI site-containing downstream primer (5′-TTC TCGAGGTACCTCACTTTCTGTGCTTCTCC-3′), into the NdeI–KpnI-linearized pFLAG°(AS)-7 vector (54) to generate pF°:AP2α-7. The AP2α-coding sequence was then excised from pF°:AP2α-7 with NdeI and EcoRI, and cloned into pF:E1-11d (55), after swapping the inserts between NdeI and EcoRI sites, to create pF:AP2α-11d. The insect cell expression plasmid, pPac-F:AP2α, was obtained by transferring the XbaI–KpnI fragment from pF:AP2α-11d to pPacPL (generous gift from C. Desplan). Other plasmids, pSG424 (56), pSGVP (57), pPacO (58) and pPacSp1 (59) were described previously.
Plasmids pGL2-TAF55(–128/+87), pGL2-TAF55(–71/+36), pGL2-TAF55(–71/+36)Sp1*-60, pGL2-TAF55(–71/+36)AP2*, pGL2-TAF55(–71/+36)Sp1*-60/AP2* and pGL2-TAF55(–71/+36)Sp1*-20 were described previously (11). The hTAFII55 promoter constructs, pGL2-TAF55(–128/+87)Sp1*-60, pGL2-TAF55(–128/+87)AP2*, pGL2-TAF55(–128/+87)Sp1*-20, pGL2-TAF55(–128/+87)Inr* and pGL2-TAF55(–128/+87)DPE* containing nucleotide substitutions in the sequence motifs of Sp1, AP2, Inr or DPE were constructed in the backbone of pGL2-TAF55(–128/+87) by incorporating the same nucleotide substitutions as shown previously (11) following the QuikChange protocol (Stratagene). Mutations on the altered sequence motif(s) were denoted with an asterisk (*) in the respective plasmid name. In plasmids pGL2-TAF55(–128/+87)Sp1*-40, pGL2-TAF55(–71/+36)Sp1*-40 and pGL2-TAF55(–71/+36)Sp1*-40/Sp1*-20, the 4 nt ‘GGGC’, spanning –45 to –42, were changed to ‘TACT’ in the backbone of pGL2-TAF55(–128/+87), pGL2-TAF55(–71/+36) and pGL2-TAF55(–71/+36)Sp1*-20, respectively. The plasmid pGL2-TAF55(–71/+36)wtSp1-60, which contains only an intact –60 Sp1-binding site, was generated by combining nucleotide substitutions at –50, –40 and –20 Sp1-binding sites. Likewise, plas mids pGL2-TAF55(–71/+36)wtSp1-50, pGL2-TAF55(–71/ +36)wtSp1-40 and pGL2-TAF55(–71/+36)wtSp1-20, each of which contains only one intact Sp1-binding site as specified, were similarly constructed in the backbone of pGL2-TAF55(–71/+36).
The 5Gal4-binding site-driven hTAFII55 core promoter plasmids, pGL2-5Gal(–16/+36)WT and pGL2-5Gal(–16/+36)Inr*, were constructed first by using pGL2-5Gal(–26/+36)WT and pGL2-5Gal(–26/+36)Inr* (11) as template for PCR amplification with an upstream primer (5′-AACTAACTGCAGCTGCGCGTCGCGACG-3′) containing adjacent PstI- and PvuII-cutting sites and a downstream primer (5′-CCTCGCCAGATCTCCGTCCGGGTCGCCT-3′) containing a BglII-cutting site. The resulting fragments were treated with PstI and BglII, and inserted into the same sites of pGL2-5Gal (11). The other heterologous promoter constructs, pGL2-5Gal(–16/+36)DPE* and pGL2-5Gal(–16/+36)Inr*DPE*, were cloned by the same strategy using pGL2-5Gal(–26/+36)DPE* and pGL2-5Gal(–26/+36)Inr*DPE* (11) as template but with a different BglII site-containing downstream primer (5′-CCTCGCCAGAT CTCATGACGGGTCGCCT-3′) that introduces mutations at the DPE. The hTAFII55 core promoter constructs, including pGL2-TAF55(–16/+36), pGL2-TAF55(–16/+36)Inr*, pGL2-TAF55(–16/+36)DPE* and pGL2-TAF55(–16/+36)Inr*DPE*, were generated by removing five Gal4-binding sites from pGL2-5Gal(–16/+36)WT, pGL2-5Gal(–16/+36)Inr*, pGL2-5Gal(–16/+36)DPE* and pGL2-5Gal(–16/+36)Inr*DPE*, respectively, by SmaI and PvuII digestion, followed by self-ligation of the remaining fragments. All constructs were confirmed by restriction enzyme digestion and DNA sequencing.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were carried out as described (60) with some modifications. Briefly, protein–DNA complexes were cross-linked in vivo by adding formaldehyde to 35 ml (∼1.1 × 106 cells/ml) of log-phase HeLa suspension cells, maintained in Joklik medium supplemented with 5% calf serum, to a final concentration of 1%. The reaction was conducted at room temperature for 10 min and then terminated by the addition of glycine to a final concentration of 0.125 M. Cells were collected by centrifugation at 300 g for 2 min, rinsed twice with ice-cold 1× PBS, resuspended in 1 ml of ice-cold cell lysis buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM DTT, 0.25 mM PMSF, plus the protease inhibitors: 1 µg/ml each of pepstatin, leupeptin and aprotinin), and kept on ice for 10 min. Cells were then collected and resuspended in 500 µl of nuclear lysis buffer (50 mM Tris–HCl, pH 8.1, 10 mM EDTA, 1% SDS, 1 mM DTT, 2.5 mM PMSF, and the protease inhibitors mentioned above). Sonication was carried out using Branson Sonifier 450 (3.2-mm tapered Micro Tip, duty cycle 50%, output level 4.5) for six 10-s bursts with 2 min on ice between each sonication. The sheared chromatin contains DNA fragments averaging between 600 and 3000 bp. For each ChIP, 100 µl of sheared chromatin was diluted to 1 ml with IP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl, pH 8.1 and 167 mM NaCl). To reduce non-specific binding, the chromatin solution was pre-cleared with 80 µl of PASDT, which contains 50% (v/v) of packed protein A–Sepharose beads (Amersham Pharmacia Biotech), 240 µg/ml of salmon sperm DNA and 240 µg/ml of tRNA, at 4°C for 30 min with rotation. After spinning at 450 g for 2 min, the pre-cleared chromatin (i.e. supernatant) was collected and incubated with 10 µl of anti-Sp1 antibodies (generous gift of J.-L. Chen and R. Tjian), 25 µl of anti-AP2 antibodies (sc-184; Santa Cruz Biotechnology), or no antibodies (as mock IP) at 4°C overnight. The immune complexes were pulled down by further incubating with 60 µl of PASDT at 4°C for 1 h. At this step, half of the supernatant from the mock IP was saved and used as the total chromatin input for later PCR analysis. The beads were then washed sequentially with 1 ml of IP dilution buffer, TSE (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl), Li buffer (100 mM Tris–HCl, pH 8.1, 500 mM LiCl, 1% NP-40, 1% deoxycholic acid) and TE (10 mM Tris–HCl, pH 8.1, 1 mM EDTA, pH 8.0), twice in each buffer. Immunocomplexes were successively eluted two times with 250 µl of 50 mM NaHCO3 plus 1% SDS at room temperature for 15 min and pooled together. Cross-links in the samples, including the chromatin input, were reversed by heating at 68°C in a water bath overnight with the addition of 5 M NaCl to a final concentration of 200 mM. All the samples were digested with 10 µg each of RNase A and proteinase K at 42°C for 1 h to remove RNA and protein. The DNA samples were purified by phenol–chloroform extraction, followed by ethanol precipitation, and finally dissolved in 20 µl of TE.
PCR analysis of immunoprecipitated DNA
PCRs were set up in duplicate in 25 µl containing 1/5 of ChIP products or 0.5% of the input chromatin, 2 U Platinum Taq DNA Polymerase (Life Technologies), 25 mM Tris–HCl, pH 8.4, 50 mM KCl, 200 µM of dNTPs, 2.5 mM MgCl2, and 15 pmol of each primer. The promoter-proximal and -distal primer pairs spanning –122 to +20 and –10 030 to –9883, relative to the hTAFII55 transcription start site, result in 142 and 148 bp products, respectively. PCR was carried out at 95°C for 45 s, 57°C for 30 s, and 72°C for 30 s for 28 cycles, which had been determined to be in the linear range of amplification (data not shown). Twelve microliters of the PCR products were resolved on an 8% polyacrylamide gel and visualized after ethidium bromide staining.
Protein purification
Recombinant FLAG-tagged human AP2α protein was expressed in and purified from BL21(DE3)pLysS bacteria harboring pF:AP2α-11d as described (61). The FLAG-tagged human Sp1 protein was purified from Sf9 insect cells infected with recombinant baculoviruses containing pVL-F:Sp1 as described previously (8).
