

Abstract
Gas-phase reactions of multiply protonated polypeptides and metal containing anions represent a new methodology for manipulating the cationizing agent composition of polypeptides. This approach affords greater flexibility in forming metal containing ions than commonly used methods, such as electrospray ionization of a metal salt/peptide mixture and matrix-assisted laser desorption. Here, the effects of properties of the polypeptide and anionic reactant on the nature of the reaction products are investigated. For a given metal, the identity of the ligand in the metal containing anion is the dominant factor in determining product distributions. For a given polypeptide ion, the difference between the metal ion affinity and the proton affinity of the negatively charged ligand in the anionic reactant is of predictive value in anticipating the relative contributions of proton transfer and metal ion transfer. Furthermore, the binding strength of the ligand anion to charge sites in the polypeptide correlates with the extent of observed cluster ion formation. Polypeptide composition, sequence, and charge state can also play a notable role in determining the distribution of products. In addition to their usefulness in gas-phase ion synthesis strategies, the reactions of protonated polypeptides and metal containing anions represent an example of a gas-phase ion/ion reaction that is sensitive to polypeptide structure. These observations are noteworthy in that they allude to the possibility of obtaining information, without requiring fragmentation of the peptide backbone, about ion structure as well as the relative ion affinities associated with the reactants.
Introduction
The nature of the charge associated with a polypeptide plays an essential role in its gas-phase ion chemistry. The numbers and identities of the cationizing species as well as their respective interactions with the polypeptides affect the chemical behavior of the ion. By changing the cationizing reagent, complementary primary sequence information can be obtained from a tandem mass spectrometry experiment. For example, while a protonated peptide usually fragments at various locations along the peptide backbone,1–4 collision-induced dissociation (CID) of a sodiated peptide can result in preferential fragmentation adjacent to the C-terminal residue.5–7 Additionally, the favored dissociation channels are highly dependent upon the charge states of the polypeptide ions.4,8,9 Since the identities and total charge of cationizing agents influence the fragmentation chemistry of peptide and protein cations in the gas phase, it is desirable to be able to manipulate both the total charge of the ion and the relative numbers of each type of cationizing reagent. While cationization of peptides within the context of structural analysis via tandem mass spectrometry is emphasized here, the ability to exchange metal ions for other types of cations, such as other metals, may also prove to be relevant in the broader context of bioinorganic chemistry.
Electrospray ionization (ESI)10,11 and matrix-assisted laser desorption ionization (MALDI)12 are the most widely employed ionization methods for peptides and proteins. Depending upon ionization and solution conditions, the polypeptide ion may be comprised of different numbers and types of cationizing reagents. In most cases, protonation is the predominant means by which peptides and proteins are ionized, with minor degrees of cationization by sodium and potassium. Commonly, metal cationized peptide/protein ions are introduced to the mass spectrometer by electrospray of solutions containing the peptide/protein of interest and a metal salt.5–7,13–21 The mass spectrum of such a mixture is comprised of peptide ions with various combinations of cationizing reagents. The presence and abundance of metal cationized peptides is highly dependent upon solution composition and pH. Thus, in some cases, it may be difficult to obtain a signal for a specific metal cationized ion of interest, and only in rare cases is it straightforward to form a specific metal cationized species exclusive of any other pseudo-molecular ions. This situation applies to all means of ionization currently in use.
In a preliminary report, we described an alternative strategy for generating gaseous metal containing peptide/protein ions that involves ion/ion reactions of multiply protonated species with metal containing anions.22 This approach allows exploration of a variety of metal/peptide interactions as well as the capability for formation of metal containing ions that may not be produced directly by ESI or MALDI of mixtures of metal salts and polypeptides. Quadrupole ion trap mass spectrometers equipped with multiple electrospray ionization sources are employed in this technique.23,24 In this method, multiply protonated peptide/protein ions are formed with one ion source, while anions of metal salts are formed from a second ion source. The ion/ion reactions occur during a mutual ion polarity storage period. The reactant ions are formed separately to allow for independent optimization of solution and electrospray conditions. Therefore, this approach avoids signal suppression25 and dilution effects, which can be observed when polypeptide and metal salts are electrosprayed from the same solution. Another advantage of forming the reactant ions separately is that it facilitates the isolation of specific ions for participation in the ion/ion reactions. Products from the ion/ion reactions of selected reactants can be mass analyzed directly or isolated and subjected to collision-induced dissociation (CID) prior to mass analysis.
Herein, factors found to affect the identities and abundances of products from ion/ion reactions intended to generate metal cationized polypeptides are described. The effect of the identity of the ligand in the anionic reactants on observed reaction products was the primary focus, although the impact of properties of the polypeptides on reaction products was also investigated. In this study, bradykinin and ubiquitin served as a model peptide and protein, respectively, since each has been well-characterized using a variety of other gas-phase methods.26–39 Silver containing anions were used in the cation-switching reactions described next. Our previous communication demonstrated the utility of cation-switching reactions to form ions cationized by other metals.22 There are several reasons for the choice of silver. For initial studies of reactions of protonated polypeptides with metal containing anions, it is desirable to use a metal that primarily exists in the +1 oxidation state and is not easily oxidized or reduced, thereby avoiding ambiguity in assignment of the product ion identities that might arise due to uncertainty in the metal oxidation state. The large shift in mass associated with the addition of silver, relative to that of many other singly charged metal ions, facilitates the identification of high mass, multiply charged reaction products using a mass analyzer with modest resolving power, such as a quadrupole ion trap. Another reason for selecting silver is the availability of a variety of silver salts. Furthermore, previous studies of the fragmentation behavior of silver cationized peptides suggest that silver cationization can be employed in strategies associated with peptide sequencing in the gas phase via CID.16,40,41 Results from the cation-switching ion/ion reactions with protonated peptide ions are presented first followed by those from the reactions with protonated ubiquitin ions.
Experimental Procedures
Bradykinin and bovine ubiquitin were obtained from Sigma (St. Louis, MO). Silver acetate, silver nitrate, silver triflate, silver hexafluorophosphate, and perfluro-1,3-dimethylcyclohexane (PDCH) were purchased from Aldrich (St. Louis, MO). Des-R1 bradykinin and des-R9 bradykinin were obtained from BACHEM (King of Prussia, PA). Aqueous peptide and protein solutions (~0.2 mg/mL) for nanospray ionization were prepared in 1% acetic acid. Metal salts (~2.0–4.0 mg/mL) were dissolved in water and nanosprayed directly. In some cases, selected peptide/protein and metal salt solutions were combined prior to nanospray ionization. In these cases, highly complex spectra typically resulted with the observation of peptide/protein ions with mixtures of metal cationizing species and various degrees of clustering with the anions. These solutions consisted of the polypeptide (~0.1 mg/mL) and the metal salt (~0.2 mg/mL) in 50:50 (v/v) methanol/water.
