

Abstract
Activated NF-κB is a critical mechanism by which lymphoma cells infected by Epstein-Barr virus (EBV/HHV-4) and Kaposi sarcoma herpesvirus (KSHV/HHV-8) are protected from apoptotic stress. Selective pharmacologic inhibition of constitutive NF-κB activity induces apoptosis in KSHV- and EBV-infected lymphoma cells. In both tumor types, pharmacologic inhibition of NF-κB in vitro induced identical mitochondrially mediated apoptosis cascades. Assessment of gene regulation by microarray analysis revealed that the inhibition of NF-κB in tumor cells results in the down-regulation of a distinct group of prosurvival genes, including cIAP-1, cIAP-2, cFLIP, and IL-6. Using EBV- and KSHV-associated lymphomas in a murine system, we demonstrated that Bay 11-7082, a selective pharmacologic inhibitor of NF-κB, prevents or delays tumor growth and prolongs disease-free survival. Inhibition of NF-κB activity and tumor growth responses were further documented using a traceable reporter KSHV-positive cell line and in vivo imaging. These findings indicate that specific NF-κB-regulated survival factors work cooperatively to protect KSHV- and EBV-infected lymphoma cells from apoptosis such that they promote the establishment and progression of KSHV- and EBV-associated lymphomas in mice. They also support the use of selective NF-κB inhibitors in the treatment of herpesvirus-associated lymphomas.
Introduction
NF-κB is the collective name for a family of transcription factors that regulate cellular proliferative and survival responses. Aberrant and increased activity of NF-κB is characteristic of some lymphoid tumors. In particular, the activation of NF-κB by viral oncogenes is a mechanism used by lymphomagenic viruses.
Kaposi sarcoma herpesvirus (KSHV) and Epstein-Barr virus (EBV) are members of the γ-herpesvirus family and can infect multiple cell types, including B cells, which provide a reservoir for latent virus.1-3 Encoded within their genomes are a number of viral transforming genes and pirated cellular homologs that subvert cellular signaling pathways, including those leading to the activation of NF-κB and survival. In EBV-positive lymphomas, expression of the viral transforming gene LMP-1 in latently infected cells provides a constitutively active receptor for the recruitment of TRAF adaptor proteins and NF-κB activation.4 In KSHV-infected cells, vFLIP interacts with TRAFs, NIK, and IKKs5-8 and thereby constitutively induces NF-κB activity in latently infected lymphoma cells.9
Animal models with targeted disruption of NF-κB subunits have demonstrated the importance of NF-κB in cellular immunity, inflammation, and lymphoid organ development.10-12 NF-κB-binding sites are present in the promoters of a multitude of genes, and the antiapoptosis activity mediated by NF-κB depends on gene induction.13 Targets of NF-κB regulation include factors that modulate signaling pathways to inhibit apoptosis, growth factors, cell cycle regulatory proteins, and proteins that further enhance NF-κB activation, thereby promoting cellular survival and growth (for a review, see Karin and Lin14). Although many genes contain NF-κB-binding sites and signaling through the NF-κB pathway has been reported to increase their transcription, this transcriptional regulation is complex and frequently depends on multiple transcription cascades in addition to NF-κB. Few studies have specifically evaluated the genes affected after NF-κB inhibition in virus-associated lymphomas. Using a phosphorylation-defective mutant of IκBα that suppresses NF-κB by sequestering it in the cytoplasm, 2 studies found down-regulation of some antiapoptosis and growth factors that include bcl-2, bcl-x, and IL-6 in EBV-infected lymphomas.15,16 Previous studies in our laboratory have shown that low-dose treatment with the NF-κB inhibitor Bay 11-7082 selectively inhibits IκBα phosphorylation and constitutive NF-κB DNA-binding activity in KSHV-infected PEL cells.17 Treatment of lymphoblastoid cell lines in vitro with Bay 11-7082 also resulted in the inhibition of NF-κB, the down-regulation of a specific subset of genes, and apoptosis.18
In this study we examined the role of NF-κB in the survival of herpesvirus-associated lymphoma cells by comparing gene expression signatures and examining the mechanism of apoptosis of KSHV- and EBV-infected lymphoma cells after NF-κB inhibition with Bay 11-7082. We also evaluated the therapeutic potential of NF-κB inhibition using mouse xenograft models of EBV-and KSHV-associated lymphomas and in vivo imaging. Treatment of mice with Bay 11-7082 resulted in NF-κB inhibition in vivo and significantly delayed the onset and development of EBV- and KSHV-infected lymphomas. These results suggest that inhibition of NF-κB may be an effective treatment for KSHV- and EBV-infected lymphomas through the down-regulation of specific prosurvival factors that protect the virally infected lymphoma cells from apoptosis.
Materials and methods
Cell lines
The following cell lines were used: BC-3, BC-1, BCBL-1, EBV-IBL, BCKN-1, and LCL 9001. BC-3 and BCBL-1 contain KSHV; EBV-IBL, BCKN-1, and LCL 9001 contain EBV; and BC-1 contains both viruses. BC-1, BC-3, and BCKN-1 were derived in our laboratory from lymphomatous effusions, and EBV-IBL was derived from an HIV-associated immunoblastic lymphoma, as described.19 BCBL-120 was obtained from the AIDS and Cancer Specimen Bank (ACSB). The lymphoblastoid cell line LCL9001 was obtained by infection of peripheral blood lymphocytes with EBV. BJAB is a Burkitt lymphoma cell line used as a control in some experiments and was obtained from the American Type Culture Collection (ATCC, Rockville, MD). Cells were grown in RPMI 1640 (Gibco BRL, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Norcross, GA) and 50 μg/mL gentamicin (Sigma, St Louis, MO).
