

Abstract
In addition to its potent effects on vasculature, it has become clear that vascular endothelial growth factor (VEGF) has effects on both neurons and glia, and recent studies suggest that it can be neuroprotective. To determine potential mechanisms underlying this neuroprotection, recombinant human VEGF was bath applied to adult rat hippocampal slices, and both extracellular and intracellular recordings were used to examine intrinsic properties and synaptic responses of hippocampal principal neurons. Initial studies in area CA1 showed that VEGF significantly reduced the amplitude of responses elicited by Schaffer collateral stimulation, without influencing membrane properties. Similar effects occurred in CA3 pyramidal cells and dentate gyrus granule cells when their major glutamatergic afferents were stimulated. Because VEGF expression is increased after seizures, effects of VEGF were also examined in rats with recurrent spontaneous seizures. VEGF reduced spontaneous discharges in slices from these rats but had surprisingly little effect on epileptiform discharges produced by disinhibition of slices from control rats. These results demonstrate a previously unknown effect of VEGF on neuronal activity and also demonstrate a remarkable potency in the epileptic brain. Based on this, we suggest that VEGF or VEGF-related targets could provide useful endpoints to direct novel therapeutic strategies for epilepsy.
Keywords: anticonvulsant, hippocampus, intracellular, neuromodulation, synaptic transmission, VEGF
Introduction
Vascular endothelial growth factor (VEGF), once considered specific for endothelial cells, is now known to also influence neurons and to do so in a number of ways. VEGF enhances neuronal proliferation (Jin et al., 2002; Zhu et al., 2003), neurite outgrowth and maturation (Rosenstein et al., 2003; Khaibullina et al., 2004), and neuronal survival (Jin et al., 2000, 2001; Svensson et al., 2002). A neuroprotective role for VEGF is supported by the demonstration that VEGF reduces excitotoxic damage to cultured hippocampal neurons (Jin et al., 2000; Matsuzaki et al., 2001; Svensson et al., 2002) and reduces damage in vivo after ischemia (Hayashi et al., 1998; Sun et al., 2003) (but see van Bruggen et al., 1999). Together with the evidence that VEGF expression increases after ischemia (Lee et al., 1999), traumatic brain injury (Chodobski et al., 2003), and seizures (Newton et al., 2003; Croll et al., 2004), it is possible that VEGF is an endogenous neuroprotective agent in the CNS. However, despite the potential importance of VEGF as a neuroprotective growth factor, few direct studies of VEGF on neurons or glia have been published to clarify its actions.
Therefore, the present study examined effects of VEGF by direct exposure to neurons using electrophysiological measures to determine effects on intrinsic properties and synaptic responses. The 165 aa splice variant of VEGF protein was chosen because it is the predominant isoform of VEGF in mammalian species (Rosenstein and Krum, 2004), and it binds to each of the known receptors for VEGF (for review, see Brockington et al., 2004; Storkebaum et al., 2004). The hippocampus was chosen as an area that would potentially benefit from neuroprotection because of its vulnerability to ischemia, traumatic injury, seizures, and excitotoxicity. VEGF is expressed in hippocampus, and its expression increases after seizures (Newton et al., 2003; Croll et al., 2004). Also, intrahippocampal infusion of VEGF appears to protect hippocampal neurons from seizure-induced damage (Croll et al., 2004).
To evaluate the effects of VEGF on synaptic responses of hippocampal neurons, we chose the major segments of the trisynaptic pathway because they are the major afferent input to principal neurons of hippocampus and because they are glutamatergic, allowing us to test the specific hypothesis that VEGF can protect neurons from excitotoxic insult by depressing glutamatergic transmission. To address the potential functional significance of seizure-induced VEGF upregulation, we also tested the ability of VEGF to depress epileptiform activity in the hippocampus of rats with chronic spontaneous seizures.
The results provide the first evidence that VEGF influences synaptic transmission in the brain and a potential explanation for the neuroprotective actions of VEGF. Remarkably, these actions of VEGF appear to be mediated not by an influence on permeability, the classic mode of action of VEGF, but by alternate mechanisms. In addition, the studies in epileptic tissue suggest a completely new role for VEGF in the brain: to decrease epileptiform activity in the epileptic brain.
Materials and Methods
Subjects
Animal care and use was in accordance with the guidelines set by the National Institutes of Health and the New York State Department of Health. Male Sprague Dawley rats were housed using a 12 h light/dark cycle and were provided food and water ad libitum. To test effects of VEGF on hippocampal neurons, slices were prepared from rats that were 27–99 d old. Many ages were chosen because one of the receptors for VEGF, Flk-1 (VEGFR2), has been reported to increase expression with age (Yang et al., 2003). However, our experiments did not demonstrate any detectable variation that correlated with age so, in Results, the data are pooled.
Pilocarpine administration
The pilocarpine model of epilepsy was used to produce chronic spontaneous seizures (Turski et al., 1989; Scharfman et al., 2000). We initiated pilocarpine induction of status epilepticus as follows: rats (mean ± SEM, 43 ± 3 d of age) received 1 mg/kg atropine methylbromide subcutaneously followed, after 30 min, by 380 mg/kg pilocarpine HCl intraperitoneally. Diazepam (5 mg/kg, i.p.) was administered 1 h after status epilepticus onset to decrease the severity of status epilepticus. Approximately 5 h later, 1 ml of castor oil was administered orally, and animals were injected subcutaneously with 2 ml of dextrose-lactate Ringer's solution. Diet was supplemented with apples, which were cut open and left at the base of the cage, for ∼1 week. Slices were made from pilocarpine-treated rats at least 4 weeks later, at a time when they had developed recurrent spontaneous seizures. In the text below, animals with a history of recurrent spontaneous seizures are referred to as “epileptic.”
Slice preparation
Animals were anesthetized with CO2 and decapitated. The brain was rapidly removed and immersed in ice-cold sucrose-based artificial CSF (sucrose-ACSF) (in mm: 126 sucrose, 5.0 KCl, 2.0 CaCl2, 2.0 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose, pH 7.4). Slices containing both the hippocampus and adjacent entorhinal cortex (400 μm) were cut in the horizontal plane using a Vibroslice (Stoelting, Kiel, WI) and transferred to a nylon net in a recording chamber (Fine Science Tools, Foster City, CA) in which they were maintained at ∼31°C, oxygenated (5% CO2, 95% O2), and immersed in sucrose-ACSF, except for the upper surface. After 30 min, sucrose-ACSF was replaced by ACSF containing NaCl substituted equimolar for sucrose (saline-ACSF), and recordings began 30 min later. The recording chamber was continually perfused with saline-ACSF at a rate of ∼1 ml/min, regulated by a peristaltic pump (Gilson, Middleton, WI).