The gas-phase ion/ion reactions were performed in quadrupole ion trap mass spectrometers equipped with multiple electrospray ionization sources, as previously described.23,24 Cationized peptides and metal containing anions were formed via nanospray ionization of the solutions mentioned previously. Typical nanospray voltages were +1.2 and −0.9 kV, respectively. PDCH was ionized by atmospheric sampling glow discharge ionization. The experimental sequence began with accumulation of positive ions (~200–600 ms) followed by ion isolation (~60 ms). The doubly charged ion of ubiquitin was formed by reacting the initial charge state distribution of ubiquitin cations with PDCH anions prior to the reaction with metal containing anions. Accumulation times of negative ions ranged from 500 to 1000 ms for anions generated by nanospray ionization. As discussed further next, the present apparatus does not allow for anion isolation after cation accumulation and isolation. These experiments, therefore, were restricted to species that yield the most abundant anions. Mutual ion storage times were typically 300–900 ms. Residual anions were ejected from the trap using an RF ramp after the mutual storage period. Isolation (~60 ms) and CID (100 ms) of selected ion/ion reaction products was performed in some cases. Mass analysis was performed using resonance ejection. Ion/ion reaction product spectra and CID spectra are the average of 100–200 scans.
High-level density functional theory (DFT) and ab initio computations were carried out to obtain theoretical structures and energies of ions and neutral species relevant to this study. Geometry optimizations, including vibrational analyses, were performed using Gaussian 9842,43 at the B3LYP/6-31+G(d)44 level for ligands and protonated species. For silver containing ions and neutrals, energies were calculated in a similar manner using the 6-311+G(d) basis sets for atoms other than silver and Hay–Wadt VDZ (n + 1) effective core potentials and valence basis sets45,46 for the description of the silver metal atom. All stationary points were found to be true minima. Single-point energy calculations were performed at the MP2(full)/6-311+G-(2d,2p) level using the optimized geometries to determine the energies of the various species. Thermal energy corrections were scaled by a factor of 0.9804.47 Energy values corresponding to optimized geometries were used to calculate the proton and silver cation affinities of the ligands. The values reported herein have been subjected to zero point energy correction.
Results and Discussion
Reaction Phenomenology
As mentioned in previous discussions of ion/ion reaction phenomenology, ion/ion reaction products can result from two major processes that occur during a single ion/ion encounter: direct charged particle transfer (e.g., proton hopping) over a distance without formation of a long-lived complex and via a long-lived complex.48,49 Charged particle transfer at a distance leads exclusively to charge transfer reaction products, whereas a wider variety of products can arise from a long-lived ion/ion complex. These products result from dissociation and/or collisional cooling of the ion/ion complex. When considering reactions of multiply protonated polypeptides and metal containing anions, proton transfer, cation switching (exchange of protons for a metal ion), and adduct products can be formed. Thus, these reactions have potential as gas-phase synthetic routes for ions that contain a metal ion, a ligand ion, or both. An indication of the major reaction products that can result from the reaction of a doubly protonated polypeptide with a singly charged anion containing both a metal (+1 oxidation state) and ligand is given in Figure 1 as they might appear on a mass-to-charge scale.
Figure 1.
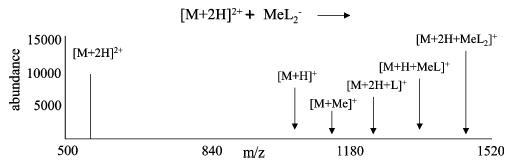
Illustration of possible products from the reaction of a doubly protonated polypeptide with a metal containing anion. The metal (Me) has an oxidation state of +1. L represents the ligand in the anion.
A diagram accounting for the formation of each reaction product in Figure 1 is shown in Scheme 1. The ion/ion reaction and dissociation steps are indicated as being irreversible, while the relaxation/excitation of complexes are indicated as reversible processes. The ion/ion reaction steps are effectively irreversible in that they are of such high exothermicity that the reversion of the recombination products to reactants of opposite polarity is highly unlikely. The decomposition reactions are essentially irreversible because the neutral product, which is formed with a very small number of densities to begin with, is not stored and hence is removed by the vacuum pumping. The associated rate constant subscripts of Scheme 1 indicate the type of reaction (i.e., the ion/ion reactions of proton hopping (denoted by the subscript hop) and complex formation (c), dissociation (d), stabilization via collisional and radiative relaxation (s), and activation (s−1)). A proton from the peptide, M, can be transferred to the anion via proton hopping, thereby providing one possible route to formation of [M + H]+. This product can also result from dissociation of a chemically bound ion/ion complex through losses of both HL and MeL, where L represents a ligand and Me represents a metal. Corresponding to the net replacement of the peptide cationizing agent, [M + Me]+ is generated via dissociation of the ion/ion complex by consecutive losses of HL. Note that there is only one pathway for the formation of the cation-switching product, [M + Me]+, while there are three routes for formation of [M + H]+. Incomplete complex dissociation products, [M + H + MeL]+ and [M + 2H + L]+, represent first generation dissociation products of the [M + 2H]2+ + MeL2−}*+ ion/ion complex and are intermediates in the formation of [M + Me]+or [M + H]+. A key factor in the observation of the [M + 2H + MeL2]+, [M + H + MeL]+, and [M + 2H + L]+ products is the presence of helium at roughly 1 mTorr, which provides a route for collisional relaxation at rates of roughly 102 s−1.50 Thus, for ion/ion reactions of a multiply protonated polypeptide with a metal containing anion to be successful in generating metal cationized polypeptides in the gas phase, an ion/ion complex must be formed during the reaction, and its subsequent dissociation rates (i.e., kd2) must be high relative to the various collisional cooling rates.
SCHEME 1.