The BC-3/NFκB-luc cell line was made by stably transfecting BC-3 cells with pcDNA/NFκB-Luc plasmid, which contains the NF-κB-regulated luciferase reporter in addition to an antibiotic resistance (neomycin) cassette. This plasmid was first constructed by removal of the PCMV promoter from pcDNA3.1(+) (Invitrogen, Carlsbad, CA) through digestion with MluI and XbaI (New England Biolabs, Beverly, MA). The MluI-XbaI fragment from pNFκB-Luc (BD Biosciences Clontech, Palo Alto, CA), containing the luciferase gene under the control of a kappa enhancer element was inserted into the modified pcDNA backbone. BC-3 cells were transfected with pcDNA/NFκB-Luc using Transfectin lipid reagent (BioRad, Hercules, CA). Cells were selected with G418 and subcloned in irradiated feeder fibroblasts. Single-cell clones were evaluated by luciferase assays, and a clone with high constitutive luciferase activity and dose-dependent reduction on treatment with Bay 11-7082 was selected (clone 6) and named BC-3/NFkB-luc (Figure 5B). This clone was confirmed to show greater than 90% decrease of luciferase activity on the elimination of vFLIP expression by RNA interference, as described.9
Figure 5.

In vivo imaging of NF-κB activity in mice with PEL. (A) NOD/SCID mice were challenged with 10 × 106 BC3/NFκB-luc traceable cells and subsequently received injections of vehicle (left) or 5 mg/kg Bay 11-7082 (right) on days 3, 5, and 7 after tumor challenge. Mice were imaged on the days indicated after injection with luciferin. ROI chosen for data acquisition is shown by the green oval over each mouse. (B) Luciferase activity was measured in the BC3/NFκB-luc cell line after treatment with Bay 11-7082 in vitro showing a reliable dose- and time-dependent response to NF-κB inhibition. Error bars represent SD. (C) Bioluminescence was quantitated and reported as relative bioluminescence units (BLU) at different time intervals for each mouse studied. All but 2 mice showed luminescence above background levels at day 3 (before drug injection); mouse 2 in the vehicle group did not develop detectable tumor until approximately day 33 of the experiment and is, therefore, not shown in panel A, and mouse 2 in the Bay 11 20-mg/kg dose group has not developed visible tumor to date. Treatment with Bay 11-7082 resulted in decreased bioluminescence by 24 hours, indicating decreased NF-κB activity. The luminescence remained low in the mice treated with Bay 11-7082 (squares), whereas it increased progressively in the mice treated with vehicle alone (triangles). Mice for which the line was interrupted before the last point in time shown represent those that developed ascites and were humanely killed.
Antibodies
Antibodies for caspase 9 were obtained from Cell Signaling Technology (Beverly, MA). IκBα, Bcl-2, TRAF2, and IL-6 were from Santa Cruz Biotechnology (Santa Cruz, CA). Caspase 3 and caspase 8 were from Upstate Biotechnology (Lake Placid, NY), PARP antibody was from Pharmingen (San Diego, CA), and Bcl-X antibody was from Transduction Laboratories (Boston, MA). This antibody recognizes long and short forms of the Bcl-X protein, but, as determined by size, only the long form was expressed in all the cell lines examined. Actin antibody was from Sigma, and cIAP-1, cIAP-2, and cFLIP antibodies were from Alexis Biochemicals (San Diego, CA).
Electrophoretic mobility shift assays and supershift analyses
Isolation of nuclear proteins was performed as described previously.17 For electrophoretic mobility shift assay (EMSA), oligonucleotides with NF-κB consensus sequences derived from the Igκ promoter (gift from Dr Hsiou-Chi Liou, Weill Medical College of Cornell University, New York, NY), and Octamer consensus sequences (Promega, Madison, WI) were end-labeled with T4 kinase and γ-[32P]ATP and purified over G-25 Sephadex columns. Before the addition of oligonucleotide probe, 4 μg nuclear protein was incubated with DNA binding buffer (2 μg poly [dI-dC], 0.25% NP-40, 5% glycerol, 10 mM Tris HCl, 50 mM KCl, 1 mM DTT, and 1 mM EDTA). A radiolabeled probe (2 × 104 cpm) was then incubated with the reaction mixture for 15 minutes, and samples were separated on 7% acrylamide gels and assessed by autoradiography.
Affymetrix oligonucleotide arrays
Total RNA (5 μg) was isolated from BC-3 and EBV-IBL cells treated with and without 5 μM Bay 11-7082 for 6 hours and subsequently were labeled with biotin (Affymetrix, Santa Clara, CA), as described. For hybridization, 50 μg cRNA was incubated with U95Av2 probe arrays (Affymetrix) for 16 hours at 45°C. Hybridized probe arrays were subsequently stained with streptavidin phycoerythrin conjugate and scanned with a Hewlett-Packard (Palo Alto, CA) GeneArray scanner at an excitation wavelength of 488 nm. Data analysis was performed with Genespring software (Silicon Genetics, Redwood City, CA) on results of 3 independent experiments. The raw data are available on-line.21
Western blot analyses
For immunoblot analyses, cytoplasmic (extracted as described using buffer A) or whole cell extracts generated using RIPA buffer (50 mM Tris-HCl, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 0.5 μg aprotinin, 1 mM Na3VO4, 1 mM NaF) were quantitated by Bradford bioassay (Bio-Rad) and separated on 8% to 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. Proteins were transferred to PVDF membranes and blocked in 5% milk-TBST for 1 hour at room temperature before primary antibody in 1% milk-TBST was added. Depending on the manufacturer's instructions, primary antibodies diluted 1:1000 were incubated either at room temperature for 1 hour or overnight at 4°C before they were washed and incubated with complimentary HRP-conjugated secondary antibodies (Amersham, Piscataway, NJ). Antibodies were diluted 1:2000 in 1% milk-TBST, and proteins were detected by enhanced chemiluminescence (Amersham).
Xenograft models of EBV and KSHV lymphoma
All animal experiments were performed in compliance with institutional guidelines and with approval of our Institutional Animal Care and Use Committee (IACUC). NOD/SCID male mice aged 6 to 8 weeks were obtained from Jackson Laboratories (Bar Harbor, ME). Animals were allowed to feed ad libitum on sterilized laboratory chow and water.
For the EBV in vivo model, LCL 9001 cells were suspended at 40 × 106 cells in 500 μL PBS and injected subcutaneously on day 1 of the experiment. Mice (n = 10 per group) subsequently received intraperitoneal injection of either vehicle or 20 mg/kg Bay 11-7082 on days 1, 3, and 5 and once a week thereafter after tumor challenge. Tumor size was measured on a weekly basis; after 4 weeks, all animals were killed and their tumors were weighed. The 2-sample t test was used to compare tumor weight between the Bay 11 and control groups. Repeated measures analysis of variance (RMANOVA) was used to compare tumor growth curves between the 2 groups. Analysis of residuals revealed good fit to the normal distribution without transformation. Accordingly, the untransformed tumor measurements were used.
