

Abstract
Recombination is a powerful evolutionary force that merges historically distinct genotypes. But the extent of recombination within many organisms is unknown, and even determining its presence within a set of homologous sequences is a difficult question. Here we develop a new statistic, Φw, that can be used to test for recombination. We show through simulation that our test can discriminate effectively between the presence and absence of recombination, even in diverse situations such as exponential growth (star-like topologies) and patterns of substitution rate correlation. A number of other tests, Max χ2, NSS, a coalescent-based likelihood permutation test (from LDHat), and correlation of linkage disequilibrium (both r2 and |D′|) with distance, all tend to underestimate the presence of recombination under strong population growth. Moreover, both Max χ2 and NSS falsely infer the presence of recombination under a simple model of mutation rate correlation. Results on empirical data show that our test can be used to detect recombination between closely as well as distantly related samples, regardless of the suspected rate of recombination. The results suggest that Φw is one of the best approaches to distinguish recurrent mutation from recombination in a wide variety of circumstances.
RECOMBINATION is a fundamental biological process that can, for example, increase viral or bacterial pathogenicity by diffusing genetic material throughout populations (Awadalla 2003). The biological mechanisms of recombination differ across organisms, but in broad terms recombination results in the creation of mosaic sequences where the evolutionary history at each site may be different. Violating this tree-like assumption of evolution can lead to serious consequences when performing phylogenetic analyses for a set of sequences. Indeed, as the evolution of the sequences cannot be described by a single tree, this can lead to overestimation or underestimation of branch lengths among other problems (Schierup and Hein 2000a,b; Posada 2001; Posada and Crandall 2002). Thus, an important question for a given set of aligned sequences is to determine whether or not recombination is likely to have occurred.
The ability of a large number of general methods to detect recombination has recently been evaluated empirically and through simulation (Crandall and Templeton 1999; Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001; Posada 2002). These studies have established that methods such as Geneconv (Sawyer 1989), Max χ2 (Maynard Smith 1992), RDP (Martin and Rybicki 2000), Phypro (Weiller 1998), RecPars (Hein 1990, 1993), and neighbor similarity score (NSS) (Jakobsen and Easteal 1996) efficiently detect recombination in a wide range of circumstances (Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001; Posada 2002). These tests infer the presence of recombination either directly through sequence comparisons or indirectly through phylogenetic means. As no underlying assumptions are made concerning the origin of the sequences, these tests can be applied to detect recombination within any set of aligned homologous sequences. Indeed, these techniques can be used to detect recombination within either closely or distantly related genotypes (Posada 2002). Moreover, these methods can be termed general since no specific assumptions concerning sample history (beyond sequence homology) are made.
In contrast to general methods for inferring recombination, there are also population-specific methods for detecting recombination, where the samples consist of genotypes from closely related individuals. Within a single population, recombination can be tested for using nonparametric approaches such as permutation tests based on summary statistics like the correlation of linkage disequilibrium with distance (Miyashita and Langley 1988; Schaeffer and Miller 1993; Awadalla et al. 1999). Linkage disequilibrium is typically measured using the statistics r2 and |D′| (Lewontin 1964; Hill and Robertson 1968).
Recently, coalescent (Kingman 1982) methods have been developed that can specifically detect (Brown et al. 2001; McVean et al. 2002) or characterize the rate of recombination (Griffiths and Marjoram 1996; Hey and Wakeley 1997; Kuhner et al. 2000; Nielsen 2000; Wall 2000; Fearnhead and Donnelly 2001; Hudson 2001; McVean et al. 2002) for a set of samples within a single population. Recombination can be modeled under either a basic crossing-over model (Hudson 1983) or a more complex model of gene conversion (Wiuf and Hein 2000). Only a few methods (Kuhner et al. 2000; Fearnhead and Donnelly 2001; McVean et al. 2002) relax the infinite-sites model (Kimura 1969) under which a site can undergo at most a single mutation. Relaxing the infinite-sites model is important for many bacterial and viral data sets, since under the infinite-sites model, high levels of recurrent mutation can cause patterns consistent with recombination (McVean et al. 2002).
The basic coalescent operates under several assumptions that include constant population size, no selection, random mating, and no population structure (Hein et al. 2005). Whereas these assumptions can be relaxed using additional parameters such as a term for population growth (Slatkin and Hudson 1991), these additional parameters are presently not accounted for in current methods that characterize and detect recombination (Kuhner et al. 2000; Fearnhead and Donnelly 2001; McVean et al. 2002). Importantly, the influence of population structure and demographic history may adversely affect the ability of coalescent methods to correctly infer the rate of recombination (McVean et al. 2002; Haydon et al. 2004).
The myriad of methods available to detect, characterize, and find recombinant sequences is somewhat bewildering. Traditionally, general approaches have been used for recombination analysis between distantly related genotypes, whereas population genetic-based approaches have been used for recombination analysis between closely related genotypes. However, in many cases the line between the approaches is blurred, and both approaches have been used to infer the presence of recombination in bacteria, viral, and animal mitochondrial data sets (McVean et al. 2002; Posada 2002; Piganeau et al. 2004).
Often, one of the primary questions for any data analysis is to determine whether recombination is likely to be present within a set of sequences at all (Awadalla et al. 1999; Maynard Smith and Smith 2002; McVean et al. 2002; Posada 2002; Piganeau et al. 2004; Tsaousis et al. 2005). Indeed, there are still open questions with regard to the extent of recombination in animal mitochondrial DNA (Maynard Smith and Smith 2002; Piganeau et al. 2004; Tsaousis et al. 2005). Moreover, if the sequences are obtained from closely related, yet distinct, organisms or from many different populations, it is inappropriate to analyze the sequences in a framework that assumes a single population, such as linkage disequilibrium or coalescent approaches (Tsaousis et al. 2005). But determining whether recombination has occurred in such circumstances is an important question that cannot be easily answered in a parametric framework. A robust nonparametric test for recombination can help distinguish between the presence and absence of recombination in such cases.