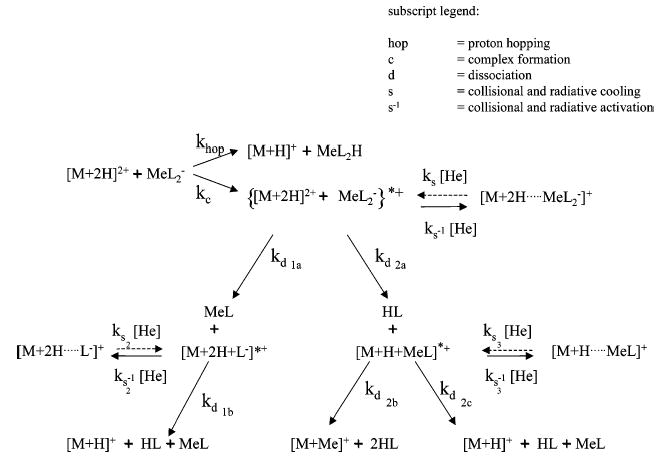
In all of the data related next, it should be recognized that the current instrumentation is limited in its ability to select with high precision reactant ions of each polarity. It is possible, for example, to accumulate the cationic reactants and mass selectively eject all other cations prior to accumulation of the negative reactants. However, it is often difficult to isolate the negative reactant once it has been accumulated in the ion trap because doing so can eject the cationic reactant. In these studies, the cation reactant was isolated prior to anion accumulation. Although efforts were made to establish conditions that maximized the signal of [AgL2]−, lower abundances of clusters of metal containing anions of the form [AgnL(n+1)]− were usually present, as were nonmetal containing anions in some cases. The cluster anions containing either no metals or more than one metal ion could give rise to minor products in the positive product ion population. The nonmetal containing anions, for example, could give rise to the observation of relatively small signals due to [M + H]+ products and [M + 2H + L]+ ions. A greater number of clusters of metal containing anions was observed from the nanospray of silver acetate and nitrate than for silver triflate and silver hexafluorophosphate. In fact, in the case of silver nitrate solutions, extensive clustering was noted such that the AgL2− ion was not the most abundanct species observed. In any case, the larger anionic clusters reacted in a manner similar to [AgL2]−. For example, in the case of the generic reaction of a doubly protonated peptide with a singly charged metal containing anion of the form [MeL2]−, the two cation transfer reaction types shown in Scheme 1 are
| (1), (2) |
In the case of the reactions of anions of larger cluster size, the reactions can be represented as:
| (3), (4) |
From the data obtained to date, the partitioning between eqs 3 and 4 for the larger anionic clusters appears to be very similar to the partitioning observed between reactions 1 and 2 for MeL2−.
In the case of a more highly charged polypeptide or protein, the reactions are further generalized as:
| (5), (6) |
where m can range to (n + 1). Detailed reaction schemes involving various intermediates, more complex but analogous to that of Scheme 1, apply to reactions 3 and 4 and reactions 5 and 6, respectively. However, the observed phenomenology can be rationalized based largely on the detailed reactions in Scheme 1.
Reactions of Bradykinin Ions with Anions Derived from a Silver Nitrate Solution
Cation-switching ion/ion reactions involving the reaction of multiply protonated bradykinin ions with silver containing anions were performed. An example of this type of reaction is shown in Figure 2. Doubly protonated bradykinin (RPPGFSPFR) ions were isolated from the initial positive ion electrospray distribution (Figure 2a). While these bradykinin ions were stored in the quadrupole ion trap after isolation, anions from a silver nitrate solution (anion mass spectrum acquired in the absence of positive ions shown in Figure 2b) were introduced into the ion trap. The metal containing anions were not isolated prior to the mutual storage period with the protonated peptide ions. Ion/ion reactions occurred during a period of simultaneous storage of doubly protonated bradykinin ions and silver nitrate anions, prior to mass analysis.
Figure 2.
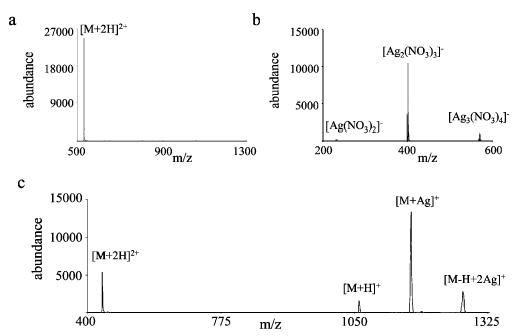
Mass spectra of the reactants for the cation-switching reaction of (a) doubly protonated bradykinin with (b) silver nitrate anions. The product ion spectrum for the reaction of ions in panels a and b is shown in panel c.
Positively charged products from this set of reactants were generated from proton transfer to yield the [M + H]+ ion, and cation switching, involving exchange of the cationizing protons for a silver ion, yielded the [M + Ag]+ polypeptide (Figure 2c). The [M + Ag]+ product ions are expected to arise via a relatively long-lived complex between the silver containing anions and protonated peptide ions (vide supra). In the complex, the transfer of a silver cation to the peptide and two protons to the nitrate anions occurs before dissociation of the complex. The net effect is the switching of one silver ion for two protons of the peptide ion. Some of the product ions in the ion/ion reaction spectra shown in Figure 2 have more than one silver ion. These products are formed from reactions with anions containing multiple metal and ligand ions (Figure 2b, see also eq 6) and are not the result of consecutive reactions of first generation ion/ion reaction products. (Consecutive reactions would yield products of lower charge states since each reaction with a singly charged anion results in products with one less charge.) As mentioned previously, the [M + H]+ product can also result from proton hopping, but it is difficult to establish conclusively that this route is significant for this set of reactants. Evidence for proton hopping has been identified unambiguously only for reactions of multiply charged cations with multiply charged anions,48,49 and it is not consistently observed for all such reactions. When proton transfer products most likely formed via proton hopping have been observed, they were noted to be present at short mutual storage times (less than 50 ms).48,49 Little difference in the relative contribution of [M + H]+ at short versus long reaction times was noted here. The generation of [M + Ag]+ products during ion/ion reactions, on the other hand, strongly implies that an ion/ion complex was formed. Once formed, there are two complex dissociation pathways that lead to [M + H]+ products (see Scheme 1). Therefore, the proton hopping process need not be invoked to explain the observance of [M + H]+ ions. While the contribution from this channel to the measured abundance of [M + H]+ cannot be determined from these data, the lack of reaction time dependence for the product ion relative abundances and the fact that some combinations of reactants show little or no proton transfer suggests that proton hopping likely plays a minor role in the reactions discussed herein.
Effect of the Anionic Ligand on Cation-Switching Reaction Products
The observation of cation-switching products in reactions of doubly protonated bradykinin with silver nitrate ions indicates that it is possible to change the cationizing agent using gas-phase ion/ion reactions. However, it is important to assess the role of the ligand in the anionic reagent in the formation of such products since, presumably, each ligand can have a different interaction with the peptide ion as well as different proton and metal ion affinities. To investigate this matter, the reactions of doubly protonated bradykinin with anions derived from negative nanospray of silver acetate, silver triflate, and silver hexafluorophosphate solutions, respectively, were performed. A diverse set of functional groups is represented by these ligands. The results from using these silver containing anions in cation-switching reactions carried out in the manner described previously for the reaction with silver nitrate anions are reported in Figures 3 and 4.
Figure 3.

Product ion spectrum from the reaction of doubly protonated bradykinin with silver acetate anions.