DNase I footprinting
The DNA fragment used for DNase I footprinting, spanning –128 to +87 of the hTAFII55 promoter region, was generated by PCR amplification using pGL2-TAF55(–1372/+87) template (11) with an upstream KpnI site-containing primer (5′-AAAACAAGGTACCGCGAAAAGATGAG-3′) and a BglII site-containing downstream primer (5′-AGCGCGAGATCT TGCCGAGAGG-3′). The PCR product was end-labeled with [γ-32P]ATP using T4 polynucleotide kinase and digested with either BglII or KpnI to remove one of the labeled ends and then purified from an 8% native polyacrylamide gel. DNase I footprinting was carried out as described previously (55), except a final concentration of 16 µM zinc acetate was included in the binding reactions to facilitate Sp1 binding.
Electrophoretic mobility shift assay (EMSA)
DNA fragments spanning –80 to –27 of either wild-type (–80/–27) or mutated hTAFII55 promoter fragments, includ ing Sp1*-60(–80/–27), AP2*(–80/–27) and Sp1*-60/AP2*(–80/–27), were prepared by PCR amplification from templates pGL2-TAF55(–128/+87), pGL2-TAF55(–128/+87)Sp1*-60, pGL2-TAF55(–128/+87)AP2* and pGL2-TAF55(–128/+87)Sp1*-60/AP2*, using primer pairs annealing to –80 to –64 and –27 to –43, respectively. The DNA fragment, Sp1*-40(–80/–27), was similarly made from pGL2-TAF55(–128/+87) template by PCR amplification using the same upstream primer and a downstream primer (5′-CGGCTGTGAGTGACAtatCCTCC-3′) with mutations at the cryptic –40 Sp1-binding site as denoted with lowercase letters. The other hTAFII55 promoter fragments spanning –80 to –27 with only one wild-type Sp1-binding site at –60, –50 and –40 (Fig. 5A), were similarly prepared using pGL2-TAF55(–128/+87)wtSp1-60, pGL2-TAF55(–128/+87)wtSp1-50 and pGL2-TAF55(–128/+87)wtSp1-40 as templates for PCR amplification. All the DNA probes used for EMSA were prepared from gel-purified PCR fragments by end-labeling with T4 polynucleotide kinase and [γ-32P]ATP. The labeled probes were then purified by passing through a Sephadex G-25 spin column (Amersham Pharmacia Biotech). Binding reactions were conducted with 10 fmol (Fig. 4) or 6 fmol (Fig. 5A) of labeled DNA, 1 µg of poly (dA–dT), 10 mM HEPES-Na (pH 7.9), 60 mM KCl, 5 mM DTT, 0.2 mM EDTA, 16 µM zinc acetate, 4 mM MgCl2, 10% glycerol and 0.1 mg/ml BSA, with or without purified proteins, in a total volume of 10 µl. The reactions were carried out at 30°C for 40 min. One microliter of anti-AP2α or anti-Sp1 antibodies, when added, was included 20 min post initial incubation. The protein–DNA complexes were resolved on a 4% native polyacrylamide gel (acrylamide:bis = 37.5:1) with electrophoresis conducted at 170 V in 0.25× TBE running buffer at 4°C.
Figure 5.

Sp1-binding sites located at the hTAFII55 promoter-proximal region are functionally involved in hTAFII55 gene expression. (A) Relative binding affinities of the three upstream Sp1-binding sites. The EMSA was performed with different amounts of FLAG-tagged Sp1 protein using equal amounts (6 fmol) of 32P-labeled hTAFII55 promoter fragments as indicated at the bottom. Brackets represent intact binding sites and ‘X’ denotes mutations at the motif. The Sp1–DNA complex is indicated by an arrow. (B) Reporter gene assay with promoter constructs containing individually mutated Sp1-binding sites. Transient transfection was performed in C-33A cells using reporter constructs pGL2-TAF55(–71/+36), pGL2-TAF55(–71/+36)Sp1*-60, pGL2-TAF55(–71/+36)AP2*, pGL2-TAF55(–71/+36)Sp1*-40, pGL2-TAF55(–71/+36)Sp1*-20 and pGL2-Basic, respectively. Reporter gene activities were normalized to that of the wild-type construct. (C) In vitro transcription assay with promoter constructs containing individually mutated Sp1-binding sites. Transcription reactions were performed in HeLa nuclear extracts with hTAFII55 promoter constructs pGL2-TAF55(–128/+87), pGL2-TAF55(–128/+87)Sp1*-60, pGL2-TAF55(–128/+87)AP2*, pGL2-TAF55(–128/+87)Sp1*-40 and pGL2-TAF55(–128/+87)Sp1*-20, which contain mutations at each of the Sp1-binding sites. The amounts of RNA synthesized were quantified by primer extension using Luc(AS)-1 primer (55). The arrow points to the major transcription start site (+1) and the bracket denotes minor start sites. Promoter constructs used in this experiment are illustrated on the right with brackets representing intact Sp1 sites, and an ‘X’ indicating a mutated site.
Figure 4.

Sp1 and AP2 are able to bind simultaneously to their juxtaposed binding sites in the hTAFII55 promoter-proximal region. EMSAs were performed with different amounts of purified FLAG-tagged Sp1 and FLAG-tagged AP2 proteins, in the absence or presence of anti-Sp1 (α-Sp1) or anti-AP2 (α-AP2) antibodies using labeled wild-type (WT) (A) or mutated (B–E) promoter fragments spanning –80 to –27, as indicated on each panel and described in Materials and Methods. The protein–DNA complexes detected are indicated on the right with different arrows and arrowheads. Individual protein-binding sites are marked by brackets with ‘X’ and asterisks (*) denoting mutations introduced at the specific motifs.
In vitro transcription
In vitro transcription was performed with HeLa nuclear extracts and analyzed by primer extension using the Luc(AS)-1 primer as described previously (55). All the primer extension products were analyzed on an 8 M urea, 5% Long Ranger polyacrylamide gel together with the dideoxynucleotide sequencing products generated with the phosphorylated form of the same primer (55).
Transient transfection and reporter gene assays
C-33A cells, which were derived from human cervical carcinoma, were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum in a humidified 5% CO2 incubator at 37°C. Transient transfection into C-33A cells were performed in 35-mm plates where each plate received 2 µg of the reporter plasmid, either alone or in conjunction with varying amounts of the Gal4–VP16 expression plasmid (pSGVP) supplemented with the cloning vector (pSG424) to a total of 250 ng, using the calcium phosphate method as described (62). Drosophila Schneider line 2 (SL2, American Type Culture Collection) cells were maintained in Schneider’s Drosophila medium (Life Technologies) supplemented with 10% heat inactivated fetal bovine serum in a 24°C incubator. Transient transfection into SL2 cells was carried out as described (63). Briefly, 4 × 106 SL2 cells were seeded onto each 60-mm plate a day prior to transfection. Each plate received 10 µg of a reporter plasmid and 5 µg of insect cell expression plasmids (pPacSp1, pPac-F:AP2α or the vector pPacO). Cells were incubated with DNA–calcium phosphate cocrystals at 24°C for 15–24 h, and then refed with fresh growth medium for an additional 24 h before harvested for reporter gene assays. The cell lysates from transfected SL2 and C-33A cells, which were collected by rubber policeman after adding 300 µl of 1× reporter lysis buffer (Promega), were prepared by freezing and thawing once in liquid N2 and 37°C water bath. Following centrifugation at 16 000 g for 5 min at 4°C, 20 µl of the supernatant was mixed with 200 µl of luciferase assay buffer containing 19.4 mM HEPES-Na (pH 7.8), 3.8 mM ATP, 11.7 mM MgSO4, and 0.22 mM d-Luciferin (Pharmingen). The luciferase activity was measured for 10 s after an initial 1-s delay on an Lmax luminometer (Molecular Devices). Transfection and reporter gene assays were performed independently at least three times, each in duplicate, and one representative result is presented.
RESULTS
Multiple transcription factor-binding sites are present in the hTAFII55 promoter-proximal region
To identify transcription factors regulating hTAFII55 gene expression, we first searched for potential transcription factor-binding sites in the hTAFII55 promoter-proximal region, spanning –281 to +87 relative to the transcription start site, which had been shown to possess potent transcription activity stronger than those exhibited by several enhancer and promoter elements derived from HIV-1, SV40 and human papillomavirus (11). The sequence analysis revealed putative binding sites for STAT, MEF2, E2F, Sp1, AP2, AREB6 and E47 (Fig. 1). These putative transcription factor-binding sites were indeed recognized by some cellular proteins, as evidenced by DNase I footprinting performed with HeLa nuclear extracts (data not shown). Clearly, no TATA box was found between –25 and –30. Instead, an Inr surrounding the transcription start site, which was mapped previously in vitro and in vivo by primer extension and RNase protection (11), and a DPE, positioned at +29 to +35, were observed in this TATA-less promoter. Interestingly, many of these cis- elements were also found in the mouse TAFII55 promoter-proximal region (data not shown), indicating that mammalian TAFII55 gene expression is likely to be combinatorially regulated by both ubiquitous and cell type-specific transcription factors.