Testing for recombination can statistically validate visual evidence of recombination obtained using, for instance, phylogenetic network approaches (e.g., Huson and Bryant 2006) or independently verify the presence of recombination if a positive estimate of the rate of recombination is inferred (e.g., McVean et al. 2002). Moreover, it is often difficult to distinguish between rate heterogeneity and recombination in many circumstances (Grassly and Holmes 1997; McGuire and Wright 2000) and thus regions that exhibit phylogenetic inconsistencies can be individually tested for recombination. Additionally, testing for recombination can be used as a prior probability for the presence of recombination when inferring the points at which infrequent recombination may have occurred (Minin et al. 2005). In this sense, testing for recombination can be used in conjunction with other methods.
Ideally, a single test could correctly determine whether recombination is present within any given set of aligned sequences, regardless of population history, demographic history, recombination rate, or mutation rate. Preferably, such a test would also minimize the production of false positives. Here we develop a new test that is powerful under many of these different situations and produces few false positives. Through simulation and empirical data analysis we characterize the performance of our test under various rates of recombination, rates of mutation, demographic histories, and sample sizes. We also show through simulation that a simple model of substitution rate autocorrelation (consistent with mutational “hot spots”) gives rise to a signal similar to recombination for two different general tests, Max χ2 and NSS, but not for our method.
METHODS
Tests for recombination based on the principle of compatibility have proved to be among the most powerful (Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001; Posada 2002). The traditional binary notion of compatibility (Le Quesne 1969) is well suited for sites with at most two alleles, but can be directly extended into a broader notion (Penny and Hendy 1986) that we term here as refined incompatibility. We then develop a new statistic to test for recombination, the Φw- (or pairwise homoplasy index, PHI) statistic that uses this notion of refined incompatibility.
Compatibility and incompatibility:
It is not obvious how to determine the genealogical history of a single site. As such, the pattern of mutation present at multiple sites must be used to infer the genealogy of the sample as a whole. One possibility is to use the observed patterns at pairs of sites, in particular the notion of compatibility (Le Quesne 1969) or the “four-gametes” test (Hudson and Kaplan 1985). Two sites i and j are compatible if and only if there is a genealogical history that can be inferred parsimoniously that does not involve any recurrent or convergent mutations (known as homoplasies as in Figure 1b). If the two sites are not compatible, they are termed incompatible. Under an infinite-sites model (Kimura 1969) of sequence evolution, the possibility of a homoplasy does not exist, and so incompatibility for a pair of sites implies that at least one recombination event must have occurred, as in Figure 1a. This can be used to estimate the minimum number of recombination events present in the sample as a whole (Hudson and Kaplan 1985; Song and Hein 1999; Myers and Griffiths 2003). Testing for compatibility can be accomplished by checking if all four combinations of {00, 01, 10, 11} are present among the sequences (Le Quesne 1969).
Figure 1.


The dual nature of incompatibility. Two possible histories for a pair of incompatible sites are shown: (a) two incompatible sites explained by a recombination event and (b) two incompatible sites explained by a convergent mutation. Mutations in the first site are indicated by open circles and mutations in the second site are indicated by solid circles. To explain the incompatibility between the pair of sites either a recombination event must be invoked or a homoplasy must have occurred in the history of one of the sites.
The traditional, binary notion of either compatibility or incompatibility treats a single homoplasy the same as many homoplasies. That is, although in some situations more than one homoplasy can be parsimoniously inferred for a pair of sites (Camin and Sokal 1965; Penny and Hendy 1986), this information is disregarded. Consider two sites i and j, with |χi| and |χj| representing the number of observed states (alleles) at each site. Let l(χi, χj) denote the minimum number of mutations required by any tree used to represent the genealogical history of both sites. Thus l(χi, χj) represents the maximum parsimony score for these two characters over all trees. Note that l(χi, χj) ≥ (|χi| − 1) + (|χj| − 1) as each state (except the ancestral state) must arise at least once in the tree. Define the refined incompatibility score of sites i and j as
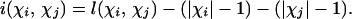 |
The refined incompatibility score relates to the traditional notion of compatibility in the following way: two sites are compatible if and only if i(χi, χj) = 0; if i(χi, χj) > 0 the two sites are incompatible. There are also two interpretations of this refined incompatibility score: in the absence of recombination, this score represents the minimum number of homoplasies that have occurred in the history of the samples for these two sites (Penny and Hendy 1986); in the absence of recurrent or convergent mutations, this score represents the minimum number of recombinations that have occurred between the two sites (T. Bruen and D. Bryant, unpublished data). This latter result depends on viewing recombinations as unrooted subtree-prune and regraft operations (see Hein et al. 2005). Importantly, this score can be calculated quickly [linear time in the number of sequences (Bruen and Bryant 2006)], which allows alignments with large numbers of sequences to be evaluated rapidly.
A parsimony informative site has at least two different alleles that are represented by at least two different sequences each (there must be at least four sequences at a site for the site to be parsimony informative) (Felsenstein 2004). A compatibility matrix (Sneath et al. 1975; Jakobsen and Easteal 1996) is traditionally used to represent compatibility between all pairs of parsimony informative sites. This matrix can also easily be extended into a refined incompatibility matrix by setting each entry (i, j) equal to the refined incompatibility score between any two sites i and j.
Sites that have the same history will tend to be more compatible than sites that have different histories (Sneath et al. 1975; Jakobsen and Easteal 1996; Drouin et al. 1999). One way to measure the extent of “clustering” in the matrix is to consider the proportion of neighboring cells in the matrix that are either compatible or incompatible. The resulting statistic is termed the NSS and has been used as a powerful test for recombination (Jakobsen and Easteal 1996; Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001; Posada 2002). However, simulations suggest that the NSS produces an excess of “false positives” in certain situations (see results and discussion) and so we have developed an alternative statistic.