Recording and stimulation
Recording. Recording electrodes made of borosilicate glass (0.75 mm inner diameter, 1.0 mm outer diameter; World Precision Instruments, Sarasota, FL) were pulled horizontally (Sutter Instruments, Novato, CA) and filled with saline-ACSF for extracellular recordings (10–15 MΩ resistance) or 1 m potassium acetate for intracellular recordings (70–110 MΩ). Electrophysiological data were collected using an amplifier with a bridge circuit (Axoclamp 2B; Axon Instruments, Union City, CA), and the bridge was balanced whenever current was passed. Recordings were monitored on a digital oscilloscope (Nicolet, Madison, WI) and DAT recorder (Sony, Tokyo, Japan) and saved on floppy disk for subsequent off-line analysis using Origin 7.0 software (OriginLab, Northampton, MA).
Stimulation. Monopolar stimulating electrodes were made from Teflon-coated wire (75 μm diameter, including Teflon; A-M Systems, Carlsborg, WA), and stimuli were triggered digitally (100 μA, 10–200 μs; Pulsemaster; World Precision Instruments) at low frequencies (<0.05 Hz).
Stimulating electrodes were placed so that they just touched the surface of the slice on the major excitatory fiber pathways of the hippocampus, 350–500 μm from the recording sites (Scharfman, 1996) (see Fig. 1A). For Schaffer collateral activation of CA1 pyramidal cells, the stimulating electrode was placed at the tip of the Schaffer collateral bundle, on the border of CA2, ∼150 μm from the pyramidal cell layer, and the recording site was CA1b. For perforant path activation of dentate granule cells, the stimulating electrode was placed near the hippocampal fissure, just beneath the subiculum, and the recording site was in the granule cell layer, either at the crest or the center of the superior blade. For mossy fiber stimulation of CA3 pyramidal cells, the stimulating electrode was placed on the border of the hilus and granule cell layer near the dentate gyrus crest, and the recordings were made from CA3b.
Figure 1.

VEGF reduces extracellularly recorded evoked potentials (“field potentials”) to afferent stimulation in the major excitatory pathways of the hippocampus. A, Schematic of the hippocampal slice showing sites for stimulation (open circles) in the perforant (1), mossy fiber (2), and Schaffer collateral (3/4) pathways, and extracellular electrode recording sites (filled circles) in the granule cell layer of the dentate gyrus (1), pyramidal cell layers of CA3 (2) and CA1 (3), and apical dendritic region of CA1 (4). B, The average change in response amplitude after VEGF administration in each of the four regions is presented as a percentage of the control value. Paired Student's t tests showed a significant reduction in response amplitude, but not response duration, in each of the four regions (for statistics and sample sizes, see Results). *p < 0.05. C, Averaged traces from each of these four recording sites show representative examples of the reduction in evoked responses from the period before VEGF exposure (Control) to the time after continuous exposure to VEGF when effects were maximal (VEGF).
For extracellular recordings in the cell layer, the recording electrode was lowered until the largest response amplitude was evoked. For extracellular recordings in the dendritic layers, the optimal response was determined by initially testing a 100–200 μm radius around the area of stratum radiatum that contained the dendrites of the pyramidal cells surrounding the recording electrode in the cell layer, i.e., ∼200 μm from the cell layer into stratum radiatum, a site that would be perpendicular to the cell layer (see Fig. 1 A). The site ultimately used was the one in which the maximal response was elicited of all of those tested.
Pharmacology
Experimental approach. To examine the effects of VEGF, slices were exposed to VEGF by adding a known concentration of VEGF to the saline-ACSF that was continuously delivered to the recording chamber holding the slices (“bath application”). In this manner, slices were exposed to a single concentration of VEGF continuously. Electrodes were maintained in the same positions, and stimulus frequency was not changed (<0.05 Hz).
To establish whether there was an effect of VEGF, electrophysiological parameters (evoked responses to afferent input, intrinsic properties, etc.) were compared between the baseline period (the 15–30 min period immediately before VEGF exposure) and a time after VEGF exposure. Slices with >10% variance in evoked responses during the baseline period were not used. To determine the maximal effect, measurements were made at intervals during the entire period of exposure to VEGF, and the time when the largest change occurred was chosen (typically 30–60 min after the start of VEGF exposure). For analysis of any given electrophysiological parameter, the mean of responses recorded during the baseline period was compared with the mean of two to three events that were recorded during VEGF bath application. To determine the reversibility of VEGF, the ACSF solution used to perfuse the slices that contained VEGF was replaced with VEGF-free ACSF. The term “Wash” refers to the period of time, after VEGF exposure, when VEGF-free ACSF was perfused.
VEGF. The 165 aa isoform of recombinant human VEGF protein was supplied by Regeneron Pharmaceuticals (Tarrytown, NY). It was supplied in 20 μl aliquots of 2.6 mg/ml solution in 15 mm citrate containing 0.02% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, pH 5.5, and maintained at –20°C until use. Aliquots were diluted with 0.05% bovine serum albumin (BSA) and added to the saline-ACSF perfusate to produce a final concentration of 200 ng/ml VEGF in <0.001% BSA, unless otherwise noted (see Results, Comparison of two concentrations of VEGF). For control experiments, BSA (at concentrations up to 0.0015%) was added to the saline-ACSF perfusate.
Bicuculline. To determine whether VEGF influenced burst discharges in the normal rat hippocampus, the GABAA receptor antagonist bicuculline methiodide (BMI) was added to slices made from naive rats to induce spontaneous burst discharges (Scharfman 1994a,b). Bicuculline was stored as a concentrated stock solution (10 mm in 0.9% saline) in aliquots at –20°C. An aliquot was thawed on the day of use and added to saline-ACSF at a final concentration of 10 μm.
Data analysis
Extracellular recordings. Population spike amplitude was measured as described previously (Alger and Teyler, 1976). The extracellularly recorded EPSP [field EPSP (fEPSP)] amplitude was measured as the voltage difference between the prestimulus potential and the peak of the fEPSP. To assess paired-pulse inhibition or paired-pulse facilitation, paired half-maximal stimuli were delivered to the Schaffer collateral pathway with a range of interstimulus intervals. Intervals of 10 and 20 ms were chosen to assess paired-pulse inhibition, whereas intervals of 40 and 80 ms were chosen to assess paired-pulse facilitation, based on empirical determination that these intervals provided strong paired-pulse inhibition and robust facilitation, respectively. The paired-pulse ratio was defined as (population spike amplitude evoked by the second stimulus)/(population spike amplitude evoked by the first stimulus).
Intracellular recordings. Intrinsic properties were measured as described previously (Scharfman, 1995; Scharfman et al., 2000). Briefly, resting potential was defined as the difference between the minimum subthreshold holding potential and the potential when the electrode was withdrawn from the cell. Input resistance was determined as the slope of the steepest part of the steady-state I–V curve based on responses to ±0.1–1.0 nA, 200 ms duration rectangular current pulses. Time constant was defined as the latency from the onset of the response to a –0.1 nA, 200 ms pulse (a pulse that did not lead to rectification or “sag”) to the point on the response that reached 63% of steady-state amplitude. Action potentials (APs) were triggered by a threshold current step (200 ms duration). AP amplitude was measured from resting potential to peak using an AP evoked at threshold. dv/dt was defined as the maximum slope of the rising phase of the AP divided by the maximum slope of the falling phase and was measured from the threshold AP.