Figure 4.
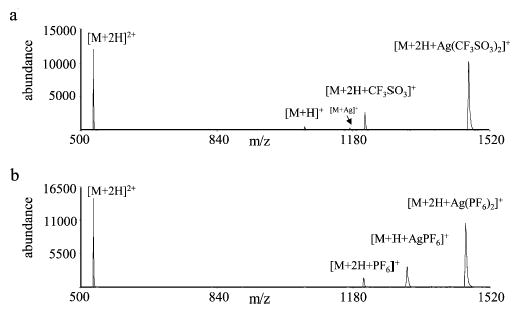
Product ion spectra for the reaction of doubly protonated bradykinin with (a) silver triflate anions and (b) silver hexafluorophosphate anions.
The product ion spectrum from the reaction of doubly protonated bradykinin and silver acetate anions is shown in Figure 3. Similar to the reaction product ion spectrum in Figure 2c for reactions featuring silver nitrate ions, products corresponding to proton transfer and cation exchange of protons for a silver ion are observed in Figure 3. There is no evidence for an acetate attachment product. The [M + H]+ product ion is more abundant than the [M + Ag]+ product ion, in sharp contrast with the products formed using silver nitrate anions. The [M + H]+/[M + Ag]+ ratio was insensitive to reaction conditions (e.g., reaction time, helium pressure, amplitude of the trapping voltage). The fact that a different ratio of [M + H]+ to [M + Ag]+ products was observed for the reactions with silver nitrate anions demonstrates the dramatic role that the ligand plays in these reactions.
While the product ion distribution is under kinetic control, it is instructive to consider the thermodynamics associated with the various competitive channels, as they can affect relative reaction rates, particularly when reactions proceed over similar energy surfaces. For the generic competitive reactions 1 and 2, the difference in the enthalpies of reaction of eqs 1 and 2 is given by
| (7) |
where PA(M) and MIA(M) are the proton and metal ion affinities of M, respectively, and PA(L−) and MIA(L−) are the respective proton and metal ion affinities of the anionic ligand. As written in eq 7, a negative ΔΔHrxn favors formation of [M + H]+, whereas a positive ΔΔHrxn favors formation of [M + Me]+. The relationship in eq 7 can be rewritten as
| (8) |
Given that proton affinities are consistently higher than other cation affinities for a given molecule,51–54 [PA(M) − MIA(M)] is likely to be positive in sign, whereas, for similar reasons, [(MIA(L−) − PA(L−)] is likely to be negative in sign. For a given metal and peptide, therefore, the outcome of the competition between eqs 1 and 2 might be expected to vary with the magnitude of [(MIA(L−) − PA(L−)]. As the magnitude of [(MIA(L−) − PA(L−)] becomes less negative, the likelihood for formation of [M + Me]+ relative to that for formation of [M + H]+ increases. The proton and metal ion affinities of ligands can be determined using DFT and ab initio computations. By comparing the proton and metal ion affinities of various ligands, it may be possible to identify ligands for which the thermochemistry is more favorable for cation switching. The proton and metal ion affinities of the ligands used in the cation-switching reactions were calculated and are summarized in Table 1. On the basis of the ligand anion thermochemical calculations, it is expected that the tendency for formation of [M + H]+ proceeds in the order of acetate > nitrate > triflate > hexafluorophosphate. This order is consistent with the results for acetate and nitrate anions (see Figures 2c and 3). However, triflate and hexafluorophosphate containing anions tend not to proceed fully to [M + H]+ and [M + Me]+ products.
TABLE 1.
Calculated Proton and Silver Ion Affinities of Ligands
| ligand (L) | proton affinitya,b | silver ion affinitya,c | silver ion affinity-proton affinitya | proton affinitya,b of Ag(L)2− |
|---|---|---|---|---|
| CH3COO− | 345.5 | 149.8 | −195.7 | 305.6 |
| NO3− | 323.1 | 134.1 | −189.0 | 282.3 |
| CF3SO3− | 299.1 | 121.6 | −177.5 | 264.9 |
| PF6− | 280.7 | 112.9 | −167.8 | 248.8 |
In units of kcal/mol.
B3LYP/6-31+G(d)//MP(2)(full)/G-311 + G(2d,2p).
Hay–Wadt VDZ (n + 1) ECP.
The results obtained for the reactions of doubly protonated bradykinin ions with anions derived from silver triflate and silver hexafluorophosphate are shown in Figure 4a,b, respectively. Attachment of the complete silver containing anion to doubly charged bradykinin was the dominant product for both silver triflate and silver hexafluorophosphate anions. This result, of course, provides direct evidence for complex formation during the cation-switching reactions. Apparently, the lifetimes of these ion/ion complexes were long enough such that most of the complex ions were cooled by collisions with the bath gas and trapped. In the case of silver triflate anions, loss of AgL from the initial adduct is observed in greater abundance than loss of HL, which is not consistent with the order discussed previously for the ligand anion thermochemistry. On the other hand, loss of HL is observed to be greater than loss of AgL for the silver hexafluorophosphate ion, as might be expected based on ligand anion thermochemistry. When the adduct species were subjected to ion trap collisional activation, losses of HL to yield the [M + Me]+ species dominated (data not shown), which is consistent with the order listed previously. The behavior of the silver triflate reaction might be due to a kinetic effect associated with the breakup of the complex. However, the possibility for ligand hopping, for which no conclusive evidence is available, cannot be precluded.
The likelihood for the observation of the intact complex is dependent upon its kinetic stability (i.e., the dissociation rate of the complex relative to the stabilization rate in the ion trap environment). Numerous complex structures might be formed. The extremes range from the attachment of the intact anion to a charged site of the peptide, whereby the components of the anionic reactant remain associated, to a complete breakup of the anion, giving rise to distinct peptide–metal and peptide–ligand interactions without interactions between the metal and the ligands. Given that ion trap collisional activation of the complexes discussed here gives rise to successive losses of HL, it is of interest to determine the strength of the interaction between L− and protonated sites in the polypeptide. To compare the relative stabilities associated with the different ligands, the guanidinium ion was used as a model for protonated arginine, and protonated methylamine was used as a model for protonated lysine and a protonated N-terminus. Table 2 lists the calculated binding strengths for these cations with nitrate, acetate, triflate, and hexafluorophosphate. The lifetimes of the complexes with respect to loss of HL are expected to increase with increasing charge site–ligand binding strength. Acetate and nitrate are significantly less strongly bound to either type of cationic site, relative to triflate and hexafluorophosphate. These results are consistent with greater kinetic stabilities for complexes containing triflate or hexafluorophosphate than for complexes containing acetate or nitrate. Hence, the identities of the ligands associated with the anionic reactants play major roles both in the likelihood for the observation of complexes as well as in the partitioning between formation of [M + H]+ and [M + Ag]+. At least as a first approximation, the thermodynamics associated with the various processes indicated in Scheme 1 appear to be of qualitative predictive value.