Test statistic (Φw):
The degree of genealogical correlation between neighboring sites is negatively correlated with the rate of recombination (Hudson and Kaplan 1985). In the case of finite levels of recombination, the genealogical correlation of sites is partially reflected by a tendency of closely linked sites to have greater compatibility than distant sites (Hagenblad and Nordborg 2002; Innan and Nordborg 2002).
To measure the similarity between closely linked sites, we propose calculating a new statistic, the pairwise homoplasy index (PHI). The idea is to calculate the mean refined incompatibility score from nearby sites by using the first k off-diagonal rows of a refined incompatibility matrix (see Figure 2). Let w denote a fixed width (measured in bases) and choose k so that it is proportional to w. Specifically, let q denote the proportion of parsimony informative sites within the alignment and set k = wq. The statistic thus measures the mean refined incompatibility score of sites up to (approximately) w bases apart. We can now formally define the Φ or PHI statistic as
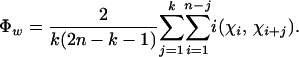 |
The term “pairwise homoplasy index” refers to the fact that the refined incompatibility score can be interpreted as the minimum number of convergent or recurrent mutations (homoplasies) necessarily present on any tree describing the history of any two sites i and j. The term k(2n − k − 1)/2 is a normalizing factor.
Figure 2.

The entries marked with a diamond in the refined incompatibility matrix represent the cells used to calculate the pairwise homoplasy index (or Φw). The cells with light shading contain the refined incompatibility score of informative site i with informative site i + 1. The cells with dark shading contain the refined incompatibility score of informative site i with informative site i + 2. In this example sites up to 2 informative bases apart are used to calculate Φw.
Clearly w should be somewhat less than the total number of sites but large enough that a number of comparisons are made. For all simulated and empirical analyses w was set to 100 and k chosen according to the above formula. Other choices of w were also considered (w = 50 and w = 150), but simulations (across different sequence lengths) suggested that w = 100 was slightly better than the other two choices (results not shown).
Significance:
Significance of the observed Φw-statistic can be obtained by using a permutation test. Under the null hypothesis of no recombination, the genealogical correlation of adjacent sites is invariant to permutations of the sites as all sites have the same history. But in the case of finite levels of recombination, the order of the sites is important, as distant sites will tend to have less genealogical correlation than adjacent sites. Let 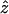 denote the observed value of the Φw-statistic on the original alignment and let Z0 denote the value of the Φw-statistic for a random permutation of the sites. Hence Z0 is distributed according to the null hypothesis of no recombination. To determine the significance of the observed value
denote the observed value of the Φw-statistic on the original alignment and let Z0 denote the value of the Φw-statistic for a random permutation of the sites. Hence Z0 is distributed according to the null hypothesis of no recombination. To determine the significance of the observed value 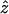 , a Monte Carlo P-value can be directly estimated by permuting the alignment many times and counting the proportion of times the Φw-statistic on a permuted alignment is less than or equal to
, a Monte Carlo P-value can be directly estimated by permuting the alignment many times and counting the proportion of times the Φw-statistic on a permuted alignment is less than or equal to 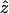 . However, computation of P-values based on permutations of the alignment is time consuming. One way to circumvent this problem is to determine the distribution of the test statistic under permutations of the alignment. The expectation (E0(Φw) = μ′) and variance (Var0(Φw) = σ2) of Φw can be calculated analytically (see appendix a for details). Moreover, initial simulations indicated that the distribution of Φw under permutations of the alignment is approximately normal (results not shown). Using these assumptions, the value of
. However, computation of P-values based on permutations of the alignment is time consuming. One way to circumvent this problem is to determine the distribution of the test statistic under permutations of the alignment. The expectation (E0(Φw) = μ′) and variance (Var0(Φw) = σ2) of Φw can be calculated analytically (see appendix a for details). Moreover, initial simulations indicated that the distribution of Φw under permutations of the alignment is approximately normal (results not shown). Using these assumptions, the value of 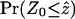 can be calculated as
can be calculated as
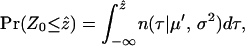 |
where n(τ | μ′, σ2) denotes a normal probability distribution function with mean μ′ and variance σ2. This alternative to the permutation test has the advantage that it can be obtained quickly and gives a more precise P-value under an assumption of normality.
The normality of the distribution of the test statistic can be explained by noting that for a large refined incompatibility matrix, calculating the Φw-statistic amounts to taking the mean of a small sample of values from the matrix. The simplest version of the central limit theorem then suggests that taking the mean of a small sample within a “large” matrix has a limiting normal distribution, if the terms are independent and identically distributed (Casella and Berger 2001). However, in this case the central limit theorem provides a guide rather than a formal equivalence.
For every data set examined (both simulated and empirical) the significance of the observed Φw-statistic was calculated using the permutation test directly as well as the normal alternative. The P-values obtained by using the permutation test are written as PP(Φw) whereas the P-values obtained by using the normal alternative are written as PN(Φw).
Simulation study:
We repeated many of the same simulations that had been performed in other studies (Posada and Crandall 2001; Wiuf et al. 2001) but expanded the parameter search space and considered the Φw-statistic as well as additional tests. The protocol followed was based on simulations from the neutral coalescent model (Kingman 1982) with recombination (Hudson 1983).
The coalescent model provides a natural foundation for simulation (Crandall and Templeton 1999; Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001). Simulations were almost all conducted using the program Treevolve (Grassly et al. 1999). For very high rates of recombination (ρ = 128), simulations were performed using the program Hudson (Schierup and Hein 2000a,b) since the program Treevolve did not run at such high rates of recombination. Mutations were added according to a Jukes–Cantor model (Jukes and Cantor 1969). Other methods of sequence evolution were also examined, including the addition of extreme rate heterogeneity (α = 0.1), which resulted in a moderate decrease in power for all methods (results not shown). For each parameter setting, 1000 replicate data sets were created, with each replicate consisting of an alignment of length 1000 (see appendix b for further details). Significance was set at the 0.05 level.