Figure 1.

Putative transcription factor-binding sites in the hTAFII55 promoter-proximal region. The major transcription start site previously determined by primer extension and RNase protection assays is indicated by a bent arrow and numbered +1 (11). Sequence motifs that match the consensus binding sites of known transcription factors are boxed.
Sp1 and AP2 bind to the hTAFII55 promoter-proximal region in vivo and in vitro
The observation that deletion of the –71 to –26 region of the hTAFII55 gene significantly impaired hTAFII55 promoter activity (11) suggests that Sp1 and AP2 may be directly involved in the regulation of hTAFII55 gene expression. To explore this possibility, we performed an in vivo ChIP assay using chromatin prepared from suspension HeLa cells. An hTAFII55 promoter-specific DNA fragment spanning –122 to +20 was significantly enriched when anti-Sp1 or anti-AP2 antibodies were used in the ChIP assay (Fig. 2, top). In contrast, no PCR products were detected in the reactions containing the same antibodies but with a different pair of primers that anneal to the coding region of an upstream protocadherin γ3A1 gene located on the same chromosome (Fig. 2, bottom). Signals were not detected from the distal primers even with additional PCR cycles (data not shown). This experiment demonstrated that Sp1 and AP2 indeed bind to the hTAFII55 promoter-proximal region in vivo.
Figure 2.
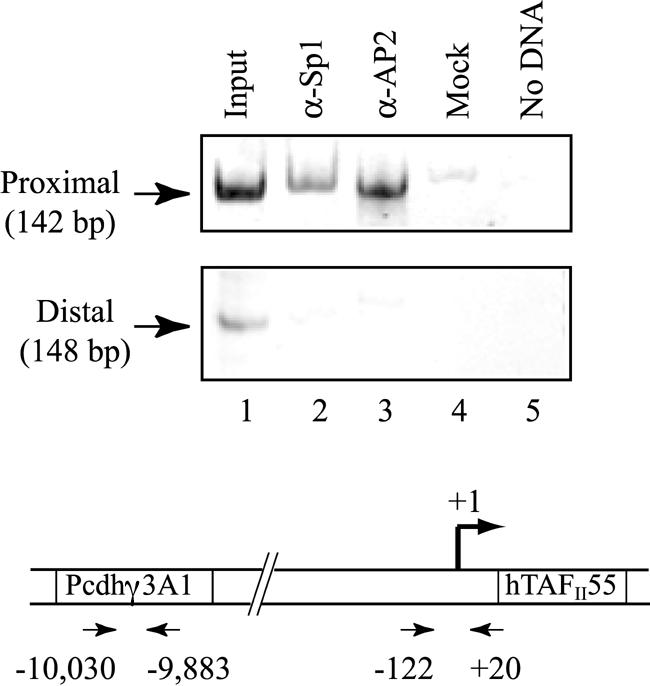
Sp1 and AP2 are associated with the hTAFII55 promoter in vivo. Cross-linked chromatin isolated from suspension HeLa cells were immunoprecipitated with α-Sp1 (lane 2), α-AP2 (lane 3), or no antibodies (lane 4). The associated chromosomal DNA fragments were amplified with an hTAFII55 promoter-specific primer pair (top), which gives rise to a 142-bp PCR product, or with primers specific to the open reading frame of the protocadherin γ3A1 gene (bottom), which result in a 148-bp fragment. The relative positions of the primer pairs are illustrated at the bottom. Chromosomal DNA input (lane 1, described in Materials and Methods) and water sample (lane 5) were subject to the same PCR amplification. PCR products were separated on an 8% polyacrylamide gel and visualized after ethidium bromide staining.
To further define the hTAFII55 sequence elements recognized by Sp1 and AP2, we conducted in vitro DNase I footprinting with purified Sp1 and AP2 proteins (Fig. 3A). Distinct DNA sequences, extending both 5′ and 3′ of the core AP2-binding site, were protected from DNase I digestion by purified AP2 protein, including hypersensitivity sites induced on the edges of the protected regions (Fig. 3B). Similarly, Sp1 also gave rise to two unequivocal protection regions (from –71 to –54 and from –50 to –6) covering –60 and –20 Sp1-binding sites (Fig. 3C). These results are summarized in Figure 3D, where thin and thick lines represent the sequences protected by Sp1 and AP2 proteins, respectively. The footprinting results suggested that different Sp1-binding sites are differentially utilized in the wild-type promoter context. Moreover, the extended footprint from –50 to –6 indicated that additional Sp1-binding sites might be present within this GC-rich hTAFII55 promoter region (see below).
Figure 3.

Sp1 and AP2 proteins bind directly to their cognate sites in the hTAFII55 promoter-proximal region. (A) Purified recombinant Sp1 and AP2 proteins. FLAG-tagged Sp1 and FLAG-tagged AP2 proteins were purified from insect cells and bacteria, respectively, as described in Materials and Methods. The proteins were then loaded on a 10% polyacrylamide–SDS gel and visualized after Coomassie blue staining. Positions of prestained protein size markers (in kDa) are indicated on the left. (B) DNase I footprinting with recombinant AP2 protein. End-labeled top and bottom strands of the hTAFII55 promoter fragment spanning –128 to +87 were incubated, respectively, with different amounts of FLAG-tagged AP2 protein as indicated. DNase I footprinting was then performed as described in Materials and Methods, and visualized after exposure to an X-ray film. The A/G footprinting markers generated by the Maxam–Gilbert sequencing method (85) were included to determine the boundaries of the protected regions (marked by vertical lines). Hypersensitive sites induced by AP2 binding are denoted by asterisks. (C) DNase I footprinting with recombinant Sp1 protein. DNase I footprinting with FLAG-tagged Sp1 was similarly performed as described above, except using only the top strand of the labeled hTAFII55 fragment and including an additional footprinting marker ‘C’ generated by the Maxam–Gilbert sequencing method. (D) Summary of the hTAFII55 promoter region protected by Sp1 and AP2. The hTAFII55 promoter sequence spanning –80 to +1 were illustrated with core sequences recognized by Sp1 and AP2 indicated by boxes. Horizontal lines mark the sequences protected by AP2 (thick bars) and Sp1 (thin bars) proteins in DNase I footprinting experiments.
Concurrent binding of Sp1 and AP2 to the hTAFII55 promoter-proximal region
The extended footprints exhibited by individual Sp1 and AP2 proteins raise an interesting possibility that both proteins may bind simultaneously to the hTAFII55 promoter. To explore this possibility, we conducted EMSA with purified Sp1 and AP2 proteins using a 32P-labeled hTAFII55 promoter fragment spanning –80 to –27. As shown in Figure 4A, AP2 formed a stable protein–DNA complex which could be supershifted by anti-AP2, but not anti-Sp1, antibodies (lanes 1–4). In contrast, two Sp1–DNA complexes, representing Sp1 binding to either the –60 or the –40 site alone (filled arrow) and to both –60 and –40 sites (filled arrowhead), were clearly detected (Fig. 4A, lanes 5–7). The –40 is a cryptic Sp1-binding site uncovered in EMSA with a mutated hTAFII55 promoter fragment (see below), whereas the –50 Sp1-binding site is a low-affinity Sp1-binding site rarely used in the context of the wild-type promoter as revealed by DNase I footprinting (see Fig. 3D) and EMSA (see below). As expected, both Sp1–DNA complexes were supershifted by anti-Sp1, but not anti-AP2, antibodies. Interestingly, a novel protein–DNA complex, distinct from individually formed Sp1–DNA and AP2–DNA complexes, was observed when both Sp1 and AP2 were incubated with the DNA probe (Fig. 4A, lane 8, indicated by an open arrowhead), suggesting that Sp1 and AP2 could bind simultaneously to the hTAFII55 promoter-proximal region. The specificity of the protein–DNA ternary complex was further confirmed by antibody supershift assays (Fig. 4A, lanes 8–10).
To confirm the identities of the two Sp1–DNA complexes formed on the wild-type probe, we also performed EMSA with mutated hTAFII55 promoter fragments. As shown in Figure 4B, mutations introduced at the –60 Sp1-binding site [changed from CCGCCC to CCGATC, corresponding to –63 to –58, with nucleotide substitutions underlined (11)] did not affect the AP2–DNA complex (lane 2) but abolished formation of the slower migrating Sp1–DNA complex detected in Figure 4A (compare Fig. 4A, lane 5 with Fig. 4B, lane 5). The Sp1–DNA complex that was still detected on the gel (Fig. 4B, lane 5) is due to Sp1 binding to the –40 site (see below). Importantly, the Sp1–AP2–DNA ternary complex, observed on the wild-type probe, was no longer seen on the mutated DNA fragment, suggesting that the ternary complex observed on the wild-type fragment (Fig. 4A, lane 8) requires an intact –60 Sp1-binding site. Binding of AP2 occludes Sp1 from binding to the –40 cognate sequence, as no ternary complexes were formed when both Sp1 and AP2 were incubated with the mutated DNA probe (Fig. 4B, compare lanes 2, 5 and 8).