TABLE 2.
Calculated Stabilities of Complexes between Ligands and Protonation Sites
| ligand (L) | stabilitya,b of the complex with CH3NH3+ | stabilitya,b of the complex with a guanidinium iona |
|---|---|---|
| CH3COO− | 10.9 | 13.8 |
| NO3− | 14.0 | 20.1 |
| CF3SO3− | 23.1 | 35.4 |
| PF6− | 33.9 | 47.0 |
In units of kcal/mol.
B3LYP/6-31+G(d)//MP(2)(full)/G-311 + G(2d,2p).
Effect of Peptide Sequence on Cation-Switching Reaction Products: PPGFSPFR versus RPPGFSPF
The product ion distribution resulting from the reactions of polypeptide cations with metal containing anions of the form MeL2− can be highly sensitive to peptide sequence, depending upon the nature of L−, as illustrated with the isomeric peptide ions derived from des-R1 and des-R9 bradykinin. The product ion spectra from the reactions with silver nitrate and silver triflate are shown in Figure 5 (compare Figures 2c and 4a for the corresponding bradykinin data). There is little difference in the appearance of the product ion spectra for reactions of the same population of silver nitrate anions with doubly protonated PPGFSPFR (Figure 5a) and RPPGFSPF (Figure 5b). The cation-switching product, [M + Ag]+, is formed for both peptides during the ion/ion reaction. The distribution of the product ions is similar to that observed in Figure 2c for reactions with bradykinin (RPPGFSPFR). Cation-switching and proton transfer products were also observed from the reaction of silver nitrate anions with peptides unrelated to bradykinin (data not shown.). In contrast to the reaction behavior of silver nitrate anions with the bradykinin variants, quite distinct relative product ion abundances are noted for des-R1 and des-R9 bradykinin from their reaction with silver triflate anions (Figure 5c,d). Various adduct products are observed in the reaction product spectra. The dominant product in the reaction spectrum for des-R1 corresponds to the addition of triflate to the doubly protonated peptide (Figure 5c). The major product from the reaction with des-R9 is [M + H + AgCF3SO3]+ (Figure 5d). Neither of these prominent product ions was the most abundant ion in reactions of silver triflate with doubly protonated bradykinin, which showed attachment of the intact metal containing anion. One commonality in these spectra is that there is little evidence for the [M + Ag]+ product. In general, a high degree of variability in relative product ion abundances is noted in reactions of silver triflate anions with diprotonated peptides (data not shown). This behavior is not restricted to bradykinin and related ions.
Figure 5.
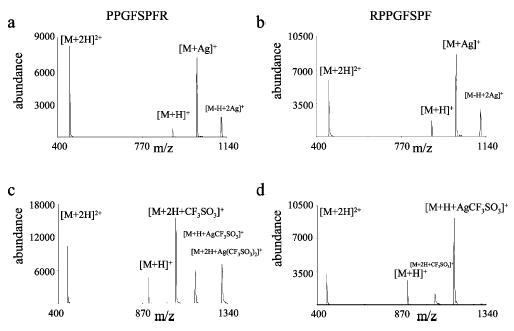
Product ion spectra from the reactions of silver nitrate anions with doubly protonated des-R1 bradykinin (a) and des-R9 bradykinin (b). The product ion spectra from the reaction of des-R1 bradykinin (c) and des-R9 bradykinin (d) with silver triflate anions.
Since the differences in the product ion spectra are primarily manifest in the presence and abundances of adduct products, rather than the presence and abundances of proton transfer and cation-switching products, the differences reflect dissimilarities in the structures of the intermediate complexes as reflected in the strengths of the relevant noncovalent interactions. It is clear from the results in Table 2 that the strengths of the interactions between the charge site and the ligand anion depend on the nature of the positive charge site. For example, the relative increase in binding strength in going from protonated methylamine to the guanidinium ion is 44% for nitrate and 53% for triflate. However, the absolute binding strengths for the two cation sites as well as the absolute difference in binding strengths are much higher for triflate than for nitrate. In the case of the nitrate containing anions, the noncovalent interaction strengths in the intermediate complexes, regardless of the sequence of the peptide, are apparently sufficiently weak that complete dissociation to the proton transfer and cation-switching products can occur. However, in the case of triflate containing anions, the strengths of the noncovalent interactions within the intermediate complexes in Scheme 1 are sufficiently strong to allow for a significant degree of collisional cooling and complex stabilization. Furthermore, the strengths of the interactions are sufficiently different in the complexes formed via the isomeric peptide reactants that a different array of products is observed. The dominance of [M + 2H + Ag(CF3SO3)2]+, where M is bradykinin, in the product ion spectrum of Figure 4a indicates that the lifetime of the ion/ion complex was longer than the lifetimes of [M + 2H + Ag(CF3SO3)2]+*, where M is des-R1 and M is des-R9 bradykinins since [M + 2H + Ag(CF3SO3)2]+ products are not dominant in the spectra of Figure 5. A difference in the stabilities of the des-R1 and des-R9 complexes is apparent from the appearance of an abundant complex for des-R1 and the absence of an intact complex for des-R9. A further significant difference between the two isomeric ions is observed in the partitioning upon dissociation of the respective ion/ion complexes. Loss of AgCF3SO3 is favored for the des-R1 bradykinin complex, while loss of HCF3SO3 is favored for the des-R9 bradykinin complex.