In addition to the Φw-statistic, four of the best nonparametric tests were computed for each parameter setting, namely the Max χ2-statistic (Maynard Smith 1992), the NSS (Jakobsen and Easteal 1996), and two measures of correlation of linkage disequilibrium (r2 and |D′|) with distance (Lewontin 1964; Hill and Robertson 1968; Miyashita and Langley 1988; Schaeffer and Miller 1993). Furthermore, results obtained from a coalescent-based likelihood permutation test (LPT) from LDHat (McVean et al. 2002) are reported as well. The Max χ2-statistic has been found to be the best general test for detecting recombination in a recent empirical study (Posada 2002), and the NSS statistic has been found to be very efficient as well (Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001; Posada 2002). Correlation of linkage disequilibrium with distance using r2 has been found to be the strongest nonparametric approach for detecting recombination within populations (McVean et al. 2002). Recently, the likelihood permutation test was introduced as a powerful alternative to methods based on linkage disequilibrium (McVean et al. 2002). For the Max χ2-statistic a fixed window size of the number of polymorphic sites divided by 1.5 was used following a previously described protocol (Posada and Crandall 2001; Posada 2002). For both measures of correlation of r2 and D′ with distance, only sites with two alleles segregating and minor allele frequencies of at least 0.1 were used, as this approach tends to maximize power (Weir and Hill 1986; McVean et al. 2002). For the likelihood permutation test, precomputed likelihood files were used on the basis of 101 grid points with a value of θ per site of either 0.001 or 0.1. For each replicate, if the expected mean sequence diversity was <10%, then a likelihood file with a θ per site value of 0.001 was used; otherwise a likelihood file with a θ per site value of 0.1 was used (under a constant-size population the expected mean sequence diversity of 10% corresponds to an expected value of θ per site of ∼0.12). The significance for each of the statistics was obtained using a permutation test. For the power determination, 1000 permutations were performed, whereas for the false positives, 200 permutations were performed.
Power:
To determine power in the presence of recombination, the recombination rate ρ (under population growth ρ†) varied among 0, 1, 2, 4, 8, 16, and 128; the expected nucleotide diversity p between any two sequences varied among 1, 5, 10, 15, and 25%; and the growth rate of the population β varied between 0 (constant-size populations) and 5000. The sample size m varied among 5, 10, 15, 25, and 50. For ρ = 128 simulations with β = 5000 were not performed since this option was not available with the program Hudson. More details explaining the protocol can be found in appendix b and elsewhere (Wiuf et al. 2001).
False positives:
Substitution rate heterogeneity across sites on a genealogy was modeled here using a Γ-distribution (Uzzell and Corbin 1971; Yang 1993). In this case, the substitution rate at each site i, Zi, is drawn from a Γ-distribution with shape parameter α and scale parameter 1/α (Yang 1993).
Autocorrelation among substitution rates was modeled assuming Markov dependence among rates (Yang 1995). To achieve this, two random variables Yi and Yi+1 were drawn from a bivariate normal distribution with correlation ρN and transformed into two marginally distributed gamma random variables Zi and Zi+1 with correlation ρG (Yang 1995). Using the bivariate normal distribution of Yi and Yi+1 (including correlation ρN), the probability distribution function of random variable Yi+1 was obtained conditional on the random variable Yi, allowing Markov-dependent substitution rates to be drawn. The substitution rates Zi and Zi+1 then represent draws from a bivariate Γ-distribution with correlation ρG. The value of ρG is positively correlated with the value ρN but not identical (Yang 1995).
Data sets were simulated using a modified version of Treevolve (Grassly et al. 1999) with a number of the sampling functions taken from PAML (Yang 1997). The correlation parameter ρN varied among 0 (no correlation), 0.3, 0.6, and 0.9; the expected nucleotide diversity p between any two sequences varied among 1, 5, 10, 15, and 25%; the value of α for the Γ-distribution varied among 0.1, 1.0, and ∞; and the growth rate of the population β varied between 0 (constant-size populations) and 5000. The sample size m varied among 5, 10, 15, 25, and 50.
Empirical data:
A number of population and species level data sets were examined. The presence of recombination in each of these data sets was debated, unknown, or suspected. The rate of recombination in these data sets ranged from rare to very frequent. In general, data sets with at least a few hundred sites were chosen.
Tests for recombination were performed using the Φw-statistic as well as the Max χ2-statistic (Maynard Smith 1992) and the NSS statistic (Jakobsen and Easteal 1996). As in the simulation studies, w was set to 100 for all analyses. One thousand permutations were performed to obtain significance. Additional results are reported for the population level data sets, using permutation tests based on r2 and |D′| (Lewontin 1964; Hill and Robertson 1968; Miyashita and Langley 1988; Schaeffer and Miller 1993) as well as a coalescent-based LPT with LDHat (McVean et al. 2002). Furthermore, an estimate of the rate of recombination was also obtained in LDHat using a model of crossing over rather than gene conversion. The maximum value of ρ was set to 100 and 100 grid points were used in LDHat. The value of Tajima's D-statistic is also reported, as it can be an indicator of population growth or selective pressure (Tajima 1989). Table 1 summarizes the data sets used. The data sets include sequences from bacteria, viruses, and fungi. Two of the data sets were from animal mitochondrial DNA (mtDNA).
TABLE 1.