Similar experiments were performed with DNA probes containing mutations at the –50 site [changed from CCGCCCGGAGG to CCGTATGGAGG, corresponding to –55 to –45 (11)] or at both –60 and –50 sites. AP2 could no longer bind to the DNA probe containing mutations at –50, which inactivates both AP2 and Sp1 binding to the –50 region (Fig. 4C, lane 2 and data not shown) but does not affect the formation of the two Sp1–DNA complexes (Fig. 4C, lanes 3–5), confirming that the –50 site is not involved in the formation of the two Sp1-containing complexes detected with the wild-type probe (Fig. 4A). Again, no ternary complexes were observed (Fig. 4C, lanes 6–8), indicating that the AP2-binding site at the –50 region is critical for the formation of the Sp1–AP2–DNA complex.
To our surprise, we still detected an Sp1–DNA complex, which could be supershifted by anti-Sp1, but not anti-AP2, antibodies, with a DNA probe containing mutations at both –60 and –50 sequences (Fig. 4D), suggesting the presence of a cryptic Sp1-binding site not identified by computer searches (see Fig. 1). Examination of the nucleotide sequence between –80 and –27 uncovered a potential Sp1-binding site [5′-GGGGCTGT-3′ (13)] spanning –46 to –39. Mutations introduced at the core sequence of this motif (5′-GTACTTGT-3′) abolished the formation of the slower migrating Sp1–DNA complex, without altering all the other species including the ternary complex (Fig. 4E). Collectively, these EMSA experiments not only uncovered a novel cryptic Sp1-binding site not found by computer searches, but also confirmed the identities of those two Sp1–DNA complexes formed on the wild-type hTAFII55 promoter-proximal region. As expected, when mutations at –40 were also introduced to the DNA probe containing mutations at both –60 and –50 sequences, no protein–DNA complex could be detected with either Sp1 or AP2 (data not shown).
Effects of different Sp1-binding sites on hTAFII55 gene expression
To address the role of –60, –50 and –40 Sp1-binding sites in regulating hTAFII55 promoter activity, we first measured the relative affinities of these neighboring Sp1-binding sites by EMSA. Using equal amounts of DNA probes containing combinations of double mutations which leave only one Sp1 site available for binding, we found that the –60 site has the highest affinity for Sp1 binding, based on the intensity of the shifted complex (Fig. 5A). The –40 site is also a strong, independent Sp1-binding site, whereas the –50 site is a relatively weak Sp1-binding site. This experiment further corroborated our in vitro DNase I footprinting and EMSA results, where the –60 Sp1-binding site was predominantly protected and the –50 Sp1-binding site was rarely used in the context of the wild-type promoter (see Figs 3C and 4).
To test whether the newly identified –40 Sp1-binding site is also involved in the regulation of the hTAFII55 promoter, we performed transient transfection in human C-33A cells with individually mutated reporter constructs (Fig. 5B). Mutations introduced at each of the –60, –50 and –40 sequences resulted in a reduction of promoter activity to a varying degree, which seems to correlate with the relative affinities of these three sites for Sp1 binding (see Fig. 5A). However, since mutations introduced at the –50 site also abolished AP2 binding (see Fig. 4C), the effect of Sp1 acting through the –50 site was in fact less than observed. Similar results were also observed by in vitro transcription/primer extension analysis performed with HeLa nuclear extracts, which directly measures the transcription activity (Fig. 5C). Taken together, these experiments demonstrated that different Sp1-binding sites contributed differentially to hTAFII55 promoter regulation, which may in part be accounted for by their relative affinities for Sp1 binding at least in the case of the upstream –60, –50 and –40 sites.
Sp1 and AP2 combinatorially stimulate hTAFII55 promoter activity
The finding that Sp1 and AP2 can bind simultaneously to the hTAFII55 promoter-proximal region (Fig. 4) suggests that these two proteins may function in a combinatorial manner to regulate hTAFII55 gene expression. To explore this possibility, we performed cotransfection and reporter gene assays in Drosophila SL2 cells, which do not contain Sp1-family proteins (64,65), thereby avoiding the complications from the endogenous and multiple forms of Sp1 family proteins typically found in mammalian cells. In this experiment, expression plasmids for Sp1 and AP2 were cotransfected, individually or together, with a wild-type or mutated hTAFII55 promoter construct into SL2 cells. As shown in Figure 6, while AP2 enhanced the wild-type hTAFII55 promoter activity ∼2-fold, Sp1 was capable of inducing 43-fold activation (compare lanes 1, 4 and 7). The low level of activation by AP2 is likely due to the presence of endogenous Drosophila AP2 protein (40), which virtually saturates the reporter activity, or due to the lack of AP2-specific mammalian coactivators in insect cells (49–52). Nevertheless, the presence of both Sp1 and AP2 resulted in a 66-fold stimulation of activity, higher than the additive effect induced by Sp1 and AP2 alone (Fig. 6, lane 10). This combinatorial stimulation exhibited by Sp1 and AP2 apparently functions through the –60 Sp1-binding site and the –50 AP2-binding site, as mutations introduced at these two sites, but not the –40 and –20 Sp1-binding sites, abolished the cooperativity (compare lanes 2, 5, 8 and 11, and lanes 3, 6, 9 and 12). These data suggest that Sp1 and AP2 can work cooperatively to regulate hTAFII55 promoter activity through concurrent binding of Sp1 to the –60 Sp1-binding site and AP2 to the –50 AP2-binding site, consistent with the EMSA results indicating that –60 and –50 site are necessary and sufficient for the formation of a ternary complex comprising Sp1, AP2 and the hTAFII55 promoter fragment (Fig. 4).
Figure 6.
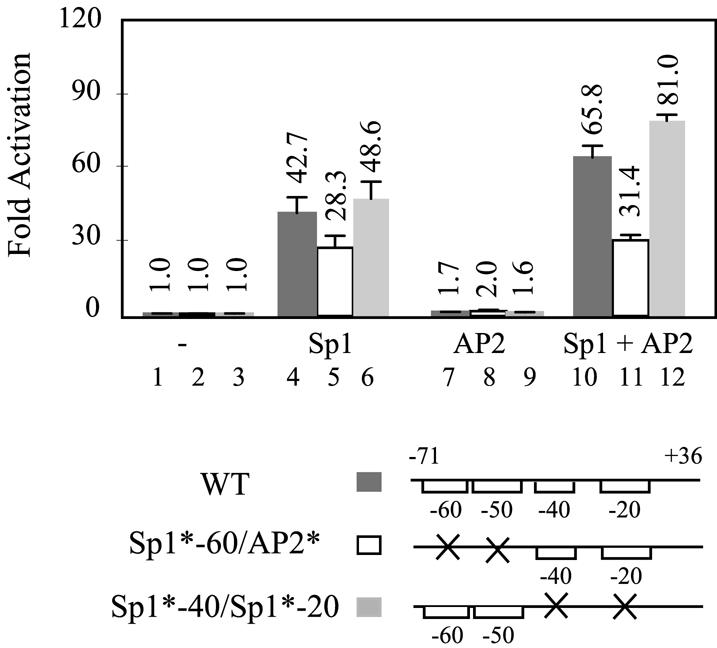
Sp1 and AP2 function cooperatively to regulate hTAFII55 promoter activity. Wild-type (WT) hTAFII55 promoter construct, pGL2-TAF55(–71/+36), and its derivatives pGL2-TAF55(–71/+36)Sp1*-60/AP2* and pGL2-TAF55(–71/+36)Sp1*-40/Sp1*-20, which contain mutations either at –50 and –60 or at –40 and –20 Sp1-binding sites, were cotransfected with Sp1 and AP2 insect expression plasmids, alone or in combination, into Drosophila SL2 cells. Reporter gene activities were normalized to that of the same reporter construct cotransfected with the cloning vector pPacO. Reporter plasmids used in the assay are depicted at the bottom, with brackets representing intact Sp1-binding sites and an ‘X’ denoting mutatations introduced at the core sequence.