Clearly, the various lifetimes of complexes and distinctive partitioning for bradykinin type peptides are related to dissimilarities in the peptide ion structure since the anionic reactant is the same in all cases. The peptide ion structure is expected to influence relative proton, metal ion, and ligand affinities of the peptide. It is interesting to speculate that since the major difference in the amino acid sequences of the peptides studied here is the number and location of arginine residues, the location of protons in the initial doubly charged peptide impacts the relative stabilities of the interactions of the peptide with silver triflate anions. Given the amino acid sequences of the peptides studied here, the probable sites of protonation are the N-terminus and the side chain of arginine. Because there are two arginine residues in bradykinin, the N-terminus is not likely to be protonated in the doubly charged ion since the proton affinity of arginine is greater than that of the N-terminus.31,55 Thus, for the doubly protonated ions of bradykinin (RPPGFSPFR) and des-R1 bradykinin (PPGFSPFR), the protons are presumably localized near the termini of the respective peptide ions. That is not a probable scenario for doubly protonated des-R9 bradykinin (RPPGFSPF).34 Interestingly, it is the reaction product ion spectrum of des-R9 bradykinin (Figure 5d) that shows little evidence for [M + 2H + Ag(CF3SO3)2]+ products while such products are prominent in the spectra for bradykinin (Figure 4a) and des-R1 bradykinin (Figure 5c). The observation of more first generation products from dissociation of the ion/ion complex for des-R1 bradykinin relative to bradykinin may imply a weaker interaction with silver triflate based on the localization of a proton on the N-terminus for des-R1 bradykinin as compared to a second arginine residue in the case of bradykinin. The overall trend in adduct ion stabilities following the order bradykinin > des-R1 bradykinin > des-R9 bradykinin noted here is consistent with measured apparent gas-phase basicities of the various (M + H)+ ions.34 The apparent gas-phase basicities are reported to be bradykinin = 225.8 ± 4.2 kcal/mol, des-R1 bradykinin = 222.8 ± 4.3 kcal/mol, and des-R9 bradykinin = 214.9 ± 2.3 kcal/mol.55
The data in Figure 5 demonstrate that the types of products observed for the reactions of doubly protonated peptides with metal containing anions can be sensitive to the amino acid sequence of the peptide. In the case of the bradykinin related ions, anions that give rise exclusively to [M + H]+ and [M + Me]+ ions did not appear to show clear differences in product ion partitioning. Nevertheless, differences might be expected if, for example, significant differences in [PA(M)-MIA(M)] between isomeric peptides exist. However, even when this is not the case, anions comprised of ligands that interact relatively strongly with cationic sites can give rise to distinct product ion distributions in reactions with isomeric peptide ions through differences in the stabilities of the intermediate complex species. For the anionic ligands studied here, acetate and nitrate are least likely to give rise to the observation of intermediate complexes, whereas triflate and hexafluorophosphate are most likely to yield complexes in the ion trap environment. These findings are significant from an ion synthesis point of view as they provide insights into anionic reagent selection for the synthesis of ions of interest (e.g., cation transfer products vs cluster ions).
Cation-Switching Reactions with Multiply Protonated Ubiquitin Ions
Cation-switching studies were extended to larger and more highly charged species to determine the generality associated with the observations described previously for doubly protonated peptide ions. Ubiquitin was chosen as the model protein for this purpose. Figure 6 shows illustrative results for relatively highly charged species, as represented by the [M + 11H]11+ ion. Figure 6a shows the results obtained from reaction with anions derived from silver nitrate, and Figure 6b shows the results obtained from reaction with anions derived from silver acetate. Consistent with the types of product ions seen in the cation-switching reaction spectra for doubly charged bradykinin, proton transfer and cation-switching products were formed in reactions with ubiquitin ions. No ligand adduct products were observed in the product ion spectra from the reaction of silver nitrate and silver acetate anions with the multiply charged protein ions. Similar to the results of reactions with bradykinin (Figure 2c), the abundance of the cation-switching reaction product is greater than that of the proton transfer product in reactions with silver nitrate. A major difference in the product ion spectra from the reaction of ubiquitin ions relative to those of reactions with bradykinin ions was the presence of consecutive reaction products. The data in Figure 6 provide evidence for consecutive proton transfer reactions and cation-switching reactions as well as proton transfer reactions followed by cation-switching reactions. In the case of the doubly charged peptide ions, reaction products from consecutive ion/ion reactions are neutral species and cannot be observed. Such reactions for doubly charged ions are expected to be minimal under the conditions of this study due to the relatively low ion/ion reaction rates associated with singly charged ions. However, in the case of highly charged reactants, reaction rates are sufficiently high to allow for consecutive ion/ion reactions to occur. Reactions of multiply protonated ubiquitin ions with silver triflate anions primarily yielded silver triflate attachment products and some consecutive reaction products (data not shown.). With the exception of the observation of consecutive reaction products, the types of products from reactions of the [M + 11H]11+ ion of ubiquitin with the various silver containing anions are similar to those noted in reactions with doubly protonated bradykinin.
Figure 6.
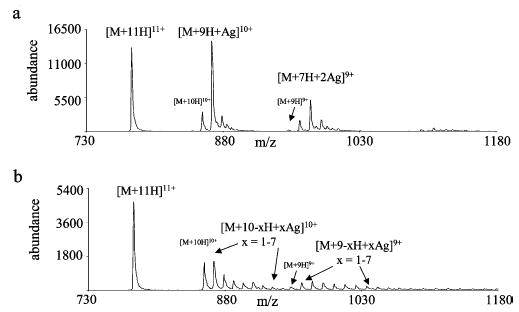
Product ion spectra from reactions of [M + 11H]11+ ubiquitin with (a) silver nitrate anions and (b) silver acetate anions.
The data for reactions with anions derived from silver acetate are particularly noteworthy in that the exchange of silver ions for protons resulting in the addition of up to seven silver ions to the formerly purely protonated ubiquitin ion population was observed. As mentioned previously, high mass cluster anions with multiple silver ions were present with negative nanospray of silver acetate. The +10 product ions therefore arise from eqs 5 and 6. In this particular case, a mixture of anions of different cluster sizes gave rise to a mixture of products with different numbers of silver ions switched for protons. An analogous reaction was also observed for doubly protonated bradykinin, as reflected by the formation of the [M − H + 2Ag]+ product (see Figure 3).
The dependence of cation-switching reaction products on the protein ion charge state was examined. Reactions of silver nitrate and silver acetate anions with the highest charge state of protonated ubiquitin, +13, as well as +8 ubiquitin yielded cation-switching products analogous to those described previously for +11 ubiquitin. It was of interest to examine whether the same types of cation-switching reaction products were observed for a very low charge state of ubiquitin. Proton transfer reactions with PDCH anions were used to reduce the charge states of ubiquitin ions present in the ESI distribution to predominantly +2. The [M + 2H]2+ ubiquitin ions were then reacted with silver nitrate, silver acetate, and silver triflate anions, respectively. In contrast to the results from reacting silver nitrate anions with [M + 11H]11+ ubiquitin, adduct products were observed, instead of proton transfer and cation-switching products, when the doubly charged ubiquitin ions were reacted with silver nitrate anions (Figure 7a). Cation-switching reactions of [M + 2H]2+ ubiquitin and silver acetate anions, yielding the [M + Ag]+ product, were noted, in analogy with the higher charge state ions of ubiquitin (Figure 7b). The partitioning between proton transfer and cation switching, however, showed some variation with ubiquitin charge state. Consistent with what was observed for the reactions of +2 bradykinin and +8 ubiquitin, the addition of the intact silver triflate reactant anion was the major product ion formed during the reaction of +2 ubiquitin and silver triflate anions.
Figure 7.