Summary of empirical data sets
| Data set | Type | No. of sequences | No. of sites | Informative sites | Observed diversity (%)a | Tajima's Db | Reference |
|---|---|---|---|---|---|---|---|
| Candida albicans | Fungi | 45 | 2553 | 58 | 0.7 | 0.936 | Anderson et al. (2001) |
| Rana | Animal mtDNA | 8 | 1143 | 257 | 14.8 | — | Sumida et al. (2000) |
| Cowdria ruminantium | Bacteria | 14 | 870 | 186 | 10.5 | 0.384 | Jiggins (2002) |
| H. pylori | Bacteria | 33 | 472 | 53 | 3.8 | −0.531 | Suerbaum et al. (1998) |
| Boletales | Fungi | 31 | 639 | 265 | 17.1 | — | Kretzer and Bruns (1999) |
| Norovirus | Virus | 25 | 1617 | 103 | 2.2 | −1.482 | Rohayem et al. (2005) |
| Apodemus | Animal mtDNA | 10 | 1140 | 275 | 14.7 | — | Martin et al. (2000) |
| Nematode Wolbachia | Bacteria | 10 | 444 | 98 | 13.0 | 0.899 | Jiggins (2002) |
Mean proportion of sites that differ between any two sequences.
Calculated on sites with only two alleles segregating.
For the Boletales data set additional analysis was performed by first estimating a neighbor-joining tree (Saitou and Nei 1987) using PAUP* (Swofford 1998). Branch lengths for the tree, a transition/transversion ratio, codon frequencies, a value of α for the substitution rate heterogeneity (Yang 1993), as well as the degree of substitution rate autocorrelation (estimated using the autodiscrete gamma model) (Yang 1995), were then estimated using a codon model in PAML (Yang 1997). A parametric bootstrap of 1000 replicates was then performed under the estimated parameters using a modified version of PAML that allowed autocorrelated substitution rates. For each replicate, a test for recombination was performed using the Max χ2-statistic, the NSS statistic, and the Φw-statistic (with 1000 permutations). Significance was set at 0.05.
RESULTS AND DISCUSSION
Simulation studies:
Analytical calculation of P-values:
Table 2 shows the proportion of times that recombination was inferred using Φw, when the rate of recombination ρ was set to 0 and there was no population growth (β = 0). Since the significance level was set to 0.05, the Φw-test is too conservative when the mean sequence diversity is ∼1% or when there are few samples (e.g., m = 5). This is partly due to the fact that there are very few informative sites or incompatibilities produced in these situations (results not shown). Table 2 also indicates that when the sequence diversity and sample size are small, obtaining significance using the permutation test (PP(Φw)) is even more conservative than obtaining significance using the normal distribution (PN(Φw)). On the other hand, Figure 3 shows that both methods for obtaining significance give very similar answers for higher amounts of sequence diversity (at least 10%), with at least 15 samples. These results suggest that it is sufficient to obtain significance for Φw using the normal distribution. For all subsequent simulations, the results quickly obtained with the Φw-statistic using the normal distribution are reported.
TABLE 2.
Proportion of times recombination inferred using Φw when ρ = 0 and β = 0 (without mutation rate correlation or substitution rate heterogeneity)
| Diversity (%)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| m | 1 | 5 | 10 | 15 | 25 | |||||
| 5 | 0.4 | 0.4 | 1.6 | 0.9 | 3.6 | 1.7 | 4.2 | 2.4 | 5.1 | 3.7 |
| 10 | 0.1 | 0.0 | 3.1 | 1.5 | 4.6 | 3.5 | 3.9 | 3.2 | 4.7 | 4.0 |
| 15 | 0.2 | 0.0 | 5.5 | 3.8 | 5.7 | 4.7 | 5.4 | 4.5 | 4.0 | 3.8 |
| 25 | 0.3 | 0.2 | 4.6 | 2.9 | 4.8 | 4.3 | 4.5 | 3.8 | 4.5 | 4.1 |
| 50 | 0.8 | 0.1 | 5.9 | 4.5 | 4.1 | 3.8 | 5.7 | 5.6 | 5.7 | 5.3 |
The columns for each parameter pair represent PN(Φw) and PP(Φw), respectively.
Figure 3.

Comparison of P-values obtained using the permutation test (horizontal axis) to analytical P-values (vertical axis) when ρ = 0 and β = 0. Points with <15 samples and <10% sequence divergence are not shown (see Table 2).
Time:
The time to calculate Φw is much faster than other population genetic methods especially for moderate numbers of sites and sequences. For instance, several simulated alignments of 25 samples with 5000 sites with moderate sequence diversity (10%), corresponding to viral genomic samples, were analyzed on a Mac G4 desktop computer. The time taken to analyze each alignment was ∼20 sec using Φw without the permutation test, 30 sec using Φw with the permutation test, 7 min with the linkage disequilibrium methods (using LDHat), and 8 hr using the likelihood permutation test of LDHat (using a precomputed likelihood file). For longer alignments, however, the permutation test becomes impractical even for Φw and in these cases analytical P-values are the only way to practically test for recombination. It is worth noting that since the power to detect recombination increases as a function of sequence length (Wiuf et al. 2001), this constitutes an important advantage for the Φw-test, since faint recombinant signals may be detectable using only very long sequences.
Power:
Figure 4 shows the power to detect recombination for Φw, Max χ2, NSS, the LPT in LDHat, and two measures of correlation of linkage disequilibrium with distance (r2 and |D′|), when the rate of recombination ρ is greater than zero, for two different sample sizes (m = 10 and m = 50). Two principal types of genealogies were created: with and without population growth. If there is population growth, the genealogies created will be more star-like with long branches at the leaves (Griffiths and Tavaré 1998; Wiuf et al. 2001). If there is no population growth, there are short branches at the tip but long branches at the root. When genealogies are more star-like, recurrent mutations will tend to mask the initial recombination, and the recombination events are best considered to be “ancestral.”
Figure 4.


Power to detect recombination for (a) m = 10 and (b) m = 50 samples for six different methods with (a and b, bottom rows) and without (a and b, top rows) population growth. The horizontal axis varies the rate of recombination whereas the vertical axis varies the amount of sequence diversity. Each cell represents the outcome of 1000 replicates with cells with lighter shading indicating increased power. The value ρ† refers to the value of ρ used to give the same expected number of recombinations under population growth.