Sp1-binding sites are not required for the core promoter activity of the TATA-less hTAFII55 promoter
The presence of Sp1-binding sites has been reported in many TATA-less promoters and is important for the function of TATA-less promoters (66,67). Since the previously characterized hTAFII55 promoter spanning –26 to +36 (11) still contains the core sequence of an Sp1-binding site and this fragment can indeed be recognized by Sp1 in EMSA (data not shown), we wondered whether this –20 Sp1-binding site might be an essential element for the function of the TATA-less hTAFII55 core promoter. To explore this possibility, we further deleted 10 nt from –26 to –17, which removed the –20 Sp1-binding site, and also introduced mutations in the Inr and DPE of this Sp1 site-deficient core promoter. Transfection of these promoter constructs into C-33A cells showed that the –16 to +36 fragment still maintains promoter activity, which is diminished when the Inr and/or DPE were mutated, similar to the results obtained with the –26 to +36 constructs (Fig. 7A). The –16 to +36 core sequence showed ∼2-fold reduction in promoter activity when compared with the –26 to +36 fragment, indicating that the –20 Sp1-binding site, although it is not required for hTAFII55 core promoter activity, indeed contributes to the promoter function.
Figure 7.

Sp1-binding sites are not essential for hTAFII55 core promoter function. (A) Reporter gene assay with different hTAFII55 core promoter constructs. Reporter constructs containing wild-type or mutated sequences at the –20 Sp1-binding site, Inr and/or DPE of the pGL2-TAF55(–26/+36) and pGL2-TAF55(–16/+36) were transfected into C-33A cells. Luciferase activities were normalized against that of pGL2-TAF55(–26/+36). Reporter constructs used in the assay are shown on the left. (B) The 52-bp sequence spanning –16 to +36, devoid of any Sp1-binding sites, can support Gal4–VP16-mediated activation in a heterologous promoter context. Transient transfection was performed in C-33A cells by cotransfecting different amounts of the Gal4–VP16 expression plasmid (pSGVP), together with either wild-type (WT) or mutated reporter constructs driven by five Gal4-binding sites as indicated.
To examine whether this hTAFII55 promoter fragment devoid of the –20 Sp1-binding site could nevertheless act as an independent core promoter module in mediating activator function in a heterologous context, we constructed a set of reporter clones containing five Gal4-binding sites linked to the –16 to +36 core promoter containing either wild-type or mutated Inr and/or DPE. The promoter activities of the resulting clones were examined in C-33A cells cotransfected with an increasing amount of a Gal4–VP16 expression plasmid. Clearly, the –16 to +36 sequence could support Gal4–VP16 transactivation, in the absence of any Sp1-binding sites (Fig. 7B), indicating that a TATA-less core promoter devoid of any Sp1-binding sites is sufficient to mediate transcriptional activation. Consistent with previous results (11), mutations introduced at Inr and/or DPE caused a significant reduction of the transcription activity, further confirming that Inr and DPE are important core promoter elements with a predominant role played by the Inr in the presence of Gal4–VP16.
Inr and DPE are independent core promoter modules that contribute to different aspects of transcription from a TATA-less promoter
To define the role of Inr and DPE in the hTAFII55 promoter containing multiple transcription factor-binding sites, we introduced mutations at the Inr and DPE in an hTAFII55 promoter construct spanning –128 to +87, which contains upstream binding sites for Sp1 and AP2 as well as putative downstream binding sites for AREB6 and E47 (see Fig. 1). When the wild-type promoter was examined by in vitro transcription performed with HeLa nuclear extracts, a cluster of transcription signals, including the major start site at +1 and several minor start sites corresponding to +3 to +6, were detected (Fig. 8A, lane 1). Surprisingly, transcription from the major start site was abolished upon Inr mutation, but those from the minor start sites were preserved (Fig. 8A, lane 3). Similar transcription patterns were observed when the 4 nt from –1 to +3 were changed from CACT to GAGC, without altering the +1 nucleotide A (data not shown), suggesting that the switch in start site preference when the Inr was mutated is not due to differences of the initiating nucleotide, but rather, the integrity of the Inr. In contrast, mutations at the DPE greatly reduced transcription signals from the minor start sites with little effect on the major start site (Fig. 8A, lane 2). This finding indicates that Inr and DPE play a different role in start site selection and appear to function independently. When these promoter constructs were examined in transfected C-33A cells by reporter gene assays, the transcription activity from each of the Inr- and DPE-mutated constructs was only reduced 2–3-fold (Fig. 8B). Taken together, these results suggest that Inr and DPE can function as independent core promoter modules to recruit transcription complexes through different mechanisms and the presence of promoter-proximal activator-binding sites in a complex promoter may mask the deleterious effects of mutations at the core promoter elements. These observations are consistent with a previous report that a complex Drosophila TATA-less promoter that presumably contains many transcription factor-binding sites still retains residual promoter activity upon mutations of the core promoter elements (68).
Figure 8.
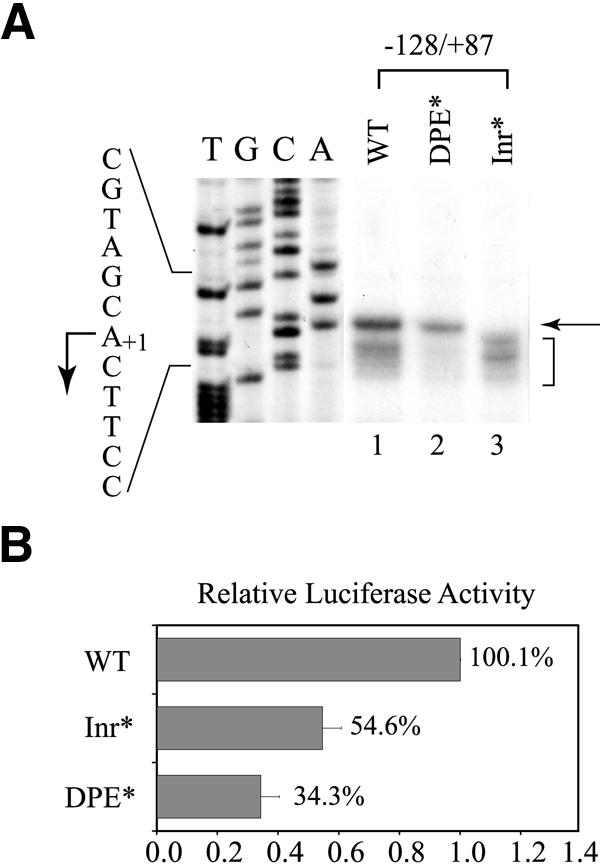
Inr and DPE contribute to different aspects of hTAFII55 promoter activity. (A) In vitro transcription performed with wild-type or mutated hTAFII55 core promoter constructs. Transcription was conducted in HeLa nuclear extracts with pGL2-TAF55(–128/+87), pGL2-TAF55(–128/+87)Inr* and pGL2-TAF55(–128/+87)DPE*. The transcripts were detected by primer extension. The arrow points to the major transcription start site, and the bracket denotes a cluster of minor start sites. DNA sequencing ladders, prepared from wild-type template with the phosphorylated form of Luc(AS)-1 primer were included as markers. The corresponding sequence is indicated on the left, with a bent arrow pointing to the A+1 position. (B) Reporter gene assay with wild-type or mutated hTAFII55 core promoter constructs. The same hTAFII55 promoter constructs were transiently transfected into C-33A cells. Luciferase activities are normalized against that of the wild-type construct.
DISCUSSION
Sp1 and AP2 are important regulators of hTAFII55 gene expression
Analysis of the hTAFII55 promoter-proximal region uncovers putative binding sites for many sequence-specific transcription factors, including ubiquitously expressed Sp1 and cell type-specific AP2. Sp1 is a zinc-finger protein originally identified in HeLa nuclear extracts important for viral and cellular promoter function (69–71). It has been implicated in the regulation of many house-keeping genes (19). Our studies show that Sp1 is an important regulator of hTAFII55 gene expression based on the following lines of evidence. First, ChIP assays demonstrated an in vivo association of Sp1 to the hTAFII55 promoter (Fig. 2). Secondly, point mutations introduced at the Sp1-binding sites, especially the –60 site, resulted in a significant decrease in promoter activity in both transfection assays (Fig. 5B) and in vitro transcription analyses (Fig. 5C). Thirdly, DNase I footprinting and EMSA proved the binding of purified Sp1 to its cognate DNA sequences (Figs 3 and 4). Finally, overexpression of Sp1 in insect cells dramatically enhanced promoter activity (Fig. 6). These data demonstrated that hTAFII55 is a newly identified Sp1-target gene. Intriguingly, hTAFII55 has been shown to interact directly with Sp1 (1), suggesting a possible autoregulatory loop for hTAFII55 gene expression, which remains to be further investigated.