In the KSHV in vivo model, BC-3 cells suspended at 10 × 106 cells in 500 μL PBS were injected intraperitoneally on day 0 of the experiment. Mice (n = 19 per group) subsequently received intraperitoneal injections 1, 3, and 5 days after BC-3 injection with vehicle (0.5% methyl cellulose) or with a fine suspension of Bay 11-7082 (5 or 20 mg/kg) in vehicle. Animals were examined every 2 or 3 days and weighed once per week. Establishment of PEL was defined as a weight gain of greater than 10% total body mass in a 1-week period. Animals were humanely killed by CO2 inhalation immediately after the development of PEL; hence, disease-free survival corresponded to the time to death. For assessment of disease-free survival, Kaplan-Meier analysis was performed and P values were determined by 2-tailed analysis with the log-rank tests.
Immunohistochemistry and in situ hybridization
Immunohistochemical staining was performed on formalin-fixed, paraffin-embedded tissue sections of tumor or cell blocks prepared from effusion cells from mice that had received injections of BC-3 and EBV-IBL cells. Staining was performed using a TechMate 500 automated immunostainer (Ventana Medical Systems, Tucson, AZ) and ChemMate ABC peroxidase secondary detection kit (Ventana Medical Systems). The following antibodies were used to demonstrate latent and lytic KSHV antigens: rat monoclonal antibody to latent nuclear antigen (LANA; ORF-73) of KSHV, mouse monoclonal antibody to early-latent protein ORF59, mouse monoclonal antibody to virion envelope glycoprotein ORF-K8.1A (Advanced Biotechnologies, Columbia, MD), and rabbit polyclonal antibody to viral IL-6 rabbit polyclonal anti-vIL-6 made in our laboratory against the PDVTPDVHDK peptide, as described.22 The following antibodies were used to demonstrate latent and lytic EBV antigens: mouse monoclonal antibody to latent membrane protein-1 (LMP-1) clones 1 to 4, mouse monoclonal antibody to BamH1 Z fragment (ZEBRA) protein, and mouse monoclonal antibody to EBNA 2 (DAKO Cytomation, Carpinteria, CA). To demonstrate KSHVLANA, biotinylated polyclonal goat anti-rat immunoglobulin (multiple adsorption) (BD Biosciences Pharmingen) was added to the secondary antibodies. Before staining, paraffin tissue sections were pretreated for 40 minutes in a water bath at 95°C using antigen retrieval (CitaPlus; BioGenex, San Ramon, CA) (KSHV-LANA); in a pressure cooker using 10 mM citrate buffer, pH 6.0 (EBNA2 and ZEBRA); in a microwave for 10 minutes using 10 mM citrate buffer, pH 6.0 (KSHV ORF 59); or with 0.2% pepsin in 50 mM Tris HCl, pH 2.0, at 37°C for 20 minutes (EBV-LMP and KSHV-ORF59). Peroxidase reaction was developed using liquid DAB substrate chromagen included in the detection kit. Negative controls with rat, mouse, or rabbit IgG were run in parallel. Sections were counterstained with hematoxylin and mounted in Permount (Fisher Scientific, Pittsburgh, PA). In situ hybridization for EBV-encoded RNA (EBER) transcripts was performed using Epstein-Barr Probe ISH kit (Novocastra, Newcastle-upon-Tyne, United Kingdom) according to the manufacturer's protocol. Images were visualized via conventional light microscopy, using a Nikon Microphot SA microscope with 10 ×/0.30 numeric aperture (NA) and 40 ×/0.70 NA Nikon objective lenses (Nikon, Melville, NY). Images were acquired using an Insight QE 4.2 digital camera from SPOT Diagnostic Instruments (Sterling Heights, MI) and SPOT software version 4.5. Subsequent image processing was carried out using Adobe Photoshop version 7.0 (Adobe Systems, San Jose, CA).
In vivo imaging
Ten million BC-3 cells were injected intraperitoneally on day 0 of the experiment. Mice (n = 12) subsequently received intraperitoneal injections 3, 5, and 7 days after BC-3 injection with vehicle (0.5% methyl cellulose, 5 mice) or with a fine suspension of Bay 11-7082 (5 mg/kg, 5 mice; 20 mg/kg, 2 mice) in vehicle. Mice were humanely killed by CO2 inhalation immediately after development of PEL, as determined by a weight gain of greater than 10% total body mass in a 1-week period. Animals were examined by in vivo imaging on days 3, 4, 6, and 10 and at approximately weekly intervals thereafter. d-luciferin firefly potassium salt was purchased from Xenogen (Alameda, CA). A 30-mg/mL stock in PBS was filtered through 0.22-μm filters before use. d-luciferin (50 μL; 75 mg/kg body weight) was injected retro-orbitally, and the mice were imaged after 3 minutes using the IVIS Imaging System (Xenogen). Data were acquired for 3 minutes. Gray-scale photographic images and bioluminescence color images were superimposed using The Living Image, version 2.20 software overlay (Xenogen). Data were analyzed with Igor Pro image analysis software (WaveMetrics, Lake Oswego, OR). A region of interest (ROI) was manually selected over signal intensity. The area of the ROI was kept constant. Data are presented as average radiance (photons/s-1·cm-1sr-1 [steradian]) within the ROI.