The top rows of Figure 4, a and b, show that without population growth (β = 0), all six methods performed similarly, although overall Φw is the most powerful method with a large number of samples. Without population growth, the power to detect recombination of all six methods generally increases as a function of both sequence diversity and the rate of recombination, similar to earlier observations (Posada and Crandall 2001; Wiuf et al. 2001). A notable exception is the LPT for which there is a slight decline in power when the mean sequence diversity reaches 10%. At this point, a likelihood file with a value of θ per site of 0.1 was used rather than a likelihood file with a value of θ per site of 0.001. However, when the sequence diversity reaches 10%, the expected value of θ per site is ∼0.12, suggesting that a value of θ per site of 0.1 is a better choice. Nonetheless, more power may be obtained by using a gross underestimate of θ, although previous work has demonstrated a relative insensitivity of the LPT to a specific estimate of θ (McVean et al. 2002).
The top rows of Figure 4, a and b, suggest that the Φw method performs similarly to the linkage disequilibrium approaches when there is very little sequence diversity (e.g., p = 1%), despite the fact that the test is too conservative in these circumstances (Table 2). For very little sequence diversity (i.e., p = 1%), the coalescent-based method LPT is the most powerful method in constant-size populations, but has about the same power as Φw for growing populations. However, the results suggest that all methods may underestimate the presence of recombination if few sequences are present with very little divergence, especially in an expanding population (or “star-like” genealogy).
By comparing the bottom rows of Figure 4, a and b, to the top rows of Figure 4, a and b, it is evident that detecting the presence of recombination under population growth (β = 5000) is a more difficult task than detecting the presence of recombination without population growth (β = 0). Of all six methods, the bottom rows of Figure 4, a and b, suggest that Φw is much better at detecting recombination under population growth than Max χ2, NSS, the coalescent-based LPT, or the linkage disequilibrium approaches. For the coalescent-based LPT, it is worth noting that population growth could be incorporated in the method in the future, possibly increasing power. The decline of linkage disequilibrium in expanding populations using r2 is consistent with previous observations (Slatkin 1994; McVean 2002), but the results suggest that the performance of the |D′| statistic is similar. The results for the Φw-test suggest that subsequent mutations do not “mask” the recombinant signal for this method. Interestingly, this is similar behavior to the RECPARS method (Hein 1993; Wiuf et al. 2001) and may be of particular importance when trying to determine ancestral recombination between diverged genotypes. The results also suggest that the Φw-statistic can be used to distinguish between star-like genealogies due to population growth and star-like genealogies due to recombination (Schierup and Hein 2000b).
A comparison of the top row of Figure 4a to the top row of Figure 4b reveals that an increase in sample size from m = 10 to m = 50 causes an increase in the ability of all six methods to infer recombination when there is no population growth (β = 0). For population growth (the bottom rows of Figure 4, a and b), the power to detect recombination for the NSS statistic for actually decreases sharply from m = 10 to m = 50. But for the other five tests, the power to detect recombination generally increases when moving from m = 10 to m = 50 even under population growth. These results expand upon some previous observations (Wiuf et al. 2001).
Under a neutral coalescent model with recombination, it is possible to use a likelihood-ratio test to determine whether the hypothesis of no recombination (ρ = 0) should be rejected at a given significance level (Kuhner et al. 2000; Brown et al. 2001). However, even when data are simulated according to the neutral coalescent with low levels of recombination, the hypothesis ρ = 0 is rejected only a limited proportion of the time (Brown et al. 2001). However, such a simulation represents an ideal situation, where the likelihood-ratio test is guaranteed to be the most powerful (Brown et al. 2001) and the model used to infer ρ is identical to the model used to generate samples. This suggests that it might be difficult for any test to correctly infer the presence of recombination for very low recombination rates. Additionally, a theoretical analysis shows that generating small sets of samples using a low rate of recombination produces only a limited number of incompatibilities (Wiuf et al. 2001). It is thus possible that full-likelihood approaches (Kuhner et al. 2000; Fearnhead and Donnelly 2001) or a phylogenetic network (Huson and Bryant 2006) approach could be particularly useful to determine whether there is any possibility of recombination when only a weak recombinant signal exists.
Table 3 demonstrates that Φw can detect recombination even under extremely high recombination rates (ρ = 128). Except for low sequence diversity (p = 1%), the presence of recombination is correctly inferred each time. But even for low sequence diversity, the presence of recombination can be inferred nearly every time by increasing the sample size from m = 10 to m = 50.
TABLE 3.
Power to detect recombination using Φw with a high rate of recombination ρ = 128
| No. of samples
|
||
|---|---|---|
| Diversity (%) | m = 10 (%) | m = 50 (%) |
| 1 | 68 | 99 |
| 5 | 100 | 100 |
| 10 | 100 | 100 |
| 15 | 100 | 100 |
| 25 | 100 | 100 |
It is worth noting that the Φw-statistic can also be calculated without the refined incompatibility score, but using only the traditional notion of compatibility. For cases without population growth (β = 0), the results are almost identical (results not shown). On the other hand, with population growth (β = 5000), there is an increase in power using the refined incompatibility score when the number of samples is large (e.g., m = 50) and there is some recurrent mutation. For a rate of recombination of ρ = 1, a sample size of 50, and exponential growth, the gains in power using the refined incompatibility score rather than the compatibility score were 2, 5, and 12% for mean pairwise sequence divergences of 10, 15, and 25%, respectively. Similar results are obtained for ρ = 2 but not for higher rates of recombination (results not shown). This suggests that the refined incompatibility score is a useful extension to the traditional notion of compatibility especially for large sample sizes with sites that experience recurrent mutations.
For no population growth, the Φw-test and the linkage disequilibrium approaches perform similarly, although Φw is more powerful for a large number of samples. However, Φw is applicable even if the samples are from different species or different populations, whereas the linkage disequilibrium and coalescent approaches are not (Tsaousis et al. 2005). Under population growth, however (β = 5000), only Φw continues to consistently infer the presence of recombination as the power of the other five methods suffers sharp declines. This suggests that, of all six methods, Φw has the greatest flexibility in detecting recombination in the different circumstances studied.