Although the activity of Sp1 is generally believed to be constitutive, Sp1 can also function synergistically with many cell type-specific transcription factors. Examples include E2F (18), HNF3 (23), RXR (24), STAT1 (25), SREBP-1 (31), GATA-1 (72), MAZ (73), NF-Y (74), NFκB (75) and AP2 (41). Interestingly, putative binding sites for STAT, E2F and AP2 have been found at the hTAFII55 promoter-proximal region (Fig. 1), raising the possibility of collaboration between Sp1 and cell type-specific transcription factors in regulating the activity of the hTAFII55 promoter. AP2 has been implicated as an important regulator of gene expression during embryogenesis. Functional AP2-binding sites have been identified in many viral and cellular promoters, including SV40 (42,43), T-cell leukemia virus type I (76), human proenkephalin (77), keratin K14 (45) and estrogen receptor (36). We provided in vivo (Fig. 2) and in vitro (Figs 3 and 4) evidence that AP2 associates with the hTAFII55 promoter. Overexpression of AP2 in insect cells only marginally enhanced hTAFII55 promoter function, suggesting that AP2 may be a very weak activator and thus may require additional coactivator(s) to achieve a high level of activation. However, AP2 further stimulated Sp1-mediated transactivation when coexpressed with Sp1 in insect cells (Fig. 6). Such stimulation requires the AP2-binding site (Fig. 6), implicating a role of AP2 in regulating hTAFII55 promoter activity.
The juxtaposed AP2- and Sp1-binding sites on the hTAFII55 promoter raises an interesting question whether the ubiquitous presence of Sp1 in most of the mammalian tissues would preclude the binding of AP2, which shows a restricted expression pattern in adult tissues (42), and thereby block the regulatory pathways mediated by AP2. Our EMSA studies demonstrated that Sp1 and AP2 can bind simultaneously to the hTAFII55 promoter through the –60 Sp1-binding site and the –50 AP2-binding site (Fig. 4). Both sites are required for functional cooperativity between Sp1 and AP2 (Fig. 6). These results indicate that the ternary complex is formed through protein binding to the adjacent core sequences and not mediated by direct Sp1–AP2 interactions, although direct interactions between Sp1 and AP2 have been reported (78–80). Our data also differ from several previous reports that favor a competition model where Sp1 and AP2 exert opposite effects in regulating gene expression and the outcome is determined by the AP2/Sp1 ratio in the specific cell type (81,82). It is tempting to speculate that AP2, which also had been shown to function together with Sp1 (41), works by recruiting transcriptional coactivator(s) that in turn facilitates Sp1 activation on the hTAFII55 promoter. On the other hand, it remains possible that AP2 and Sp1 function cooperatively by modulating binding affinity in a chromatin context, since transfected DNA may assemble into chromatin-like structures.
Our studies also indicate that not all Sp1-binding sites are functionally equivalent. Among the four Sp1-binding sites located at –60, –50, –40 and –20 of the hTAFII55 promoter-proximal region, the –60 site showed the highest affinity for Sp1 binding, as illustrated by DNase I footprinting (Fig. 3C) and EMSA (Fig. 5A, lanes 5–8). The –60 Sp1-binding site is required for concurrent binding of Sp1 and AP2 (Fig. 4) as well as the functional cooperativity with AP2 (Fig. 6). In contrast, the –50 Sp1-binding site, which overlaps with the binding sequence recognized by AP2, has a low affinity for Sp1 (Fig. 5A, lanes 9–12). It is probable that these two overlapping binding sites provide the basis for differential regulation by Sp1 and AP2 proteins in different cellular environments. In the current study, we uncovered a novel Sp1-binding site at –40 that was not initially identified by computer database searches (Fig. 4). The –40 sequence is also a high-affinity Sp1-binding site which plays a functional role in regulating hTAFII55 promoter activity (Fig. 5). The –20 Sp1-binding site also contributes to the hTAFII55 promoter activity, as deletion of this Sp1-binding site showed an ∼2–3-fold reduction of hTAFII55 promoter activity (Fig. 7). However, since nucleotide substitutions at the –20 region in a longer hTAFII55 promoter construct only exhibited a marginal effect on hTAFII55 transcription (Fig. 5B and C), we could not rule out a potential involvement of other cellular factors acting through sequences surrounding the –20 Sp1-binding site. This possibility remains to be further investigated.
Sp1 is not required for the hTAFII55 core promoter function
Our finding that Sp1-binding sites are not required for the core promoter activity of the hTAFII55 promoter is surprising, as Sp1 is thought to be an essential factor in recruiting TFIID and the rest of the general transcription machinery to TATA-less promoters (14). Our report pointed out the possibility of alternative pathways by which a pre-initiation complex assembles on a TATA-less promoter. Such pathways do not require Sp1 as an anchoring factor, and may depend on other cellular proteins, such as NC2 (83) which has been shown to activate transcription specifically from DPE-driven, but not TATA-containing, promoters, to facilitate pre-initiation complex assembly. It is likely that dramatic differences do exist between TATA-containing and TATA-less promoters in their mechanisms to direct transcription initiation. Our current studies also show that the hTAFII55 core promoter consists of an Inr and a DPE, both of which are essential for core promoter function (Fig. 7A) but can function independently in the presence of upstream transcription factor-binding sites (Fig. 8A). These data suggest a differential requirement of the DPE in directing hTAFII55 promoter activity, depending on the surrounding transcription factor-binding sites. In the shortest core promoter construct (–16 to +36), devoid of any activator-binding sites, the DPE plays a less significant role than the Inr (Fig. 7A). This property remains unchanged in the presence of Gal4–VP16. However, in the context of the native hTAFII55 promoter, the DPE is able to act as an independent core promoter element in the absence of the Inr (Fig. 8). In light of the recent finding that TATA and DPE showed different responses to enhancer function in Drosophila embryos (84), we speculate that some transcriptional activators, such as Sp1 and AP2, may work preferentially through the DPE. This possibility is currently under investigation. Taken together, our results suggest that Inr and DPE of the hTAFII55 gene play a non-redundant role in the function of a TATA-less core promoter and may respond differentially to distinct groups of transcriptional regulators.
Acknowledgments
ACKNOWLEDGEMENTS
We thank C. Desplan for pPacPL, T. Williams for the SPRSV-AP2 construct, and J.-L. Chen and R. Tjian for pPacO and pPacSp1 plasmids as well as anti-Sp1 antibodies. We are grateful to P. deHaseth, P. Kaur, M. C. Thomas and S.-Y. Wu for insightful discussion, and S. Y. Hou, M. C. Thomas and S.-Y. Wu for critical reading of the manuscript. This work was supported in part by Grant 9950106N from the American Heart Association and in part by Grants GM59643 and CA81017 from the National Institutes of Health. C.-M.C. is a Mt Sinai Health Care Foundation Scholar.