Results
Inhibition of NF-κB induces apoptosis in KSHV- and EBV-infected lymphoma cells
Previous studies in our laboratory have shown that low-dose treatment with the inhibitor Bay 11-7082 selectively inhibits constitutive NF-κB in KSHV-infected PEL cells. This agent triggers apoptosis in KSHV-infected lymphoma cells but not in uninfected B lymphoma cells.17 A similar effect was reported for lymphoblastoid cell lines,18 and we confirmed this effect in an EBV-infected cell line derived from an HIV-positive patient with an immunoblastic lymphoma, EBV-IBL. These cells were treated with Bay 11 for 24 hours and were evaluated for the selective inhibition of NF-κB/DNA binding and for the induction of apoptosis. Figure 1A demonstrates that treatment of EBV-IBL cells resulted in the inhibition of NF-κB/DNA binding, similar to that observed for KSHV-infected BC-3 cells. In both cell lines, the inhibition of NF-κB/DNA binding, visible after 1 hour of treatment, was maintained throughout the 24-hour assay. In contrast, no change was observed in the binding of nuclear proteins to control octamer consensus DNA sequences. In EBV-IBL and BC-3 cells, significant induction of apoptosis occurred on treatment with Bay 11 (Figure 1B). Results from apoptosis measured by Annexin V binding were supplemented with cell viability measurements by trypan blue exclusion. Analysis of other KSHV- and EBV-infected cell lines showed similar findings on treatment with Bay 11, demonstrating that selective inhibition of constitutive NF-κB activity in KSHV- and EBV-infected lymphoma cells resulted in the apoptosis of these cells within 24 hours.
Figure 1.

Induction of mitochondrial-mediated apoptosis in EBV- and KSHV-infected lymphoma cells after selective inhibition of NF-κB. (A) EBV-IBL and BC-3 cells were treated with 5 μM Bay 11 and assessed for NF-κB and octamer/DNA binding by EMSA. Nuclear proteins were extracted from cells 1 hour and 24 hours after treatment, and EMSA was performed using NF-κB or octamer-specific radiolabeled oligonucleotide probes. Cell lines demonstrated the inhibition of NF-κB throughout the 24-hour assay but no concomitant inhibition of octamer/DNA binding. (B) EBV-IBL and BC-3 cells were treated with Bay 11 and assessed for apoptosis after 24-hour treatment. For analysis, cells were labeled with FITC-conjugated Annexin-V at room temperature and were analyzed by flow cytometry. Reported results are the percentages of apoptosis cells from 4 independent experiments for untreated ( ) and Bay 11-treated (▪) cells. Error bars represent SD. (C) EBV-IBL and BC-3 cells were cultured at 7.5 × 105 cells/mL and treated with Bay 11-7082 for 24 hours. After 1, 3, 6, and 12 hours, whole cell extracts were generated and evaluated for the expression of caspases 9, 3, and 8 and PARP by Western blot analyses. Antibodies for caspase 9 and 3 recognized the full-length and cleaved caspase isoforms, which are indicated. Antibody for caspase 8 detected only the full-length or procaspase isoform, so activation was assessed by attenuated detection of the full-length protein. After initial probe, blots were stripped and reprobed with an actin control. Results shown are for BC-3 but are representative of both cell types in terms of caspase activation pattern and timing.
) and Bay 11-treated (▪) cells. Error bars represent SD. (C) EBV-IBL and BC-3 cells were cultured at 7.5 × 105 cells/mL and treated with Bay 11-7082 for 24 hours. After 1, 3, 6, and 12 hours, whole cell extracts were generated and evaluated for the expression of caspases 9, 3, and 8 and PARP by Western blot analyses. Antibodies for caspase 9 and 3 recognized the full-length and cleaved caspase isoforms, which are indicated. Antibody for caspase 8 detected only the full-length or procaspase isoform, so activation was assessed by attenuated detection of the full-length protein. After initial probe, blots were stripped and reprobed with an actin control. Results shown are for BC-3 but are representative of both cell types in terms of caspase activation pattern and timing.
Inhibition of NF-κB induces mitochondrially mediated apoptosis in KSHV- and EBV-infected lymphoma cells
To characterize the mechanism of apoptosis in KSHV- and EBV-infected cells after the inhibition of NF-κB, caspase activation was assessed by Western blot analyses. Time-course evaluations were performed with proteins extracted from BC-3 and EBV-IBL cells 1, 3, 6, and 12 hours after Bay 11 treatment. For caspases 9 and 3, activation was defined by the presence of active caspase cleavage proteins, generated by cleavage of the inactive procaspase precursors into smaller active proteases. For BC-3 and EBV-IBL cells, the caspase activation pattern was identical, with caspase 9 activated maximally within 3 hours of Bay 11 treatment (Figure 1C). With caspase 3, accumulation of the cleavage products was observed starting at 3 hours and steadily increasing to the 12-hour time point. Activation of these caspases was concomitant with the onset of morphologic and structural changes, indicative of the execution phase of apoptosis and evaluated by PARP cleavage. Combined with the lack of significant caspase 8 activation before 12 hours and an absence of Bid cleavage (data not shown), this suggested a pattern of death characteristic of the mitochondrial-mediated apoptosis pathway.
Down-regulation of common prosurvival factors in KSHV- and EBV-infected lymphoma cells after NF-κB inhibition
Although a large number of NF-κB-regulated genes have been established in a variety of experimental systems, it is unknown which genes are controlled by this transcription factor in herpesvirus-associated lymphomas. Hence, microarray analyses were performed using Affymetrix gene chip array technology. Expression of more than 12 500 human genes was evaluated in BC-3 and EBV-IBL cells after Bay 11 treatment or control using UA95v2 arrays. Data analysis with Genespring software (Silicon Genetics) showed 2-fold or greater decreases on NF-κB inhibition in 232 and 162 genes in BC-3 and EBV-IBL cells, respectively. Gene clustering demonstrated that these decreases were consistent in each of 3 independent experiments performed per cell line. We further evaluated commonality of NF-κB-regulated genes among EBV- and KSHV-infected lymphoma cells because these are more likely to be essential for tumor cell survival. Overlapping of gene lists revealed only 25 genes common to BC-3 and EBV-IBL that changed by more than 2-fold after NF-κB inhibition (Figure 2). Among these were the antiapoptosis genes cIAP-2 and A20, the signaling gene IκBα, and the growth factor IL-6. Many of the genes affected by inhibition of NF-κB in the lymphoma cell lines, including cFLIP and cIAP, are the same as those previously identified on NF-κB inhibition in lymphoblastoid cell lines using a Lymphochip cDNA microarray,18 confirming that EBV-associated immunoblastic lymphoma cell lines are similar in their NF-κB dependence and survival signals as in vitro-established lymphoblastoid cells.
Figure 2.