The amplitude of a synaptic potential was defined as the voltage difference between the prestimulus potential and the peak of the synaptic potential. Maximum slope was defined as the maximum rate of change during the rising phase of the synaptic potential, determined by a derivative function of the potential waveform, computed by Origin 7.0. EPSP half-duration was determined as the time from the EPSP onset to the time during the decay phase when the amplitude had declined to 50% of its peak amplitude.
To evaluate amplitudes of depolarizing and hyperpolarizing components of the synaptic potential, a stimulus was triggered at the midpoint of a 200 ms current pulse that depolarized the cell to a membrane potential at which the depolarizing and hyperpolarizing components were easily distinguished. To evaluate reversal potential of the hyperpolarizing component, amplitudes were plotted as a function of membrane potentials using ±0.1–0.5 nA current steps and a fixed stimulus that was subthreshold at all potentials. Amplitudes were calculated at a fixed delay after the stimulus that was chosen to sample the peak of the IPSP, as well as a time immediately after the peak of the IPSP (Scharfman and Sarvey, 1988). The most negative reversal potential was always the latter and was used to define reversal potential for a given cell, because it was assumed that the earlier, more positive reversal potentials were confounded by overlapping glutamatergic components of the synaptic response.
Epileptiform burst discharges. Spontaneous burst discharges from bicuculline-treated slices of normal rats and spontaneous burst discharges from untreated slices of epileptic rats were measured identically. Measurements included frequency, amplitude, half-duration, and coastline bursting index. Burst frequency was measured as the average number of bursts per minute based on a minimum of three 1 min recordings. Burst amplitude was determined as the maximum difference from the average pre-event voltage to the peak of the largest part of the burst. Half-duration was measured as the length of time from the event onset to the time during the decay of the event when the difference from baseline was reduced to 50% of the maximum difference. The coastline bursting index was slightly modified from Korn et al. (1987). It was calculated using the following formula:
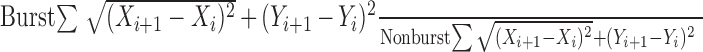 |
where distances between each pair of consecutive data points were determined by summing the squared differences between their coordinates and taking the square root. The sum of all pairs of consecutive points during the burst period was then divided by the sum of the distances between each pair of consecutive data points for an equivalent length of time when the slice was not bursting.
In slices from epileptic rats, some slices exhibited small burst discharges, making it important to clearly distinguish epileptiform events from noise. Therefore, we defined burst discharges in epileptic tissue as follows: repetitive clusters of population spikes superimposed on a positive-going envelope that was >1.5 mV in amplitude, measured from baseline to the peak of the positive-going envelope.
Statistical analysis
For all comparisons of evoked and spontaneous events, a paired Student's t test was used to compare events in a single recording location before and after bath application of VEGF (p = 0.05). For t test results, the sample size is indicated by the reported degrees of freedom in the format t(df) = x (i.e., df = sample size –1).
Results
VEGF depresses transmission throughout the trisynaptic circuit
Evoked field responses
The effect of bath-applied VEGF on synaptic transmission in the normal hippocampus was first examined using extracellular recordings of evoked responses to half-maximal stimulation of the major afferent pathways: (1) perforant path input to the dentate gyrus granule cell layer (Fig. 1, region 1), (2) mossy fiber input to area CA3 pyramidal cell layer (Fig. 1, region 2), and (3) Schaffer collateral input to area CA1 pyramidal cells (Fig. 1, region 3). In addition to evoked population spikes, the fEPSP was recorded in area CA1 by recording in stratum radiatum (Fig. 1, region 4).
Figure 1 shows a summary of the effect of VEGF on evoked response amplitude. In all regions, VEGF significantly reduced population spike amplitude (dentate gyrus, t(3) = 3.80, p = 0.032; CA3, t(3) = 4.48, p = 0.021; CA1 pyramidal cell layer, t(9) = 3.57, p = 0.006) or fEPSP amplitude (CA1 stratum radiatum, t(6) = 2.69, p = 0.036).
Time course of VEGF action
Figure 2 shows the time course of the effects of VEGF on population spike amplitude in the pyramidal cell layer in area CA1. The population spike recorded from the pyramidal cell layer was evoked by a half-maximal stimulus to the Schaffer collateral pathway, as shown in Figure 1A. Measurements were made every 10 min during the 70 min period of exposure to VEGF. Measurements of population spike amplitude showed a rapid and progressive decrease in amplitude relative to baseline (10 min, t(5) = 3.57, p = 0.016; 20 min, t(5) = 3.33, p = 0.020; 30 min, t(5) = 4.93, p = 0.04; 40 min, t(5) = 4.54, p = 0.006; 50 min, t(5) = 2.57, p = 0.049; 60 min, t(5) = 3.39, p = 0.019; 70 min, t(5) = 3.33, p = 0.021) (Fig. 2). For comparison, BSA was added to the perfusate (0.001–0.0015% final concentration) (Fig. 2). There were no significant changes at any time relative to baseline amplitudes (e.g., 60 min, t(4) = 0.55, p = 0.610).
Figure 2.

The amplitude of the population spike (Pop. Spike) recorded in the pyramidal cell layer of area CA1 steadily decreased with time after VEGF administration. Measurements were made every 10 min during continuous VEGF exposure (n = 6). Paired t test comparisons to baseline amplitude showed a significant reduction in population spike amplitude at each time point after the start of VEGF exposure. For comparison, slices were exposed to vehicle (BSA) and did not show any significant changes relative to baseline values at any time (n = 5). *p < 0.05.
Paired-pulse inhibition and facilitation in area CA1
Paired pulses were triggered to the Schaffer collaterals while recording in the CA1 pyramidal cell layer to assess effects of VEGF on paired-pulse inhibition (interstimulus interval, 10 or 20 ms) or facilitation (40 or 80 ms) of the population spike. Figure 3 shows the paired-pulse ratios as a function of VEGF exposure. VEGF did not change the paired-pulse ratio significantly at any interstimulus interval (10 ms, t(3) = –0.99, p = 0.393; 20 ms, t(3) = –0.21, p = 0.850; 40 ms, t(3) = 1.79, p = 0.215; 80 ms, t(3) = 0.48, p = 0.676). These data suggest that the effects of VEGF are unlikely to be mediated by selective effects on recurrent inhibition or to be related to presynaptic mechanisms.
Figure 3.

The ratio of population spikes recorded in the CA1 pyramidal cell layer to a pair of identical, half-maximal stimuli delivered to the Schaffer collateral pathway was unchanged by exposure to VEGF. Comparison of the paired-pulse ratio before VEGF administration (Control) and at the time during VEGF exposure when responses were maximally depressed (VEGF) showed no statistically significant change in paired-pulse inhibition (10 and 20 ms interstimulus intervals) or paired-pulse facilitation (40 and 80 ms interstimulus intervals; n = 5).