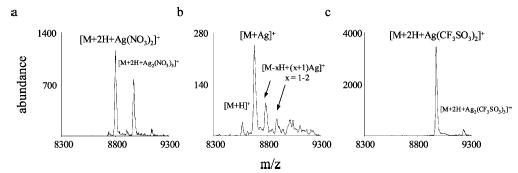
Product ion spectra for reactions of [M + 2H]2+ ubiquitin with (a) silver nitrate anions, (b) silver acetate anions, and (c) silver triflate anions.
The most striking difference in the behavior of the +2 ubiquitin ion relative to the behaviors of more highly charged ubiquitin ions and the doubly protonated bradykinin ion was in the reaction with silver nitrate anions. Essentially no complex formation was noted for the highly charged ubiquitin ions and +2 bradykinin, whereas almost complete adduct formation was observed for +2 ubiquitin. This contrasting behavior can readily be rationalized on the basis of the lifetime of the ion/ion collision complex. The two protons in +2 ubiquitin are likely to be sited on arginine residues (there are four arginines in ubiquitin). Furthermore, given the presence of many relatively basic sites within the protein, these charges are likely to be stabilized further via intramolecular interactions. The interaction strength between a nitrate anion and the guanidinium ion is significantly greater than that with protonated methylamine (see Table 2). The stabilization of charge associated with intramolecular interactions is likely to increase the strength of the interaction with a nitrate anion. Therefore, relative to ions with total charge greater than the number of arginine residues in the protein, the binding of nitrate anions for the +2 ubiquitin ion is expected to be higher. Relative to +2 bradykinin, the size of the protein, with its far greater number of degrees of freedom, is expected to extend the lifetime of the ion/ion complex for the protein relative to that for the peptide. Furthermore, there are fewer possibilities for intramolecular stabilization interactions of the protonated arginine residues in bradykinin than for the protonated arginine residues in +2 ubiquitin. Therefore, the lifetime of the ion/ion complex with +2 ubiquitin is expected to be longer than that of the complex with +2 bradykinin.
Conclusions
Metal cations can be transferred to multiply protonated polypeptides via reactions with metal containing anions of the form MeL2−, thereby providing a gas phase route to the formation of metal cationized polypeptides. A variety of reaction products can be observed, and their identities and abundances can be largely rationalized on the basis of the fate of an initially formed ion/ion complex. Metal ion transfer, in conjunction with proton transfer, and proton transfer to the metal containing anion are competitive processes in the sequential loss of ligands from the ion/ion complex. Furthermore, adduct ions that are intermediates in the proton and metal ion transfer reactions can also be observed. For a given metal, the nature of the ligand, the sequence and composition of the polypeptide, and the charge state of the polypeptide are factors in determining the identities and relative abundances of the product ions. Anions with ligands that interact strongly with protonation sites tend to give rise to adduct ions. Anions with ligands that interact weakly with cationic sites, such as the acetate anion, tend to lead primarily to proton transfer and cation transfer products. The nature of the ligands in the metal containing anion affects the competition between proton transfer and cation switching via the difference between metal ion affinity and proton affinity of the ligand anion, [(MIA(L−) − PA(L−)]. As [(MIA(L−) − PA(L−)] becomes less negative, the tendency for formation of the cation-switching product, [M + Me]+, increases. The overall reaction behavior of a multiply charged protein shares commonalities with that of a doubly charged peptide for a given anion reactant. However, differences in size, composition, charge state, etc. can give rise to differences in the extent of adduct ion formation and in the degree of proton transfer versus cation switching. As a rule, adduct formation can be expected to be more likely with a protein than with a peptide due to the larger numbers of degrees of freedom associated with the protein, the generally larger number of strongly basic sites (e.g., arginines), and the greater likelihood for extensive charge solvation.
The degree to which ion/ion product distributions vary with the sequence and composition of a polypeptide is anionic ligand dependent. For example, in the case of RPPGFSPF versus PPGFSPFR, little difference in the product ion spectra was noted in the ion/ion reactions of the doubly protonated ions with anions derived from silver nitrate. However, anions derived from silver triflate showed quite distinct product ion spectra for these isomeric polypeptide cations. Differences in the stabilities of the adduct species formed via use of different ligands with the metal ion of interest apparently account for the distinct ion/ion reaction product ion spectra. This observation indicates that ion/ion reactions of this kind might be useful as a structural probe. Metal containing anions with ligands that interact strongly with protonation sites appear to be most useful for this purpose, whereas those with ligands that interact weakly with protonation sites are best suited for cation switching.
Acknowledgments
This research was sponsored by the Division of Chemical Sciences and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy under Award DE-FG02-00ER15105 and the National Institutes of Health under Grant GM45372.
References
- 1.Biemann K, Martin SA. Mass Spectrom Rev. 1987;6:1–79. [Google Scholar]
- 2.Hunt DF, Yates JR, III, Shabanowitz J, Winston S, Hauer CR. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polce MJ, Ren D, Wesdemiotis C. J Mass Spectrom. 2000;35:1391–1398. doi: 10.1002/1096-9888(200012)35:12<1391::AID-JMS85>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Wysocki VH, Tsaprails G, Smith LL, Breci LA. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 5.Lin T, Payne AH, Glish GL. J Am Soc Mass Spectrom. 2001;12:497–504. doi: 10.1016/S1044-0305(01)00234-3. [DOI] [PubMed] [Google Scholar]