Inhibition of NF-κB-regulated antiapoptosis gene expression in EBV- and KSHV-infected lymphoma cells. Total RNA (5 μg) was isolated from BC-3 and EBV-IBL cells treated with and without 5 μM Bay 11-7082 for 6 hours and subsequently was labeled and hybridized to Affymetrix UA95v2 probe arrays. Data analysis was performed with Genespring software based on results of 3 independent experiments. Genes with greater than 2-fold change in both cell types are shown. In both cell types, treatment with Bay 11 resulted in more than 2-fold down-regulation of IL-6, cIAP-2, A20, and IκBα.
To verify down-regulation at the protein level and to further evaluate the expression of other potentially relevant proteins, Western blot analyses were performed. As in the apoptosis assays, time course evaluations were performed in treated and control BC-3 and EBV-IBL cells. Significant decreases in cIAP-1, cIAP-2, cFLIPL, IκBα, TRAF2, and IL-6 protein expression occurred within 3 hours of NF-κB inhibition, consistent with caspase activation and the onset of apoptosis (Figure 3A). The reported mechanism of action of Bay 11 is as an irreversible inhibitor of IκBα phosphorylation, leading to its stabilization and to the retention of NF-κB in the cytoplasm.23 Consistent with this, increased levels of IκBα protein were found 1 hour after treatment with Bay 11 (Figure 3A). However, by 3 hours, the levels of IκBα protein were decreased, most likely as a result of the transcriptional down-regulation seen in the microarray data (Figure 2) and consistent with a previously reported delayed negative feedback loop.24,25 Interestingly, the down-regulation of Bcl-2 and Bcl-XL protein was not observed in either BC-3 or EBV-IBL over 12 hours, though these have been suggested to be NF-κB dependent in other cell systems. Additional KSHV- and EBV-infected lymphoma cell lines were assessed for protein down-regulation after the inhibition of NF-κB by Bay 11. As shown in Figure 3B, the down-regulation of IκBα, cIAP-2, cFLIP, IL-6, and TRAF2 proteins also occurred in the cell lines BC-1, BCBL-1, BCKN-1, and LCL 9001 on Bay 11 treatment, suggesting that in KSHV- and EBV-infected lymphocytes, constitutive NF-κB activity regulates a specific set of common prosurvival genes critical for the survival of infected lymphoma cells.
Figure 3.

Down-regulation of NF-κB-regulated antiapoptosis, signaling, and growth factors is concomitant with the onset of apoptosis in EBV- and KSHV-infected lymphoma cells. (A) EBV-IBL and BC-3 whole cell extracts, generated as described in the legend to Figure 2, were evaluated by Western blot analyses for expression of the indicated proteins. After initial probe, blots were stripped and reprobed with an actin control. Results shown are for BC-3 but are representative of both cell types in terms of down-regulation and timing. (B) Evaluation of protein expression in KSHV-infected BC-1 and BCBL-1 cells and EBV-infected BCKN-1 and LCL-9001 cell lines. Protein expression was evaluated by Western blot analyses using whole cell extracts from cell lines cultured at 7.5 × 105 cells/mL and treated with Bay 11-7082 for 6 hours. After initial probe, blots were stripped and reprobed with actin control.
Inhibition of NF-κB delays the development of EBV-associated lymphoproliferation in vivo
Using an established murine model for EBV-associated immunodeficiency-associated lymphoproliferative disorders, we gave NOD/SCID mice subcutaneous injections with the lymphoblastoid cell line LCL 9001 and evaluated these mice for the development of solid tumors.26 After injecting the tumor cells, we gave the mice intraperitoneal injections with vehicle alone or with 20 mg/kg Bay 11 on days 1, 3, and 5 and then once weekly and measured the tumors each week. After 4 weeks, we humanely killed all animals with EBV-associated tumors and excised and weighed the tumors. The mean tumor area was greater in untreated mice than in Bay 11-treated mice at all time points (Figure 4A). Although the mean tumor size increased over time in both and control groups, tumor growth curves differed significantly (P < .035 by repeated-measures analysis of variance [RMANOVA]). Concordantly, the mean weight of excised tumors was greater in untreated mice (1.53 g) than in the mice that received Bay 11 (0.88 g; P < .027 by 2-sample t test).
Figure 4.
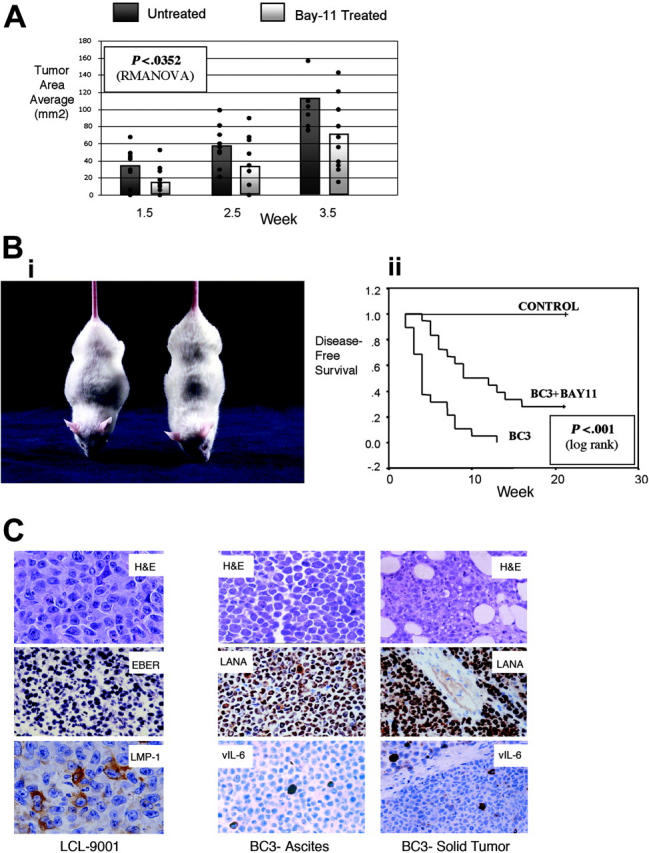
Treatment of NOD/SCID mice with Bay 11-7082 delays the development of EBV- and KSHV-associated lymphomas in vivo. (A) NOD/SCID mice (n = 10/group) were challenged with 40 × 106 LCL 9001 cells and subsequently received injections of vehicle or 20 mg/kg Bay 11 on days 1, 3, and 5 after tumor challenge and at weekly intervals thereafter. Tumors were measured on a weekly basis; tumor area for each mouse is shown by a dot, and average size is illustrated by the bar graph for each group. Differences evaluated by RMANOVA revealed significant differences in tumor size (P < .0352) between vehicle and Bay 11-treated mice averaged over all time points. (B) (i) Murine model of primary effusion lymphoma. The mouse on the left received injection of BC-3 cells and developed lymphomatous ascites, as evidenced by abdominal distention. The mouse on the right was a healthy control mouse. (ii) Disease-free survival curves. NOD/SCID mice (n = 19/group) were challenged with 10 × 106 BC-3 cells and then received injections of vehicle or Bay 11-7082 (5 or 20 mg/kg) on days 1, 3, and 5 after tumor injection. Disease-free survival evaluated by Kaplan-Meier and log rank test showed significant differences in tumor development in Bay 11 and vehicle-treated mice (P < .001 for 5 and 20 mg/kg groups). There was no major difference among the curves for the 5- and 20-mg/kg dose groups, so only the 5-mg/kg data are shown. The control group represents mice that did not receive injections of tumor cells. (C) Cells from ascites and solid tumors in BC-3- and LCL 9001-challenged mice were evaluated for morphology and viral antigen expression after tumor development. Cells were stained with hematoxylin and eosin (H&E); for KSHV, they were further evaluated with antibodies to LANA and vIL-6, and for EBV they were further evaluated with antibodies to LMP-1 and by in situ hybridization for EBER. Original magnification, 100 ×, except LCL H&E and LMP-1, for which original magnification was 400 ×.