Intracellular recordings from CA1 neurons
Intrinsic properties
Intrinsic properties of hippocampal neurons were measured to test the possibility that VEGF suppresses responses to synaptic activation by, for example, a decline in input resistance that would arise from a change in neuronal membrane permeability. This was a consideration given the potent permeabilizing effects of VEGF on endothelial cells (Dvorak et al., 1999; Yancopoulos et al., 2000). CA1 pyramidal cells were recorded for at least 10 min before VEGF exposure to obtain baseline values and to confirm stability of membrane properties. VEGF was bath applied until there was an effect on the evoked responses to afferent stimulation (at least 30 min). At the time when there was a maximal effect on the evoked response, measurements of intrinsic properties were made and compared with baseline values.
Table 1 compares the intrinsic properties measured at two times: (1) the baseline period, and (2) the time after VEGF was bath applied when there was a decreased evoked response to afferent input. There was no statistically significant effect of VEGF on resting membrane potential (t(9) = 1.05, p = 0.313) input resistance (t(8) = 0.23, p = 0.821), time constant (t(8) =–0.54, p = 0.599), action potential amplitude (t(7) = 1.05, p = 0.313), slope (t(7) = 0.70, p = 0.499), or duration (t (7) = 1.20, p = 0.250) at a time when synaptic potentials were depressed (see below). The absence of effects of VEGF on intrinsic properties is exemplified in Figure 4 and quantified in Table 1. Figure 4B shows the maximal initial responses and average steady-state responses of eight cells to a series of current pulses (–0.05 to +0.04 nA, 200 ms). Comparison of the maximum slope of this relationship (between –0.1 and +0.1 nA) showed no significant difference for the initial (t(7) = 1.90, p = 0.092) or steady-state (t(7) = 1.60, p = 0.165) responses. These data indicate that the VEGF-related reductions in evoked population spikes and evoked fEPSPs, reported above, were unlikely to be mediated by intrinsic changes in CA1 pyramidal cells. Together with the evidence that effects of VEGF can reverse (see below), the results confirm that the effects of VEGF are not attributable to a general deterioration of the slice.
Table 1.
Comparison of intrinsic properties of CA1 pyramidal cells before and after VEGF bath application
|
|
n |
Control |
VEGF |
|---|---|---|---|
| Membrane properties | |||
| RMP (mV) | 10 | -62.0 ± 1.3 | -61.9 ± 1.3 |
| RIN (MΩ) | 9 | 42.9 ± 5.2 | 47.9 ± 5.1 |
| τ(ms) | 9 | 11.0 ± 1.9 | 8.5 ± 1.2 |
| Action potential amplitude | |||
| Total (mV) | 8 | 77.6 ± 1.9 | 72.7 ± 3.4 |
| From threshold (mV) | 8 | 68.7 ± 2.1 | 66.6 ± 2.8 |
| Action potential slope | |||
| Maximum rise (V/s) | 8 | 362.5 ± 39.3 | 342.5 ± 45.6 |
| Maximum decay (V/s) | 8 | 80.7 ± 14.6 | 71.4 ± 13.1 |
| dv/dt ratio | 8 | 5.3 ± 0.7 | 5.9 ± 0.9 |
| Action potential duration | |||
| Total (ms) | 8 | 2.9 ± 0.7 | 2.6 ± 0.4 |
| Half-duration (ms) |
8 |
0.76 ± 0.3 |
0.44 ± 0.1 |
Measurement of intrinsic properties showed no significant changes after bath application of VEGF. Measurements were made after at least 30 min of exposure to VEGF, at a time when synaptic potentials were maximally depressed (mean ± SEM; paired Student's t test, p > 0.05). RMP, Resting membrane potential; RIN, input resistance; τ, time constant; dv/dt ratio, ratio of the maximal slope of the rising phase/maximal slope of the decay phase.
Figure 4.
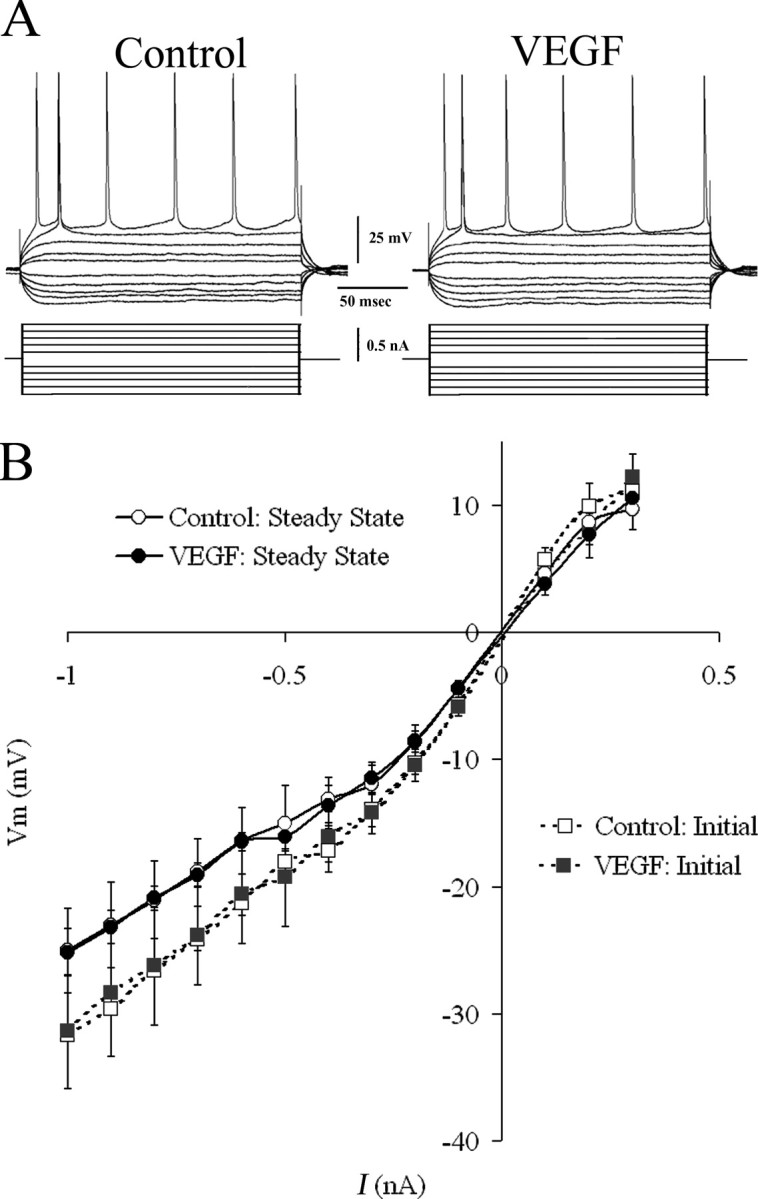
Effects of VEGF on CA1 pyramidal cell intrinsic properties. A, Representative traces from a CA1 pyramidal cell injected with pulses of hyperpolarizing and depolarizing direct current show no evident change in input resistance, time constant, or firing behavior during exposure to VEGF, at a time when synaptic responses were depressed. B, Composite I–V curve based on responses from eight cells includes measurements of peak (initial) and plateau (steady state) responses to injected current before and during 30–60 min of exposure to VEGF. Comparison of maximal slopes of these curves showed no significant effect of VEGF (paired Student's t test, p > 0.05; n = 8). I, Intracellularly injected current; Vm, membrane voltage.