REFERENCES
- 1.Chiang C.-M. and Roeder,R.G. (1995) Cloning of an intrinsic human TFIID subunit that interacts with multiple transcriptional activators. Science, 267, 531–536. [DOI] [PubMed] [Google Scholar]
- 2.Lavigne A.-C., Mengus,G., May,M., Dubrovskaya,V., Tora,L., Chambon,P. and Davidson,I. (1996) Multiple interactions between hTAFII55, and other TFIID subunits. Requirements for the formation of stable ternary complexes between hTAFII55 and the TATA-binding protein. J. Biol. Chem., 271, 19774–19780. [DOI] [PubMed] [Google Scholar]
- 3.Wieczorek E., Brand,M., Jacq,X. and Tora,L. (1998) Function of TAFII-containing complex without TBP in transcription by RNA polymerase II. Nature, 393, 187–191. [DOI] [PubMed] [Google Scholar]
- 4.Gegonne A., Weissman,J.D. and Singer,D.S. (2001) TAFII55 binding to TAFII250 inhibits its acetyltransferase activity. Proc. Natl Acad. Sci. USA, 98, 12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavigne A.-C., Mengus,G., Gangloff,Y.-G., Wurtz,J.-M. and Davidson,I. (1999) Human TAFII55 interacts with the vitamin D3 and thyroid hormone receptors and with derivatives of the retinoid X receptor that have altered transactivation properties. Mol. Cell. Biol., 19, 5486–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moqtaderi Z., Yale,J.D., Struhl,K. and Buratowski,S. (1996) Yeast homologues of higher eukaryotic TFIID subunits. Proc. Natl Acad. Sci. USA, 93, 14654–14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibuya T., Tsuneyoshi,S., Azad,A.K., Urushiyama,S., Ohshima,Y. and Tani,T. (1999) Characterization of the ptr6+ gene in fission yeast: a possible involvement of a transcriptional coactivator TAF in nucleocytoplasmic transport of mRNA. Genetics, 152, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu S.-Y., Thomas,M.C., Hou,S.Y., Likhite,V. and Chiang,C.-M. (1999) Isolation of mouse TFIID and functional characterization of TBP and TFIID in mediating estrogen receptor and chromatin transcription. J. Biol. Chem., 274, 23480–23490. [DOI] [PubMed] [Google Scholar]
- 9.Dantonel J.-C., Murthy,K.G., Manley,J.L. and Tora,L. (1997) Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature, 389, 399–402. [DOI] [PubMed] [Google Scholar]
- 10.Proudfoot N.J., Furger,A. and Dye,M.J. (2002) Integrating mRNA processing with transcription. Cell, 108, 501–512. [DOI] [PubMed] [Google Scholar]
- 11.Zhou T. and Chiang,C.-M. (2001) The intronless and TATA-less human TAFII55 gene contains a functional initiator and a downstream promoter element. J. Biol. Chem., 276, 25503–25511. [DOI] [PubMed] [Google Scholar]
- 12.Purrello M., Di Pietro,C., Viola,A., Rapisarda,A., Stevens,S., Guermah,M., Tao,Y., Bonaiuto,C., Arcidiacono,A., Messina,A., Sichel,G., Grzeschik,K.-H. and Roeder,R. (1998) Genomics and transcription analysis of human TFIID. Oncogene, 16, 1633–1638. [DOI] [PubMed] [Google Scholar]
- 13.Kriwacki R.W., Schultz,S.C., Steitz,T.A. and Caradonna,J.P. (1992) Sequence-specific recognition of DNA by zinc-finger peptides derived from the transcription factor Sp1. Proc. Natl Acad. Sci. USA, 89, 9759–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pugh B.F. and Tjian,R. (1990) Mechanism of transcriptional activation by Sp1: evidence for coactivators. Cell, 61, 1187–1197. [DOI] [PubMed] [Google Scholar]
- 15.Philipsen S. and Suske,G. (1999) A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res., 27, 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birnbaum M.J., van Wijnen,A.J., Odgren,P.R., Last,T.J., Suske,G., Stein,G.S. and Stein,J.L. (1995) Sp1 trans-activation of cell cycle regulated promoters is selectively repressed by Sp3. Biochemistry, 34, 16503–16508. [DOI] [PubMed] [Google Scholar]
- 17.Lin S.-Y., Black,A.R., Kostic,D., Pajovic,S., Hoover,C.N. and Azizkhan,J.C. (1996) Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol. Cell. Biol., 16, 1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlseder J., Rotheneder,H. and Wintersberger,E. (1996) Interaction of Sp1 with the growth- and cell cycle-regulated transcription factor E2F. Mol. Cell. Biol., 16, 1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandeis M., Frank,D., Keshet,I., Siegfried,Z., Mendelsohn,M., Nemes,A., Temper,V., Razin,A. and Cedar,H. (1994) Sp1 elements protect a CpG island from de novo methylation. Nature, 371, 435–438. [DOI] [PubMed] [Google Scholar]
- 20.Macleod D., Charlton,J., Mullins,J. and Bird,A.P. (1994) Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev., 8, 2282–2292. [DOI] [PubMed] [Google Scholar]
- 21.Marin M., Karis,A., Visser,P., Grosveld,F. and Philipsen,S. (1997) Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell, 89, 619–628. [DOI] [PubMed] [Google Scholar]
- 22.Pasceri P., Pannell,D., Wu,X. and Ellis,J. (1998) Full activity from human β-globin locus control region transgenes requires 5′HS1, distal β-globin promoter, and 3′ β-globin sequences. Blood, 92, 653–663. [PubMed] [Google Scholar]
- 23.Braun H. and Suske,G. (1998) Combinatorial action of HNF3 and Sp family transcription factors in the activation of the rabbit uteroglobin/CC10 promoter. J. Biol. Chem., 273, 9821–9828. [DOI] [PubMed] [Google Scholar]
- 24.Krey G., Mahfoudi,A. and Wahli,W. (1995) Functional interactions of peroxisome proliferator-activated receptor, retinoid-X receptor, and Sp1 in the transcriptional regulation of the acyl-coenzyme-A oxidase promoter. Mol. Endocrinol., 9, 219–231. [DOI] [PubMed] [Google Scholar]
- 25.Look D.C., Pelletier,M.R., Tidwell,R.M., Roswit,W.T. and Holtzman,M.J. (1995) Stat1 depends on transcriptional synergy with Sp1. J. Biol. Chem., 270, 30264–30267. [DOI] [PubMed] [Google Scholar]
- 26.Jackson S.P., MacDonald,J.J., Lees-Miller,S. and Tjian,R. (1990) GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell, 63, 155–165. [DOI] [PubMed] [Google Scholar]
- 27.Jackson S.P. and Tjian,R. (1988) O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell, 55, 125–133. [DOI] [PubMed] [Google Scholar]
- 28.Hoey T., Weinzierl,R.O., Gill,G., Chen,J.-L., Dynlacht,B.D. and Tjian,R. (1993) Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell, 72, 247–260. [DOI] [PubMed] [Google Scholar]
- 29.Gill G., Pascal,E., Tseng,Z.H. and Tjian,R. (1994) A glutamine-rich hydrophobic patch in transcription factor Sp1 contacts the dTAFII110 component of the Drosophila TFIID complex and mediates transcriptional activation. Proc. Natl Acad. Sci. USA, 91, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanese N., Saluja,D., Vassallo,M.F., Chen,J.-L. and Admon,A. (1996) Molecular cloning and analysis of two subunits of the human TFIID complex: hTAFII130 and hTAFII110. Proc. Natl Acad. Sci. USA, 93, 13611–13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Näär A.M., Beaurang,P.A., Zhou,S., Abraham,S., Solomon,W. and Tjian,R. (1999) Composite co-activator ARC mediates chromatin-directed transcriptional activation. Nature, 398, 828–832. [DOI] [PubMed] [Google Scholar]
- 32.Ryu S., Zhou,S., Ladurner,A.G. and Tjian,R. (1999) The transcriptional cofactor complex CRSP is required for activity of the enhancer-binding protein Sp1. Nature, 397, 446–450. [DOI] [PubMed] [Google Scholar]
- 33.Doetzlhofer A., Rotheneder,H., Lagger,G., Koranda,M., Kurtev,V., Brosch,G., Wintersberger,E. and Seiser,C. (1999) Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol., 19, 5504–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams T., Admon,A., Lüscher,B. and Tjian,R. (1988) Cloning and expression of AP-2, a cell-type-specific transcription factor that activates inducible enhancer elements. Genes Dev., 2, 1557–1569. [DOI] [PubMed] [Google Scholar]
- 35.Moser M., Imhof,A., Pscherer,A., Bauer,R., Amselgruber,W., Sinowatz,F., Hofstädter,F., Schüle,R. and Buettner,R. (1995) Cloning and characterization of a second AP-2 transcription factor: AP-2β. Development, 121, 2779–2788. [DOI] [PubMed] [Google Scholar]
- 36.McPherson L.A., Baichwal,V.R. and Weigel,R.J. (1997) Identification of ERF-1 as a member of the AP2 transcription factor family. Proc. Natl Acad. Sci. USA, 94, 4342–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao F., Satoda,M., Licht,J.D., Hayashizaki,Y. and Gelb,B.D. (2001) Cloning and characterization of a novel mouse AP-2 transcription factor, Ap-2δ, with unique DNA binding and transactivation properties. J. Biol. Chem., 276, 40755–40760. [DOI] [PubMed] [Google Scholar]
- 38.Winning R.S., Shea,L.J., Marcus,S.J. and Sargent,T.D. (1991) Developmental regulation of transcription factor AP-2 during Xenopus laevis embryogenesis. Nucleic Acids Res., 19, 3709–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meier P., Koedood,M., Philipp,J., Fontana,A. and Mitchell,P.J. (1995) Alternative mRNAs encode multiple isoforms of transcription factor AP-2 during murine embryogenesis. Dev. Biol., 169, 1–14. [DOI] [PubMed] [Google Scholar]
- 40.Monge I. and Mitchell,P.J. (1998). DAP-2, the Drosophila homolog of transcription factor AP-2. Mech. Dev., 76, 191–195. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell P.J., Wang,C. and Tjian,R. (1987) Positive and negative regulation of transcription in vitro: enhancer-binding protein AP-2 is inhibited by SV40 T antigen. Cell, 50, 847–861. [DOI] [PubMed] [Google Scholar]