Inhibition of NF-κB delays the development of KSHV-infected lymphomas in vivo
For evaluation of KSHV-associated PEL responses to NF-κB inhibition, we challenged male NOD/SCID mice with BC-3 cells injected intraperitoneally and then gave the mice intraperitoneal injections with 5 or 20 mg/kg Bay 11 or with vehicle alone on days 1, 3, and 5 after injection with tumor cells. For experimental purposes, we defined the establishment of PEL according to 2 criteria, the presence of ascites and greater than 10% increased body weight in a 1-week period. We killed the mice that developed PEL according to protocol and evaluated disease-free survival by the Kaplan-Meier method. Strikingly, all mice that received injections of BC-3 cells acquired PEL, with evident distention and ascites in the peritoneal cavity (Figure 4B). Some mice also acquired solid tumor near the site of injection. Mice that received injections of Bay 11 had delayed tumor development compared with mice injected with vehicle alone (P < .001; Kaplan-Meier analysis, with treatment arms compared by log-rank test). Approximately one third of the mice treated with 5 or 20 mg/kg Bay 11-7082 never acquired lymphoma. By contrast, each mouse (n = 19) challenged with BC-3 cells and untreated or treated with vehicle alone developed malignant ascites within the 3-month period (Figure 4B). There was no difference between mice treated with 5 mg/kg or 20 mg/kg Bay 11-7082, supporting our previous in vitro observations that the low dose is effective. The lowest lethal dose (50 mg/kg) was 3 injections of Bay 11-7082 given at 2-day intervals, reflecting a potentially wide therapeutic window for this drug.
Histologic and immunohistochemical characterization of KSHV- and EBV-associated lymphomas
We collected tumors and ascites and examined them by immunohistochemistry and in situ hybridization to confirm that the malignancies were of correct histology and contained viral RNA and proteins. In both tumor models, we found sheets of large tumor cells consistent with large-cell non-Hodgkin lymphoma (NHL) (Figure 4C), mixed with areas of necrosis in untreated and Bay 11-treated mice. As expected, all tumor cells from mice that received injections were positive for latent viral proteins (EBV EBER or KSHV LANA), and a minority of cells were positive for EBV LMP-1 and EBNA-2 or KSHV vIL-6. This is consistent with the latent nature of viral infection in these tumors. Finally, to determine whether treatment with Bay 11 induces lytic replication in vivo, we examined the tumor cells for the presence of KSHV vIL-6, ORF59, and K8.1 or EBV LMP-1, EBNA-2, and Zebra proteins. We found no differences in presence of these proteins between the Bay 11-7082-treated and untreated groups, supporting that NF-κB inhibition in vivo does not induce viral lytic replication.
In vivo imaging of mice with primary effusion lymphoma shows inhibition of NF-κB and tumor responses
To further document NF-κB inhibition in vivo and to develop a better method to evaluate tumor responses to treatment, we developed a traceable reporter PEL cell line, BC-3/NFκB-luc. This cell line expresses luciferase under the control of NF-κB and constitutively, with decreased activity on inhibition of NF-κB by treatment with Bay 11-7072 (Figure 5B). This cell line was injected intraperitoneally into groups of NOD-SCID mice that subsequently were given 3 injections on alternative days (days 3, 5, and 7 after tumor inoculation) of vehicle alone or Bay 11-7082. Imaging 3 days after tumor inoculation showed bioluminescence levels above background in all except 2 mice, indicating the establishment of tumors in most animals (Figure 5A). Bioluminescence was decreased in all the mice with visible tumors treated with Bay 11-7082 by 24 hours, indicating effective in vivo NF-κB inhibition by this drug. Although tumors continued to grow in the mice that received injections of vehicle alone, as reflected by increasing bioluminescence, all mice treated with Bay 11-7082, in both dose groups, showed tumor responses as seen in Figure 5 and by the lack of development of ascites. Figure 5A shows imaging of 4 mice treated with vehicle only and 5 mice treated with 3 injections of 5 mg/kg Bay 11-7082. Bioluminescence of all the mice was quantitated and plotted against time in Figure 5C, showing clear progression in the group treated with vehicle only and regression with a delay of recurrence in all the mice treated with Bay 11-7082.
Discussion
In this study we demonstrated that the inhibition of NF-κB is a viable therapeutic approach for the treatment of EBV- and KSHV-associated lymphomas. Results of functional and gene expression analyses provided insight into the mechanism of apoptosis and revealed which genes are actually controlled by NF-κB in these tumor cells.