Synaptic potentials
Figure 5 demonstrates the effect of VEGF to depress evoked synaptic potentials. For each CA1 pyramidal cell, a stimulus was delivered to the Schaffer collateral pathway at a strength that elicited the maximal subthreshold synaptic potential at –60 mV. Figure 5, A and B, shows examples of synaptic responses at –72 mV and at hyperpolarized (–82 mV) and depolarized (–62 mV) potentials. At –70 ± 2 mV, the synaptic potential was depolarizing, and VEGF reduced the peak amplitude of the depolarization (Fig. 5B, point 3) by 35 ± 6.6%, which was significant (t(7) = 2.97, p = 0.021). BSA (0.001% final concentration) did not change the peak amplitude of the depolarizing response evoked at –70 mV (t(5) = 0.95, p = 0.386). The depolarization evoked at a hyperpolarized potential (–80 ± 2 mV) (Fig. 5B, point 4) was also significantly reduced by VEGF (29 ± 4.8%; t(8) = 3.01, p = 0.029). When the same stimulus was delivered at more depolarized potentials, the synaptic potential began with a depolarization (presumably an EPSP), followed by a hyperpolarization (presumably an IPSP). At –60 ± 2 mV, VEGF produced a significant (35 ± 8.9%) reduction in the amplitude of the depolarizing component (t(7) = 2.51, p = 0.041) (Fig. 5B, point 1). VEGF also produced a significant (34 ± 8.8%) reduction in the amplitude of the hyperpolarizing component (t(7) = –3.06, p = 0.018) (Fig. 5B, point 2). In addition, the maximum slope of the depolarizing phase of the synaptic potential (at –70 ± 2 mV) was reduced by 40 ± 8.8% (t(7) = 2.92, p = 0.022).
Figure 5.
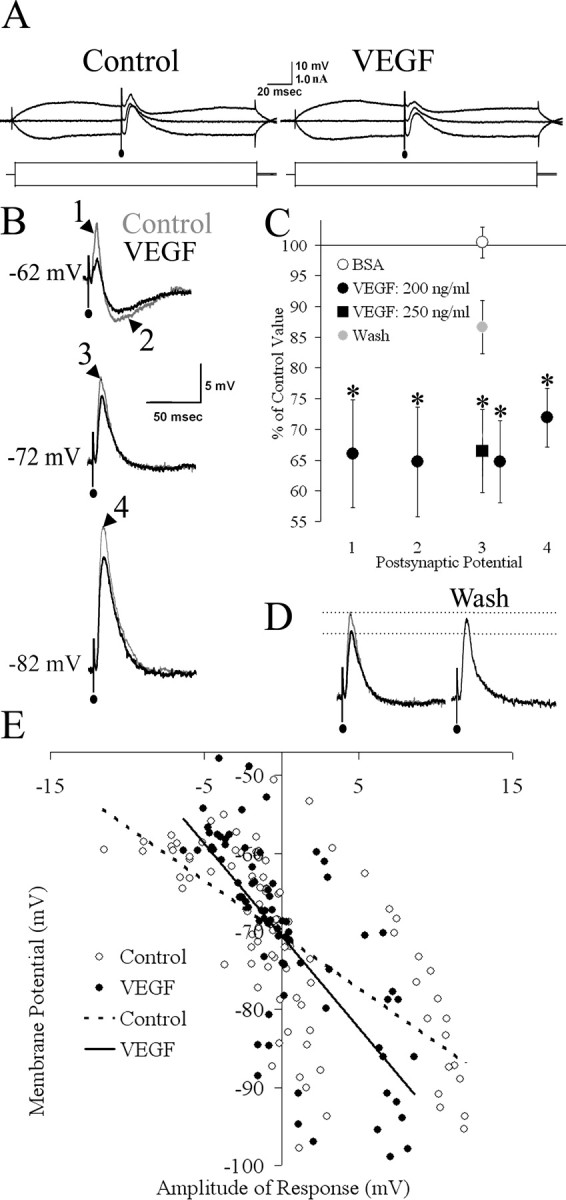
VEGF reduces EPSPs and IPSPs evoked in area CA1 pyramidal cells. A, Averages of representative traces from a CA1 pyramidal cell with synaptic potentials evoked on depolarizing and hyperpolarizing current pulses and at resting potential (–72 mV). B, Magnification of the traces shown in A. For each cell, the synaptic potential was measured in four ways: the amplitudes of depolarizing (EPSP) and hyperpolarizing (IPSP) phases of the potential when the cell was depolarized to –60 mV (points 1 and 2, respectively), the amplitude to peak of the depolarization when the cell was at –70 mV (point 3), and the peak amplitude of the depolarizing potential elicited from –80 mV (point 4). C, The mean postsynaptic potential amplitude after VEGF administration at each of the four points in B is represented as a percentage of the control value. Exposure of eight slices to the concentration of VEGF used to produce changes in field potentials but not intrinsic intracellular properties (200 ng/ml) produced a significant reduction in amplitude of both the EPSP component (point 1) and the IPSP component (point 2) when the stimulus was triggered at –60 ± 2 mV, as well as the peak of the depolarization when the cell was maintained at –70 ± 2 mV (point 3) or –80 ± 2 mV (point 4). When VEGF was removed from the perfusate in a subset of these recordings (Wash; n = 4), evoked postsynaptic potential amplitude at –70 ± 2 mV increased significantly from the amplitude during VEGF exposure, demonstrating reversibility of the VEGF-related reduction. For comparison, the effect of a higher concentration of VEGF (250 ng/ml) on the amplitude of the peak depolarization at –70 ± 2 mV was measured in six slices. This concentration also produced a significant reduction in amplitude (*p < 0.05). When BSA was applied in place of VEGF (BSA; n = 6), the amplitude of the peak depolarization at –70 ± 2 mV did not differ significantly from baseline values. D, Representative traces demonstrating the reversal of the amplitude of the depolarizing postsynaptic potential of a cell at –70 mV. When VEGF was removed from the perfusate (Wash), the amplitude of the postsynaptic potential recovered to near baseline. E, The polarity and amplitude of evoked postsynaptic potentials was plotted as a function of holding potential. There was no significant effect of VEGF on reversal potential (n = 8; see Results).
Comparison of two concentrations of VEGF
To determine whether the concentration of VEGF used to generate the results reported above (200 ng/ml) was maximal, we tested a higher concentration (250 ng/ml). The change in peak amplitude of the depolarizing postsynaptic potential of CA1 pyramidal cells maintained at –70 ± 2 mV was measured after at least 30 min of VEGF exposure, at the time when the reduction in amplitude was maximal. Comparison of maximal effects of both concentrations showed a similar reduction in the response amplitude (200 ng/ml, 34 ± 6.6% reduction, n = 8; 250 ng/ml, 35 ± 6.8% reduction, n = 6) (Fig. 5C), suggesting that the 200 ng/ml concentration was likely to be a maximal concentration.