False positives:
Of particular concern for any test for recombination is the effect of confounding processes such as substitution rate heterogeneity and autocorrelated substitution rates. Autocorrelation of substitution rates implies that the rate of substitution of one site is not independent of the rate of substitution of a neighboring site and can create “mutational hot spots” within a sequence. This can potentially create the same patterns as recombination.
Figure 5 shows the proportion of false positives for Max χ2 and NSS when there is no recombination (ρ = 0) but “mosaic” sequences are artificially induced by using a range of autocorrelated substitution rates. Figure 5 shows that both Max χ2 and NSS falsely infer the presence of recombination >50% of the time in certain cases. The results for the linkage disequilibrium, likelihood permutation test, and Φw are omitted from Figure 5 since these methods did not falsely infer recombination >7% of the time, although Table 4 shows this information for Φw. Table 4 shows that the Φw-statistic did not infer recombination >6% of the time when recombination was falsely inferred >50% of the time using both Max χ2 and NSS. Although the global model of substitution rate autocorrelation employed by this study is quite simple since it ignores codon positions and substitution rate correlation within local patterns of substitution (McVean 2001), it nonetheless provides a guide to the effect of autocorrelated substitution rates.
Figure 5.



Percentage of false positives for (a) m = 10 samples (with β = 5000), (b) m = 50 samples (with β = 0), and (c) m = 50 samples (with β = 5000), for Max χ2 and NSS, with extreme rate heterogeneity (top row) and moderate rate heterogeneity (bottom row). The horizontal axis varies the substitution rate correlation whereas the vertical axis varies the amount of sequence diversity. Each cell represents the outcome of 1000 replicates with cells with lighter shading indicating a higher percentage of false positives. The results for Φw, r2, and |D′| are omitted since these approaches did not falsely infer recombination >7% of the time for any of the conditions, but Table 4 shows a number of these results for Φw.
TABLE 4.
Proportion of times recombination is falsely inferred using Φw with substitution rate heterogeneity α = 0.1, mutation rate correlation, and sample size m = 50
| Mutation rate correlation
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Diversity (%) | 0 | 0.3 | 0.6 | 0.9 | ||||
| 1 | 2.0 | 3.6 | 2.5 | 3.6 | 2.6 | 3.9 | 1.1 | 3.8 |
| 5 | 4.9 | 4.7 | 5.8 | 4.5 | 4.7 | 3.3 | 3.0 | 1.0 |
| 10 | 4.1 | 5.6 | 4.7 | 4.6 | 4.8 | 3.0 | 1.8 | 1.5 |
| 15 | 4.9 | 4.0 | 4.5 | 4.7 | 3.8 | 4.5 | 2.9 | 1.8 |
| 25 | 5.3 | 4.0 | 3.7 | 3.5 | 4.1 | 3.9 | 3.4 | 2.1 |
The columns for each parameter pair represent the outcomes for β = 0 and β = 5000, respectively.
The problem of false positives in NSS and Max χ2 is most severe for large sample sizes (e.g., m = 50), both under constant-size populations (Figure 5b) and under population growth (Figure 5c). Although the problem is in general greater for higher substitution heterogeneity (Figure 5, top rows) it is also a problem with lower substitution rate heterogeneity (Figure 5, bottom rows).
The level of false positives of both NSS and Max χ2 suggests caution in interpreting evidence for recombination, especially when autocorrelated rates are an issue. For instance, inferring the presence of recombination in mitochondrial DNA should be done cautiously as substitution rate correlation is known (Yang 1995; Nielsen 1997).
The results using Φw contrast strongly with the results using the NSS (which is also compatibility based). This is likely due to the difference in the statistics themselves. The Φw-statistic uses compatibility between closely linked sites directly whereas the NSS statistic measures clustering within a compatibility matrix. As the clustering can be caused by substitution rate correlation, and not only by recombination, this might explain the difference between the two statistics. For Max χ2 the problem is possibly due to pairs of sequences that differ greatly on one side of a site (due to high mutation) but share a great degree of similarity on the other side of a site (due to low mutation). Local “bursts” of mutation (McVean 2001) likely exacerbate the problem, especially for linkage disequilibrium approaches that are based on allele frequencies at different sites.
Empirical data:
The general information concerning the empirical data sets is summarized in Table 1. Tables 5 and 6 show the results of tests for recombination on all the empirical data sets. In addition to the results obtained using the Φw-statistic, results using Max χ2 (Maynard Smith 1992), NSS (Jakobsen and Easteal 1996), correlation of r2 and |D′| with distance (Lewontin 1964; Hill and Robertson 1968), and a LPT (McVean et al. 2002) are shown. The estimates of ρ for the population level data sets were obtained using LDHat (McVean et al. 2002). Tests for recombination within populations (i.e., r2, |D′|, and LPT) were not applied to data sets that contained individuals from different species.
TABLE 5.
Analysis of suspected recombinant data sets
| Data set | ρa | Φwbc | χ2 | NSS | r2ad | |D′|ad | LPTade |
|---|---|---|---|---|---|---|---|
| Candida | 16 | 2.4 × 10−15* (0.000*) | 0.000* | 0.000* | 0.000* (0.000*) | 0.122 (0.001) | 0.000* (0.000*) |
| Rana | — | 5.5 × 10−31* (0.000*) | 0.000* | 0.000* | — | — | |
| Cowdria | 17 | 3.8 × 10−5* (0.000*) | 0.041* | 0.001* | 0.167 (0.039*) | 0.043* (0.029*) | 0.000* (0.001*) |
| H. pylori | ≥100 | 9.3 × 10−3* (0.004*) | 0.158 | 0.330 | 0.125 (0.000*) | 0.536 (0.003*) | 0.000* (0.000*) |
P < 0.05.
Calculated on sites with only two alleles segregating with LDHat.
Each pair shows P-values calculated analytically and using a permutation test, respectively.
w was set to 100 for all tests.