Coordinated expression of NF-κB-regulated proteins protects KSHV- and EBV-infected lymphoma cells from apoptosis
Our results confirm that in KSHV- and EBV-infected lymphoma cells, apoptosis is initiated with the inhibition of constitutive NF-κB activity. In these cells, the onset of molecular changes—such as PARP cleavage, indicative of cellular breakdown—correlates with the activation of caspases 9 and 3. This activation occurs in the absence of overwhelming caspase 8 activation, suggesting that in these cells caspase-dependent apoptosis occurs through a mitochondrial-dependent pathway.
Here we demonstrate that the inhibition of NF-κB results in the down-regulation of specific antiapoptosis, signaling, and growth-related proteins, which we suggest collectively mediate similar survival pathways in KSHV- and EBV-infected lymphoma cells. Evaluation of antiapoptosis factors revealed high expression of cIAP-1, cIAP-2, and cFLIP genes that were down-regulated at the RNA and protein levels within 3 hours of NF-κB inhibition in KSHV- and EBV-infected cells. This correlated with the activation of caspases and the onset of PARP cleavage, suggesting that loss of these proteins contributes to the apoptosis of these cells. LMP-1 has been shown to inhibit p53-mediated apoptosis by activating NF-κB and in turn inducing A20.27 We could not confirm that the mechanism of apoptosis after the elimination of NF-κB is p53 mediated because Bay 11 treatment did not affect the expression of p53 or p53-induced proapoptosis genes, such as Apaf-1, Fas, or Bax. No significant changes were observed in Bcl-2 and Bcl-xL proteins when PARP cleavage was evident, indicating that these proteins were insufficient to protect the cells from apoptosis. These observations demonstrate that the inhibition of NF-κB does not result in the nonspecific down-regulation of all antiapoptosis proteins or the up-regulation of proapoptosis genes.
In addition to antiapoptosis proteins, the inhibition of NF-κB resulted in the down-regulation of signaling factors involved in the NF-κB activation pathway, including IκBα and TRAF2. By inducing the expression of signaling factors used by viral activating stimuli such as vFLIP and LMP-1, NF-κB ensures the constitutive expression of proteins necessary for its continuous activation. NF-κB inhibition in KSHV- and EBV-infected lymphoma cells also resulted in the consistent down-regulation of IL-6. This cytokine promotes B-cell growth and differentiation and is important for the survival of multiple myeloma, KSHV-infected PEL, and EBV-infected lymphoma cells,28-30 suggesting that the regulation of IL-6 by NF-κB is important for the survival of KSHV- and EBV-infected cells by promoting their proliferation in response to autocrine growth factors. However, the addition of exogenous IL-6 did not rescue BC-3 cells from apoptosis after treatment with Bay 11 (S.A.K., unpublished observation, May 2002), supporting our view that survival is based on the coordinated expression of multiple survival genes.
Together these findings provide a model by which NF-κB protects KSHV- and EBV-infected lymphoma cells from apoptosis. As illustrated in Figure 6, constitutive activation of NF-κB regulates expression of the antiapoptosis proteins cIAP-1, cIAP-2, and cFLIPL, which protect cells from stimuli activating death receptor and mitochondrial-dependent death pathways. Transient activation of apoptosis pathways initiated by normal stress stimuli is kept in check by the cIAP proteins, which bind to activated caspases 3 and 7 and prevent further induction of the apoptosis cascade. Further protection is afforded by growth factors that are up-regulated by NF-κB and that promote cell proliferation and survival. After the inhibition of NF-κB, cells are sensitized to apoptosis stimuli by down-regulation of these protective factors, and they undergo spontaneous apoptosis. Most likely, normal stress stimuli or ligation of death receptors initiates the activation of caspase 3, which is no longer inhibited by cIAP proteins and thus proceeds to activate subsequent apoptosis effector cascades. This may or may not be sufficient for complete activation of apoptosis in the cells but, coupled with the down-regulation of growth factors and signaling molecules necessary for the activation of NF-κB, ensures cell demise. Inhibition of growth factors such as IL-6 would abrogate cell proliferation signals and ultimately result in DNA damage. This, in turn, would initiate an apoptosis loop whereby the mitochondrial events initiated by other stress stimuli would be enhanced by the stress of DNA damage and would result in further activation of caspases 9 and 3.
Figure 6.

Common NF-κB-mediated prosurvival mechanisms protect KSHV- and EBV-infected lymphoma cells from apoptosis. In KSHV- and EBV-infected lymphoma cells, apoptosis events are prevented by antiapoptosis, cell signaling, and growth factors regulated by NF-κB (i; NF-κB target genes shown in red). When abrogation of constitutive NF-κB activity occurred (ii), KSHV- and EBV-infected lymphoma cells were sensitized to apoptosis stimuli, and spontaneous apoptosis proceeded through activation of mitochondrial-dependent apoptosis events. Activation of caspases most likely occurred by normal stress stimuli but was enhanced by DNA damage induced in the cells as a result of growth factor deprivation.
Establishment of viral lymphoma in vivo can be prevented by pharmacologic inhibition of NF-κB
In these studies, the injection of KSHV-infected PEL cells into NOD/SCID mice induced effusion lymphoma, comprised almost entirely of latently infected lymphoma cells and reminiscent of that seen in humans.31,32 Bay 11-7082 treatment prevented or delayed the establishment of PEL. In mice that received injections of EBV-infected lymphoblastoid cells, Bay 11-7082 also significantly reduced the rate of tumor growth over time. Notably, treatment with Bay 11-7082 did not result in the induction of EBV or KSHV viral lytic replication, which might have been a potential complication of NF-κB inhibition as a therapeutic approach. We also developed a new NF-κB-dependent reporter PEL cell line and showed that Bay 11-7082 inhibited NF-κB in vivo and clearly resulted in tumor responses.