Reversibility of the effects of VEGF
The ability to reverse the effects of VEGF was tested on the postsynaptic depolarizing potential at –70 ± 2 mV in four CA1 pyramidal cells. Figure 5D shows an example of an evoked postsynaptic potential that demonstrated reversal of VEGF-induced depression after 60 min of exposure to VEGF-free ACSF (Wash). In the four cells, VEGF significantly reduced the amplitude of the depolarizing potential to 66 ± 1.2% of the control amplitude (t(3) = 8.87, p = 0.003). The amplitude was restored to 87 ± 4.3% of the control value after 60 min of wash, a significant recovery (t(3) = –3.66, p = 0.035) (Fig. 5C). Therefore, the effects of VEGF are reversible.
Reversal potential
Although VEGF did not increase paired-pulse inhibition, hyperpolarize the resting potential to ECl–, or decrease input resistance, suggesting that increased GABAergic inhibition could not explain the depression of synaptic potentials by VEGF, we also examined the reversal potential of the inhibitory component of the IPSP to further evaluate a role of GABAergic inhibition in the effects of VEGF. Figure 5E shows the reversal potentials of the hyperpolarizing component of synaptic potentials evoked by Schaffer collateral stimuli, which were not statistically different after bath application of VEGF (–70.3 ± 2.9 mV pre-VEGF; –70.5 ± 1.8 post-VEGF; t(7) =–0.20, p = 0.848).
Spontaneous burst discharges
Bicuculline-induced spontaneous discharges
Given the ability of VEGF to dampen synaptic transmission in the hippocampus, we asked whether VEGF could also suppress hippocampal epileptiform activity. To test this hypothesis, we first examined epileptiform activity induced in slices from normal rats that were disinhibited by exposure to the GABAA antagonist BMI (10 μm). BMI was added to seven slices from five normal male rats. This concentration of BMI produced spontaneous epileptiform bursts in the CA3 pyramidal cell layer with a mean amplitude of 11.3 ± 1.5 mV and a frequency of 14.3 ± 1.75 bursts/min. Surprisingly, the same concentration of VEGF that inhibited synaptic potentials (200 ng/ml) did not significantly alter the amplitude, frequency, coastline bursting index, or half-duration of BMI-induced bursts (amplitude, t(6) = 0.27, p = 0.979; frequency, t(6) = 0.23, p = 0.821; coastline bursting index, t(6) = –0.21, p = 0.839; half-duration, t(6) =–0.45, p = 0.665) (Fig. 6).
Figure 6.

Differential effects of VEGF on spontaneous epileptiform burst discharges recorded in area CA3 in slices from epileptic and normal rats. A, Representative example of spontaneous burst discharges recorded from slices made from normal adult rats that were exposed to the GABAA receptor antagonist bicuculline (10 μm) demonstrates the similarity of epileptiform bursts before and after VEGF application. B, Examples of spontaneous CA3 burst discharges in a slice made from an epileptic rat (slices were made during the period of recurrent seizures after pilocarpine-induced status epilepticus; see Materials and Methods) show reduced burst discharges after the slice was exposed to VEGF.C, One-minute-long recordings made in a slice from an epileptic rat demonstrate reduced burst frequency after bath application of VEGF. D, The average change in burst amplitude frequency, duration, and intensity (measured using the coastline bursting index; see Materials and Methods) after VEGF administration is represented as a percentage of control. VEGF significantly reduced amplitude, frequency, and intensity but not the duration of the bursts (n = 11). *p < 0.05. However, paired Student's t tests did not show a significant reduction in any of these measures when epileptiform bursts were induced by bicuculline bath application (n = 7).
Spontaneous discharges in pilocarpine-treated rats with recurrent seizures (epileptic rats)
We have shown previously that spontaneous burst discharges can be recorded in hippocampal slices within the CA3 region of epileptic rats several weeks after pilocarpine-induced status epilepticus, and that these bursts discharges do not require exposure to bicuculline or other convulsants (Scharfman et al., 2000, 2001). Furthermore, we have shown that VEGF protein expression appears to be increased in hippocampus in this animal model of epilepsy (Croll et al., 2004). These data suggested that VEGF might have greater effects in an epileptic rat relative to a normal rat that had been disinhibited. Therefore, we examined the effects of VEGF on spontaneous CA3 burst discharges recorded in slices from epileptic rats.
Slices (n = 11) from seven rats that were examined 1–4 months after status epilepticus exhibited spontaneous bursts in the CA3 pyramidal cell layer. The mean amplitude of the underlying positive wave [on which spikes are superimposed (Fig. 6)] was 3.9 ± 0.5 mV, and the mean burst frequency was 21.7 ± 11.1 bursts/min.
Figure 6 shows a comparison of spontaneous bursts before and after VEGF bath application. In all slices, VEGF reduced the amplitude and frequency of spontaneous bursts and, in two slices, stopped them completely. When pooling all experiments, mean burst amplitude was significantly reduced by 19 ± 5.6% (t(10) = 4.40, p = 0.001). The frequency of bursts was also reduced significantly by 35 ± 10.9% (t(8) = 2.80, p = 0.022). VEGF reduced the coastline bursting index by 39 ± 8.8%, which was also significant (t(10) = 3.04, p = 0.012). However, VEGF did not significantly reduce the half-duration of these bursts (t(10) = 0.13, p = 0.892).
In summary, VEGF decreased evoked potentials in the hippocampus of adult male rats. Although there was no evident effect on epileptiform bursts that were produced by blockade of GABAA receptor-mediated inhibition, VEGF did suppress epileptiform bursts that were recorded in slices from animals with spontaneous recurrent seizures.
Discussion
Summary
The results of this study demonstrate, for the first time, that VEGF produces robust alterations in hippocampal synaptic physiology. Extracellular recordings from normal male rats showed that VEGF reduced evoked population spike amplitudes in each of the major excitatory pathways in the hippocampus and fEPSP amplitude in the Schaffer collateral pathway. Intracellular recordings from CA1 pyramidal cells showed reduced amplitude of depolarizing and hyperpolarizing postsynaptic potentials and decreased EPSP slope, consistent with extracellular recordings. The stability of intrinsic cellular properties indicated that the effects on evoked responses were unlikely to be caused by changes in neuronal membrane permeability or other effects on membrane properties.
This study also demonstrated an increased efficacy of VEGF in epileptic tissue relative to normal tissue. VEGF suppressed spontaneous burst discharges in slices from epileptic rats but had relatively little effect on burst discharges induced by disinhibition of normal rat slices. Given that VEGF is upregulated after seizures (Newton et al., 2003; Croll et al., 2004), it is possible that VEGF upregulation after seizures is an endogenous compensatory mechanism to reduce excitability. Indeed, after ischemia and traumatic brain injury, VEGF appears to be neuroprotective (Jin et al., 2000, 2001). However, it is currently unclear whether the pharmacological studies of VEGF conducted to date, including the data presented here, reflect endogenous actions of VEGF.