- 6.Lin T, Glish GL. Anal Chem. 1998;70:5162–5165. doi: 10.1021/ac980823v. [DOI] [PubMed] [Google Scholar]
- 7.Lin T, Payne AH, Glish GL. Analyst. 2000;125:635–640. doi: 10.1039/a908950k. [DOI] [PubMed] [Google Scholar]
- 8.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Anal Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 9.Engel BJ, Pan P, Reid GE, Wells JM, McLuckey SA. Int J Mass Spectrom. 2002;219:171–187. [Google Scholar]
- 10.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 11.Smith RD, Loo JA, Edmonds CG, Barinaga CJ, Udseth HR. Anal Chem. 1990;62:882–899. doi: 10.1021/ac00208a002. [DOI] [PubMed] [Google Scholar]
- 12.Karas M, Hillenkamp F. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 13.Yu X, Wojciechowski M, Fenselau C. Anal Chem. 1993;65:1355–1359. doi: 10.1021/ac00058a010. [DOI] [PubMed] [Google Scholar]
- 14.Loo JA, Hu P, Smith RD. J Am Soc Mass Spectrom. 1994;5:959–965. doi: 10.1016/1044-0305(94)80014-6. [DOI] [PubMed] [Google Scholar]
- 15.Hu P, Loo JA. J Am Chem Soc. 1995;117:11314–11319. [Google Scholar]
- 16.Chu IK, Cox DM, Guo X, Lau TC, Siu KWM. Anal Chem. 1999;71:2364–2372. doi: 10.1021/ac981351n. [DOI] [PubMed] [Google Scholar]
- 17.Rodriquez CF, Fournier R, Chu IK, Hopkinson AC. Int J Mass Spectrom. 1999;192:303–317. [Google Scholar]
- 18.Chu IK, Rodriquez CF, Lau TC, Hopkinson AC, Siu KWM. J Phys Chem B. 2000;104:3393–3397. [Google Scholar]
- 19.Anbalagan V, Silva ATM, Rajagopalachary S, Bulleigh K, Talaty ER, Van Stipdonk MJ. J Mass Spectrom. 2004;39:495–504. doi: 10.1002/jms.610. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Siu KWM, Guevremont R, Le Blanc JCY. J Am Soc Mass Spectrom. 1997;8:781–792. [Google Scholar]
- 21.Kish MM, Wesdemiotis C. Int J Mass Spectrom. 2003;227:191–203. [Google Scholar]
- 22.Newton KA, McLuckey SA. J Am Chem Soc. 2003;125:12404–12405. doi: 10.1021/ja036924e. [DOI] [PubMed] [Google Scholar]
- 23.Wells JM, Chrisman PA, McLuckey SA. J Am Soc Mass Spectrom. 2002;13:614–622. doi: 10.1016/S1044-0305(01)00364-6. [DOI] [PubMed] [Google Scholar]
- 24.Badman ER, Chrisman PA, McLuckey SA. Anal Chem. 2002;74:6237–6243. doi: 10.1021/ac026020w. [DOI] [PubMed] [Google Scholar]
- 25.Mirza UA, Chait BT. Anal Chem. 1994;66:2898–2904. doi: 10.1021/ac00090a017. [DOI] [PubMed] [Google Scholar]
- 26.Valentine SJ, Counterman AE, Clemmer DE. J Am Soc Mass Spectrom. 1997;8:954–961. doi: 10.1016/S1044-0305(99)00079-3. [DOI] [PubMed] [Google Scholar]
- 27.Wyttenbach T, Bowers MT. J Am Soc Mass Spectrom. 1999;10:9–14. [Google Scholar]
- 28.Wyttenbach T, Kemper PR, Bowers MT. Int J Mass Spectrom. 2001;212:13–23. [Google Scholar]
- 29.Szilagy Z, Drahos L, Vekey K. J Mass Spectrom. 1997;32:689–696. [Google Scholar]
- 30.Strittmatter EF, Williams ER. J Phys Chem A. 2000;104:6069–6076. doi: 10.1021/jp000038y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell S, Rodgers MT, Marzluff EM, Beauchamp JL. J Am Chem Soc. 1994;116:9765–9766. [Google Scholar]
- 32.Purves RW, Barnett DA, Ells B, Guevremont R. J Am Soc Mass Spectrom. 2000;11:738–745. doi: 10.1016/S1044-0305(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 33.Cotter RJ, Kovtoun S, Doroshenko V, Prasad D, Prieto M, Berkout V. Adv Mass Spectrom. 2001;15:399. [Google Scholar]
- 34.Ewing NP, Pallante GA, Zhang X, Cassady CJ. J Mass Spectrom. 2001;36:875–881. doi: 10.1002/jms.188. [DOI] [PubMed] [Google Scholar]
- 35.Breuker K, Oh HB, Horn DM, Cerda BA, McLafferty FW. J Am Chem Soc. 2002;124:6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 36.Clipston NL, Jainhuknan J, Cassady CJ. Int J Mass Spectrom. 2003;222:363–381. [Google Scholar]
- 37.Badman ER, Myung S, Clemmer DE. Anal Chem. 2002;74:4889–4894. doi: 10.1021/ac020300u. [DOI] [PubMed] [Google Scholar]
- 38.Laskin J, Beck KM, Hache JJ, Futrell JH. Anal Chem. 2004;76:351–356. doi: 10.1021/ac0351116. [DOI] [PubMed] [Google Scholar]
- 39.Delobel A, Halgand F, Laffranchise-Gosse B, Snijders H, Laprevote O. Anal Chem. 2003;75:5961–5968. doi: 10.1021/ac034532k. [DOI] [PubMed] [Google Scholar]
- 40.Barr JM, Van Stipdonk MJ. Rapid Comm Mass Spectrom. 2002;16:566–578. doi: 10.1002/rcm.612. [DOI] [PubMed] [Google Scholar]
- 41.Chu IK, Cox DM, Guo X, Kireeva I, Lau TC, McDermott JC, Siu KWM. Anal Chem. 2002;74:2072–2082. doi: 10.1021/ac0111006. [DOI] [PubMed] [Google Scholar]
- 42.Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98, Revision A.7; Gaussian, Inc.: Pittsburgh, PA, 1998.
- 43.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 44.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 45.Basis sets were obtained from the Extensible Computational Chemistry Environment Basis Set Database, www.emsl.pnl.gov/forms/basisform.htmlVersion, 02/25/04, as developed and distributed by the Molecular Science Computing Facility, Environmental and Molecular Sciences Laboratory that is part of the Pacific Northwest Laboratory, P.O. Box 999, Richland, WA 99352 and funded by the U.S. Department of Energy.
- 46.Wadt WR, Hay PJ. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 47.Foresman, J. B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, 1996.
- 48.Wells JM, Chrisman PA, McLuckey SA. J Am Chem Soc. 2001;123:12428–12429. doi: 10.1021/ja0170403. [DOI] [PubMed] [Google Scholar]
- 49.Wells JM, Chrisman PA, McLuckey SA. J Am Chem Soc. 2003;125:7238–7249. doi: 10.1021/ja035051l. [DOI] [PubMed] [Google Scholar]
- 50.Goeringer DE, McLuckey SA. Int J Mass Spectrom. 1998;177:163–174. [Google Scholar]
- 51.Kish MM, Ohanessian G, Wesdemiotis C. Int J Mass Spectrom. 2003;227:509–524. [Google Scholar]
- 52.Shoeib T, Gorelsky SI, Lever ABP, Siu KWM. Inorg Chim Acta. 2001;315:236–239. [Google Scholar]
- 53.Shoeib T, Siu KWM, Hopkinson AC. J Phys Chem A. 2002;106:6121–6128. [Google Scholar]
- 54.Cerda BA, Wesdemiotis C. J Am Chem Soc. 1995;117:9734–9739. [Google Scholar]
- 55.Wu Z, Fenselau C. Rapid Comm Mass Spectrom. 1992;6:403–405. [Google Scholar]