- 42.Mitchell P.J., Timmons,P,M., Hébert,J.M., Rigby,P.W. and Tjian,R. (1991) Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev., 5, 105–119. [DOI] [PubMed] [Google Scholar]
- 43.Imagawa M., Chiu,R. and Karin,M. (1987) Transcription factor AP-2 mediates induction by two different signal-transduction pathways: protein kinase C and cAMP. Cell, 51, 251–260. [DOI] [PubMed] [Google Scholar]
- 44.Lüscher B., Mitchell,P.J., Williams,T. and Tjian,R. (1989) Regulation of transcription factor AP-2 by the morphogen retinoic acid and by second messengers. Genes Dev., 3, 1507–1517. [DOI] [PubMed] [Google Scholar]
- 45.Leask A., Byrne,C. and Fuchs,E. (1991) Transcription factor AP2 and its role in epidermal-specific gene. Proc. Natl Acad. Sci. USA, 88, 7948–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng Y.-X., Somasundaram,K. and El-Deiry,W.S. (1997) AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nature Genet., 1, 78–82. [DOI] [PubMed] [Google Scholar]
- 47.Batschè E., Muchardt,C., Behrens,J., Hurst,H.C. and Crèmisi,C. (1998) RB and c-Myc activate expression of the E-cadherin gene in epithelial cells through interaction with transcription factor AP-2. Mol. Cell. Biol., 18, 3647–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mertens P.R., Alfonso-Jaume,M.A., Steinmann,K. and Lovett,D.H. (1998) A synergistic interaction of transcription factors AP2 and YB-1 regulates gelatinase A enhancer-dependent transcription. J. Biol. Chem., 273, 32957–32965. [DOI] [PubMed] [Google Scholar]
- 49.Kannan P. and Tainsky,M.A. (1999) Coactivator PC4 mediates AP-2 transcriptional activity and suppresses ras-induced transformation dependent on AP-2 transcriptional interference. Mol. Cell. Biol., 19, 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kannan P., Yu,Y., Wankhade,S. and Tainsky,M.A. (1999) PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Res., 27, 866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bamforth S.D., Braganca,J., Eloranta,J.J., Murdoch,J.N., Marques,F.I.R., Kranc,K.R., Farza,H., Henderson,D.J., Hurst,H.C. and Bhattacharya,S. (2001) Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nature Genet., 29, 469–474. [DOI] [PubMed] [Google Scholar]
- 52.Braganca J., Swingler,T., Marques,F.I.R., Jones,T., Eloranta,J.J., Hurst,H.C., Shioda,T. and Bhattacharya,S. (2002) Human CREB-binding protein/p300-interacting transactivator with ED-rich tail (CITED) 4, a new member of the CITED family, functions as a co-activator for transcription factor AP-2. J. Biol. Chem., 8, 8559–8565. [DOI] [PubMed] [Google Scholar]
- 53.Williams T. and Tjian,R. (1991) Analysis of the DNA-binding and activation properties of the human transcription factor AP-2. Genes Dev., 5, 670–682. [DOI] [PubMed] [Google Scholar]
- 54.Wu S.-Y. and Chiang,C.-M. (1996) Establishment of stable cell lines expressing potentially toxic proteins by tetracycline-regulated and epitope-tagging methods. BioTechniques, 21, 718–725. [DOI] [PubMed] [Google Scholar]
- 55.Hou S.Y., Wu,S.-Y., Zhou,T., Thomas,M.C. and Chiang,C.-M. (2000) Alleviation of human papillomavirus E2-mediated transcriptional repression via formation of a TATA binding protein (or TFIID)-TFIIB-RNA polymerase II-TFIIF preinitiation complex. Mol. Cell. Biol., 20, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadowski I. and Ptashne,M. (1989) A vector for expressing GAL4(1–147) fusions in mammalian cells. Nucleic Acids Res., 17, 7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sadowski I., Ma,J., Triezenberg,S. and Ptashne,M. (1988) GAL4–VP16 is an unusually potent transcriptional activator. Nature, 335, 563–564. [DOI] [PubMed] [Google Scholar]
- 58.Krasnow M.A., Saffman,E.E., Kornfeld,K. and Hogness,D.S. (1989) Transcriptional activation and repression by Ultrabithorax proteins in cultured Drosophila cells. Cell, 57, 1031–1043. [DOI] [PubMed] [Google Scholar]
- 59.Courey A.J. and Tjian,R. (1988) Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell, 55, 887–898. [DOI] [PubMed] [Google Scholar]
- 60.Kuo M.-H. and Allis,C.D. (1999) In vitro cross-linking and immunoprecipitation for studying dynamic protein: DNA associations in a chromatin environment. Methods, 19, 425–433. [DOI] [PubMed] [Google Scholar]
- 61.Chiang C.-M. and Roeder,R.G. (1993) Expression and purification of general transcription factors by FLAG epitope-tagging and peptide elution. Peptide Res., 6, 62–64. [PubMed] [Google Scholar]
- 62.Chiang C.-M., Broker,T.R. and Chow,L.T. (1991) An E1M∧E2C fusion protein encoded by human papillomavirus type 11 is a sequence-specific transcription repressor. J. Virol., 65, 3317–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Nocera P.P. and Dawid,I.B. (1983) Transient expression of genes introduced into cultured cells of Drosophila. Proc. Natl Acad. Sci. USA, 80, 7095–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagen G., Müller,S., Beato,M. and Suske,G. (1994) Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J., 13, 3843–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pascal E. and Tjian,R. (1991) Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev., 5, 1646–1656. [DOI] [PubMed] [Google Scholar]
- 66.Pugh B.F. and Tjian,R. (1991) Transcription from a TATA-less promoter requires a multisubunit TFIID complex. Genes Dev., 5, 1935–1945. [DOI] [PubMed] [Google Scholar]
- 67.Smale S.T. (2001) Core promoters: active contributors to combinatorial gene regulation. Genes Dev., 15, 2503–2508. [DOI] [PubMed] [Google Scholar]
- 68.Burke T.W. and Kadonaga,J.T. (1997) The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of Drosophila. Genes Dev., 11, 3020–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dynan W.S. and Tjian,R. (1983) The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell, 35, 79–87. [DOI] [PubMed] [Google Scholar]
- 70.Jones K.A., Yamamoto,K.R. and Tjian,R. (1985) Two distinct transcription factors bind to the HSV thymidine kinase promoter in vitro. Cell, 42, 559–572. [DOI] [PubMed] [Google Scholar]
- 71.Jones K.A., Kadonaga,J.T., Luciw,P.A. and Tjian,R. (1986) Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science, 232, 755–759. [DOI] [PubMed] [Google Scholar]
- 72.Merika M. and Orkin,S.H. (1995) Functional synergy and physical interactions of the erythroid transcription factor GATA-1 with the Krüppel family proteins Sp1 and EKLF. Mol. Cell. Biol., 15, 2437–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parks C.L. and Shenk,T. (1997) Activation of the adenovirus major late promoter by transcription factors MAZ and Sp1. J. Virol., 71, 9600–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sorensen P. and Wintersberger,E. (1999) Sp1 and NF-Y are necessary and sufficient for growth-dependent regulation of the hamster thymidine kinase promoter. J. Biol. Chem., 274, 30943–30949. [DOI] [PubMed] [Google Scholar]
- 75.Pazin M.J., Sheridan,P.L., Cannon,K., Cao,Z., Keck,J.G., Kadonaga,J.T. and Jones,K.A. (1996) NF-κB-mediated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes Dev., 10, 37–49. [DOI] [PubMed] [Google Scholar]
- 76.Nyborg J.K. and Dynan,W.S. (1990) Interaction of cellular proteins with the human T-cell leukemia virus type I transcriptional control region. Purification of cellular proteins that bind the 21-base pair repeat elements. J. Biol. Chem., 265, 8230–8236. [PubMed] [Google Scholar]
- 77.Hyman S.E., Comb,M., Pearlberg,J. and Goodman,H.M. (1989) An AP-2 element acts synergistically with the cyclic AMP- and phorbol ester-inducible enhancer of the human proenkephalin gene. Mol. Cell. Biol., 9, 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pena P., Reutens,A.T., Albanese,C., D’Amico,M., Watanabe,G., Donner,A., Shu,I.-W., Williams,T. and Pestell,R.G. (1999) Activator protein-2 mediates transcriptional activation of the CYP11A1 gene by interaction with Sp1 rather than binding to DNA. Mol. Endocrinol., 13, 1402–1416. [DOI] [PubMed] [Google Scholar]
- 79.Vergeer W.P., Sogo,J.M., Pretorius,P.J. and de Vries,W.N. (2000) Interaction of Ap1, Ap2, and Sp1 with the regulatory regions of the human pro-α(I) collagen gene. Arch. Biochem. Biophys., 377, 69–79. [DOI] [PubMed] [Google Scholar]
- 80.Xu Y., Porntadavity,S. and St Clair,D.K. (2002) Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2). Biochem. J., 362, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen T.-T., Wu,R.-L., Castro-Munozledo,F. and Sun,T.-T. (1997) Regulation of K3 keratin gene transcription by Sp1 and AP-2 in differentiating rabbit corneal epithelial cells. Mol. Cell. Biol., 17, 3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Novikov D.K. and Kamps,M.E. (2001) Characterization of the promoter region of the human peroxisomal multifunctional enzyme type 2 gene. Biochem. Biophys. Res. Commun., 284, 226–231. [DOI] [PubMed] [Google Scholar]
- 83.Willy P.J., Kobayashi,R. and Kadonaga,J.T. (2000) A basal transcription factor that activates or represses transcription. Science, 290, 982–985. [DOI] [PubMed] [Google Scholar]
- 84.Butler J.E.F. and Kadonaga,J.T. (2001) Enhancer-promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev., 15, 2515–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.