VEGF suppression of synaptic transmission
VEGF decreased the evoked responses of hippocampal neurons to synaptic stimulation in each of the major glutamatergic pathways of the trisynaptic circuit. These findings suggest that VEGF has the ability to dampen glutamatergic pathways independent of the afferent system or target cell, at least in hippocampal principal cells. Whether other cells are influenced by VEGF will require additional examination, but, if true, it would be a potentially effective means to protect the brain from excitotoxic injury. VEGF has been shown previously to protect susceptible neuronal populations from cell death in a number of animal models in which excitotoxicity is thought to play a role, including stroke/hypoxia, seizures/excitotoxicity, amyotrophic lateral sclerosis, and Parkinson's disease (Hayashi et al., 1998; Jin et al., 2000; Matsuzaki et al., 2001; Lambrechts et al., 2003, Sun et al., 2003; Yasuhara et al., 2004).
Several possible mechanisms underlying the suppression of transmission can be ruled out. First, a general deterioration of neurons is unlikely given that the effects of VEGF on postsynaptic potentials could reverse. Similarly, the lack of apparent effects on membrane properties at a time when synaptic responses were already depressed argues against the possibility that cells had become permeabilized or destabilized in other ways. Finally, the lack of change in paired-pulse facilitation suggests that VEGF does not alter transmitter release at the presynaptic terminal, at least with regard to its ability to induce facilitation of hippocampal synaptic transmission.
The lack of change in paired-pulse inhibition suggests that the depression of synaptic transmission in the major glutamatergic pathways is not a result of increased recurrent inhibition. Other changes in GABAergic inhibition are also unlikely to explain the effects of VEGF, because there was no change in resting potential (if GABA release was enhanced, resting potential would be likely to hyperpolarize to ECl–) and no change in input resistance (tonic activation of postsynaptic GABAA receptors would be likely to open chloride channels and hence decrease input resistance). Finally, there was no change in the reversal potential of the hyperpolarizing component of the synaptic potential. Although these are robust benchmarks of GABAergic inhibition, they do not address all of the aspects of GABA-mediated actions in hippocampus, and it is still possible that some effects of VEGF influence actions not studied here. One example would be effects on GABAergic transmission to distal dendrites, which would be difficult to detect with somatic electrodes, or extracellular recordings from dendritic regions. In addition, there could be an influence of VEGF on GABAB receptor-mediated transmission or GABAergic transmission between GABAergic neurons.
There are several possible receptors that could mediate the effects of VEGF that were observed because the isoform that was used, the 165 aa splice variant of VEGFA protein, binds to all known VEGF receptors in the brain: the two primary signal transducing receptors VEGFR1 (also known as Flt-1) and VEGFR2 (also known as Flk-1 or KDR) and the neuropilins Npn-1 and Npn-2.
It is unlikely that the effects observed were mediated by VEGFR1, although very little is currently understood about physiological effects of VEGFR1 in hippocampus. What has been shown is that VEGFR1-expressing cells in culture have decreased potassium currents (specifically the delayed rectifier) after exposure to VEGF (Xu et al., 2003), and a decrease in the delayed rectifier would be more likely to increase excitability rather than decrease it. Regarding VEGFR2, there is evidence that VEGFR2 influences hippocampal function (Cao et al., 2004) but no published data, to our knowledge, of the effects of VEGFR2 activation on hippocampal synaptic transmission.
Evidence to date suggests that neuropilins in the normal adult rat brain are primarily neuronal (Kawakami et al., 1996; Sahay et al., 2005). Effects of VEGF that we observed cannot easily be explained by neuropilin-1 activation because activation of this receptor appears to increase, rather than decrease, hippocampal glutamatergic transmission (Sahay et al., 2005). Little physiological information is currently available about neuropilin-2.
Effects of VEGF could be glial, not neuronal. This would be consistent with the lack of evidence for presynaptic versus postsynaptic actions of VEGF and the lack of evidence that effects on EPSPs were distinct from effects on IPSPs. This hypothesis is consistent with data showing the key role of glia in regulating synaptic transmission in hippocampal slices (Keyser and Pellmar, 1994; Hülsmann et al., 2003). There are also several additional reasons to suggest a role of glia in the effects of VEGF to depress synaptic transmission. First, the expression pattern of VEGF in normal hippocampus suggests a pattern of immunoreactivity that surrounds glial-like cells (Croll et al., 2004). One of the receptors that may be involved is VEGFR2, which appears to be expressed by glia in the adult brain (Lennmyr et al., 1998; Croll and Wiegand 2001; Krum et al., 2002), although there could be neuronal expression in hippocampus, particularly under certain conditions. VEGF has been shown to act on glia by increasing glial hypertrophy (Krum et al., 2002). We speculate that hypertrophy, should it occur in hippocampus, could allow glia to invaginate the synaptic cleft and increase sequestration of neurotransmitter from the synapse. This explanation for the suppressive effect of VEGF on synaptic transmission is admittedly speculative but would be consistent with the data presented here, e.g., the similarity of effects on EPSP and IPSP amplitude.
VEGF and epileptiform activity
The ability of VEGF to suppress epileptiform activity in epileptic rats, but not disinhibited slices, was remarkable. Why might this be the case? A possible explanation is suggested by the finding that immunoreactivity for VEGF protein is increased on glial-like cells after chronic seizures that follow pilocarpine-induced status epilepticus (Croll et al., 2004). This increase in VEGF protein, presumably associated with glial membranes, could occur if there is increased receptor density on glia or increased affinity of VEGF at its glial receptors. There is evidence of increased expression of VEGF receptors on glial cells after ischemia (Lennmyr et al., 1998; Croll and Wiegand, 2001; Fujita et al., 2001; Zhang et al., 2001; Beck et al., 2002), although, to date, it is unclear whether VEGF receptors also change expression patterns in epileptic animals. In summary, VEGF may be particularly effective at depressing burst discharges in epileptic tissue because of the changes in the epileptic brain that enhance VEGF action. The fact that the increase in expression appears to be associated with glia further directs our working hypothesis for VEGF action toward a glial mechanism.
Conclusion
In conclusion, the present work adds an important new facet to the growing list of pleiotropic effects of growth factors in the CNS. VEGF has the ability to regulate hippocampal synaptic transmission by dampening the response to afferent input on principal cells and appears extremely potent in epileptic tissue. Although effects of VEGF on spontaneous seizures in vivo have yet to be addressed, the present study provides the first clue that VEGF receptors or signaling pathways could have merit as novel targets for anticonvulsant drug development
Footnotes
This work was supported by National Institutes of Health Grants NS 35762 and NS 41490 (H.E.S.), the Epilepsy Foundation, and the Helen Hayes Hospital Foundation. We thank Regeneron Pharmaceuticals for their generous gift of VEGF. We also thank Nicholas Makarenko, Neil MacLusky, Jamee Nicoletti, Sachin Shah, and Kerry Stormes for their help throughout this project.
Correspondence should be addressed to Dr. Daniel P. McCloskey, Center for Neural Recovery and Rehabilitation Research, Helen Hayes Hospital, Route 9 West, West Haverstraw, NY 10993-1195. E-mail: mccloskeyd@helenhayeshosp.org.