Our data provide proof of concept that a selective NF-κB inhibitor can prevent, delay the appearance of, reduce the tumor burden of, or slow the growth of EBV- and KSHV-associated lymphoma in vivo. Although current clinical studies and treatment of humans with broad proteosomal inhibitors are already under way for a number of lymphoid tumors, the Bay 11-7082 compound is potentially advantageous because it is more specific23 and effective17 in its inhibition of constitutively active NF-κB, at least for preclinical and proof-of-principle studies. Clinical trials may be appropriate to weigh the relative benefits of broad-acting proteosomal inhibitors that achieve NF-κB down-regulation among their effects with more selective NF-κB-targeted therapies, such as Bay 11-7082 or the newer IKK inhibitors. Using this sort of therapeutic modality in addition to conventional chemotherapy may further enhance the therapeutic efficacy of either modality alone. Inhibitors that specifically target LMP-1 and vFLIP, the viral genes responsible for initiating signals leading to NF-κB activation, would represent an ideal mode of therapy for malignancies associated with EBV or KSHV. We have developed the first traceable system to study signaling and lymphomagenesis in vivo, which should provide a useful method to test new approaches for the treatment of viral lymphomas.
Acknowledgments
We thank Yi Fang Liu for her excellent assistance with immunohistochemistry.
Prepublished online as Blood First Edition Paper, December 27, 2005; DOI 10.1182/blood-2005-07-2730.
Supported by National Institutes of Health grant R01-CA68939 and by Leukemia and Lymphoma Society Translational Research grant 6183-02.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Mesri EA, Cesarman E, Arvanitakis L, et al. Human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J Exp Med. 1996;183: 2385-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyashita EM, Yang B, Lam KMC, Crawford DH, Thorley-Lawson DA. A novel form of Epstein-Barr virus latency in normal B cells in vivo. Cell. 1995;80: 593-601. [DOI] [PubMed] [Google Scholar]
- 3.Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity. 1998;9: 395-404. [DOI] [PubMed] [Google Scholar]
- 4.Devergne O, Hatzivassiliou E, Izumi KM, et al. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-κB activation. Mol Cell Biol. 1996;16: 7098-7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaudhary PM, Jasmin A, Eby MT, Hood L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene. 1999;14: 5738-5746. [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. The human herpes virus 8 encoded viral FLICE inhibitory protein physically associates with and persistently activates the IκB kinase complex. J Biol Chem. 2002;277: 13745-13751. [DOI] [PubMed] [Google Scholar]
- 7.Field N, Low W, Daniels M, et al. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003;116: 3721-3728. [DOI] [PubMed] [Google Scholar]
- 8.Guasparri I, Wu H, Cesarman E. The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signaling. EMBO Rep. 2006;7: 114-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004;199: 993-1003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-kappa B RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185: 953-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kontgen F, Grumont RJ, Strasser A, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;15: 1965-1977. [DOI] [PubMed] [Google Scholar]
- 12.Weih F, Carrasco D, Durham SK, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80: 331-340. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z-G, Hu H, Goeddel D, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis, while NF-κB activation prevents cell death. Cell. 1996;87: 565-576. [DOI] [PubMed] [Google Scholar]
- 14.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3: 221-227. [DOI] [PubMed] [Google Scholar]
- 15.Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc Natl Acad Sci U S A. 2000;97: 6055-6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feuillard J, Schuhmacher M, Kohanna S, et al. Inducible loss of NF-κB activity is associated with apoptosis and Bcl-2 down-regulation in Epstein-Barr virus-transformed B lymphocytes. Blood. 2000;95: 2068-2075. [PubMed] [Google Scholar]
- 17.Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-κB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96: 2537-2542. [PubMed] [Google Scholar]
- 18.Cahir-McFarland ED, Carter K, Rosenwald A, et al. Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J Virol. 2004;78: 4108-4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cesarman E, Moore PS, Rao P, Inghirami G, Knowles DM, Chang Y. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995;86: 2708-2714. [PubMed] [Google Scholar]
- 20.Renne R, Zhong W, Herndier B, et al. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2: 342-346. [DOI] [PubMed] [Google Scholar]
- 21.Cesarman E. Inhibition of NF-κB in KSHV- and EBV-infected lymphomas: primary hybridization data. Available at: http://www.cornellpathology.com/cesarman/data.html. Accessed December 6, 2005.
- 22.Moore PS, Boschoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science. 1996;274: 1739-1744. [DOI] [PubMed] [Google Scholar]
- 23.Pierce JW, Schoenleber R, Jesmok G, et al. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272: 21096-21103. [DOI] [PubMed] [Google Scholar]
- 24.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259: 1912-1915. [DOI] [PubMed] [Google Scholar]
- 25.Nelson DE, Ihekwaba AE, Elliott M, et al. Oscillations in NF-κB signaling control the dynamics of gene expression. Science. 2004;306: 704-708. [DOI] [PubMed] [Google Scholar]
- 26.Rowe M, Young LS, Crocker J, Stokes H, Henderson S, Rickinson AB. Epstein-Barr virus associated lymphoproliferative disease in the SCID mouse model: implication for the pathogenesis of EBV-positive lymphoma in man. J Exp Med. 1991;173: 147-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fries KL, Miller WE, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J Virol. 1996;70: 8653-8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fassone L, Gaidano G, Ariatti C, et al. The role of cytokines in the pathogenesis and management of AIDS-related lymphomas. Leuk Lymphoma. 2000;38: 481-488. [DOI] [PubMed] [Google Scholar]
- 29.Jones KD, Aoki Y, Chang Y, Moore PS, Yarchoan R, Tosato G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi's sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood. 1999;94: 2871-2879. [PubMed] [Google Scholar]
- 30.Tanner JE, Alfieri C. Interactions involving cyclosporine A, interleukin-6, and Epstein-Barr virus lead to the promotion of B-cell lymphoproliferative disease. Leuk Lymphoma. 1996;21: 379-390. [DOI] [PubMed] [Google Scholar]
- 31.Said JW, Chien K, Takeuchi S, et al. Kaposi's sarcoma-associated herpesvirus (KSHV or HHV 8) in primary effusion lymphoma: ultrastructural demonstration of herpesvirus in lymphoma cells. Blood. 1996;87: 4937-4943. [PubMed] [Google Scholar]
- 32.Boshoff C, Gao SJ, Healy LE, et al. Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in NOD/SCID mice. Blood. 1998;91: 1671-1679. [PubMed] [Google Scholar]