DOI:10.1523/JNEUROSCI.2577-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258889-09$15.00/0
References
- Alger BE, Teyler TJ (1976) Long-term and short-term plasticity in the CA1, CA3 and dentate regions of the rat hippocampal slice. Brain Res 110: 463–480. [DOI] [PubMed] [Google Scholar]
- Beck H, Acker T, Puschel AW, Fujisawa H, Carmeliet P, Plate KH (2002) Cell type-specific expression of neuropilins in an MCA-occlusion model in mice suggests a potential role in post-ischemic brain remodeling. J Neuropathol Exp Neurol 61: 339–350. [DOI] [PubMed] [Google Scholar]
- Brockington A, Lewis C, Whaerton S, Shaw PJ (2004) Vascular endothelial growth factor and the nervous system. Neuropathol Appl Neurobiol 30: 427–446. [DOI] [PubMed] [Google Scholar]
- Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, During MJ (2004) VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet 36: 827–835. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Chung I, Kozniewska E, Ivanenko T, Chang W, Harrington JF, Duncan JA, Szmydynger-Chodobska J (2003) Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience 122: 853–867. [DOI] [PubMed] [Google Scholar]
- Croll SD, Wiegand SJ (2001) Vascular growth factors in cerebral ischemia. Mol Neurobiol 23: 121–135. [DOI] [PubMed] [Google Scholar]
- Croll SD, Goodman JH, Scharfman HE (2004) Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword? Adv Exp Med Biol 548: 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM (1999) Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol 237: 97–132. [DOI] [PubMed] [Google Scholar]
- Fujita H, Zhang B, Sato K, Tanaka J, Sakanaka M (2001) Expressions of neuropilin-1, neuropilin-2 and semaphorin 3A mRNA in the rat brain after middle cerebral artery occlusion. Brain Res 914: 1–14. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Abe K, Itoyama Y (1998) Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J Cereb Blood Flow Metab 18: 887–895. [DOI] [PubMed] [Google Scholar]
- Hülsmann S, Straub H, Richter DW, Speckmann EJ (2003) Blockade of astrocyte metabolism causes delayed excitation as revealed by voltage-sensitive dyes in mouse brainstem slices. Exp Brain Res 150: 117–121. [DOI] [PubMed] [Google Scholar]
- Jin K, Mao XO, Batteur SP, McEachron E, Leahy A, Greenberg DA (2001) Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience 108: 351–358. [DOI] [PubMed] [Google Scholar]
- Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA (2002) Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci USA 99: 11946–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA (2000) Vascular endothelial growth factor: direct neuroprotective effect in vitro ischemia. Proc Natl Acad Sci USA 97: 10242–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami A, Kitsukawa T, Takagi S, Fujisawa H (1996) Developmentally regulated expression of a cell surface protein, neuropilin, in the mouse nervous system. J Neurobiol 29: 1–17. [DOI] [PubMed] [Google Scholar]
- Keyser DO, Pellmar TC (1994) Synaptic transmission in the hippocampus: critical role for glial cells. Glia 10: 237–243. [DOI] [PubMed] [Google Scholar]
- Khaibullina AA, Rosenstein JM, Krum JM (2004) Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res 148: 59–68. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R (1987) Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol 57: 325–340. [DOI] [PubMed] [Google Scholar]
- Krum JM, Mani N, Rosenstein JM (2002) Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience 110: 589–604. [DOI] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, Van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, et al. (2003) VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet 34: 383–394. [DOI] [PubMed] [Google Scholar]
- Lee MY, Ju WK, Cha JH, Son BC, Chun MH, Kang JK, Park CK (1999) Expression of vascular endothelial growth factor mRNA following transient forebrain ischemia in rats. Neurosci Lett 265: 107–110. [DOI] [PubMed] [Google Scholar]
- Lennmyr F, Ata KA, Funa K, Olsson Y, Terent A (1998) Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and FLK-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol 57: 874–882. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M (2001) Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J 15: 1218–1220. [PubMed] [Google Scholar]
- Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, Duman RS (2003) Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci 23: 10841–10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein JM, Krum JM (2004) New roles for VEGF in nervous tissue—beyond blood vessels. Exp Neurol 187: 246–253. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Khaibullina A, Krum JM (2003) Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci 23: 11036–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Kim CH, Sepkutty JP, Cho E, Huganir RL, Ginty DD, Kolodkin AL (2005) Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci 25: 3613–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1994a) Synchronization of area CA3 hippocampal pyramidal cells and non-granule cells of the dentate gyrus in bicuculline-treated rat hippocampal slices. Neuroscience 59: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1994b) EPSPs of dentate gyrus granule cells synchronized with epileptiform bursts of dentate hilar mossy cells and area CA3 pyramidal cells in disinhibited rat hippocampal slices. J Neurosci 14: 6041–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1995) Electrophysiological diversity of pyramidal shaped neurons at the granule cell layer hilus border of the rat dentate gyrus recorded in vitro. Hippocampus 5: 287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1996) Conditions required for polysynaptic excitation of dentate granule cells by area CA3 pyramidal cells in rat hippocampal slices. Neuroscience 72: 655–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sarvey JM (1988) Physiological correlates of responses to gamma aminobutyric acid (GABA) recorded from rat visual cortical neurons in vitro. Synapse 2: 619–626. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL (2000) Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci 20: 6144–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Smith KS, Goodman JH, Sollas AL (2001) Survival of dentate hilar mossy cells after pilocarpine-induced seizures and their synchronized burst discharges with area CA3 pyramidal cells. Neuroscience 104: 741–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Carmaliet P (2004) VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. BioEssays 26: 943–954. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111: 1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson B, Peters M, Konig H-G, Poppe M, Levkau B, Rothermundt M, Arolt V, Kogel D, Prehn JH (2002) Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J Cereb Blood Flow Metab 22: 1170–1175. [DOI] [PubMed] [Google Scholar]
- Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA (1989) Rev: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse 3: 154–171. [DOI] [PubMed] [Google Scholar]
- van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, Tumas D, Gerlai R, Williams SP, Campagne ML, Ferrara N (1999) VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest 104: 1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JY, Zheng P, Shen DH, Yang SZ, Zhang LM, Huang YL, Sun FY (2003) Vascular endothelial growth factor inhibits outward delayed-rectifier potassium currents in acutely isolated hippocampal neurons. Neuroscience 118: 59–67. [DOI] [PubMed] [Google Scholar]
- Yancopoulos G, Davis S, Gale N, Rudge J, Wiegand S, Holash J (2000) Vascular-specific growth factors and blood vessel formation. Nat Genet 407: 242–248. [DOI] [PubMed] [Google Scholar]
- Yang SZ, Zhang LM, Huang YL, Sun FY (2003) Distribution of Flk-1 and Flt-1 receptors in neonatal and adult rat brains. Anat Rec A Discov Mol Cell Evol Biol 274A: 851–856. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Shingo T, Kobayashi K, Takeuchi A, Yano A, Muraoka K, Matsui T, Miyoshi Y, Hamada H, Date I (2004) Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson's disease. Eur J Neurosci 19: 1494–1504. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Tasang W, Zhang L, Powers C, Chopp M (2001) Up-regulation of neuropilin-1 in neovasculature after focal cerebral ischemia in the adult rat. J Cereb Blood Flow Metab 21: 541–549. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Jin K, Mao XO, Greenberg DA (2003) Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. FASEB J 17: 186–193. [DOI] [PubMed] [Google Scholar]