Terms in parentheses show results on sites with minor allele frequencies >0.1.
Denotes the value of a likelihood permutation test calculated in LDHat.
TABLE 6.
Analysis of possibly recombinant data sets
| Data set | ρa | Φwbc | χ2 | NSS | r2ad | |D′|ad | LPTade |
|---|---|---|---|---|---|---|---|
| Norovirus | 23 (21) | 0.002* (0.003*) | 0.025* | 0.237 | 0.029* (0.574) | 0.868 (0.340) | 0.022* (0.026*) |
| Apodemus | — | 0.135 (0.151) | 0.274 | 0.006* | — | — | — |
| Boletales | — | 0.934 (0.931) | 0.003* | 0.000* | — | — | — |
| Wolbachia | 0 (2) | 0.086 (0.103) | 0.566 | 0.108 | 0.049* (0.019*) | 0.286 (0.204) | 0.709 (0.090) |
P < 0.05.
Calculated on sites with only two alleles segregating.
Each pair shows P-values calculated analytically and using a permutation test, respectively.
w was set to 100 for all tests.
Terms in parentheses show results on sites with minor allele frequencies >0.1.
Denotes the value of a likelihood permutation test calculated in LDHat.
Recombinant examples:
Table 5 shows that the null hypothesis of no recombination is rejected by all tests for most of the suspected recombinant data sets, including the Candida example that had very little sequence diversity (0.7%). Whereas a lack of sequence diversity in the simulations made recombination harder to detect, this may be partially overcome by using longer alignments, such as that for the Candida example, which had 2553 sites. Interestingly, the null hypothesis of no recombination was not universally rejected for two of the bacterial data sets: Cowdria and Helicobacter pylori. For these two bacterial examples, evidence for recombination was found using the Φw-statistic as well as the coalescent-based likelihood permutation test. However, recombination was detected in the Cowdria example using the correlation of distance with r2 only after sites with minor alleles were removed. Moreover, in the H. pylori data set neither NSS nor Max χ2 found significant evidence for recombination. This could be due to the high suspected rate of recombination in the H. pylori example, which has conditions approaching linkage equilibrium (Suerbaum et al. 1998). The linkage disequilibrium methods seem to be highly sensitive to sites with low allele frequencies and consistent results are obtained only after the removal of these sites.
Possibly recombinant examples:
The results obtained from the data sets for which the status of recombination is debated are quite interesting (Table 6). For the Norovirus example, evidence of recombination is found using Φw, Max χ2, and the LPT. There is some evidence of recombination found with r2, but after sites with minor allele frequencies <0.1 are removed no further evidence is found by the linkage disequilibrium methods. Since the samples came from a number of different cities, it could be that evidence of recent recombination is weakened by removing these sites. However, the LPT finds evidence of recombination regardless of whether or not these sites are removed.
For the bacterial symbiont nematode Wolbachia, there is little prior reason to suspect recombination (Jiggins 2002). Nonetheless, evidence for recombination is found using correlation of r2 with distance and marginal evidence for recombination is found by using the likelihood permutation test when sites with minor alleles frequencies <0.1 are removed. The results obtained using the Φw-statistic also suggest that there is marginal evidence for recombination with Wolbachia. The possible presence of recombination in Wolbachia should be tested further using more data.
Recombination in the animal mitochondrial DNA of Apodemus was first proposed (Ladoukakis and Zouros 2001) and then disputed (Maynard Smith and Smith 2002). Tests for recombination using Φw and Max χ2 indicate that there is little evidence for recombination, although the NSS statistic does find evidence for recombination. The evidence for recombination within Apodemus using the Max χ2-test is even weaker here than in previous studies (Maynard Smith and Smith 2002), possibly due to the fact that this implementation of the Max χ2-test uses a “fixed window size.” Given the high level of false positives of NSS, the results suggest that evidence for recombination within Apodemus is lacking.
For the fungal Boletales, results using the Φw-statistic are quite distinct from the results obtained using both the NSS and the Max χ2-statistic. The Φw-based tests find no evidence for recombination whereas both other tests find strong evidence for recombination. Interestingly, although most other methods for detecting recombination find evidence for recombination within this data set, Geneconv (Sawyer 1989), another powerful sequence-based test for recombination, does not (Posada 2002).
One possibility for the Boletales data set is that the Φw-statistic is too conservative and produced a type II error (“false negative”). The Boletales data set is a saturated data set with a strong A + T bias (Kretzer and Bruns 1999). The strong A + T bias results in an estimated transition/transversion ratio of 0.4. Simulations show, however, that even under such conditions, there is reason to believe that recombination will still create distinct patterns of compatibility and incompatibility that should be detectable using the Φw-statistic (results not shown). Moreover, simulations indicate that the Φw-statistic appears to be more powerful than the NSS statistic (which is also compatibility based), suggesting that a type II error for the Φw-statistic, but not for the NSS statistic, is unlikely.
Another possibility for the Boletales example is that both Max χ2 and the NSS statistic are producing type I errors, which, according to the simulations, autocorrelated substitution rates might induce. To test this, a parametric bootstrap with 1000 replicates simulating codons (with no recombination) was performed using a substitution rate heterogeneity of 1.31 and global substitution rate correlation ρG = 0.35 as estimated from the data set. Figure 6 shows the distribution of estimated P-values obtained on the 1000 replicates using the Max χ2-statistic, NSS statistic, and the Φw-statistic. Recombination was inferred 5.7% of the time using the Φw-statistic, 8.5% of the time with the Max χ2-statistic, and 37.5% of the time using the NSS statistic. Since none of the replicates contained recombination, the P-values for each of the three methods should follow a uniform distribution. Figure 6 shows that the parametric bootstrap creates conditions similar to recombination for both Max χ2 and NSS [a one-sided Kolmogorov–Smirnov test (Massey 1951) rejects the uniform distribution at a significance level of 10−7 for both Max χ2 and NSS but fails to find any evidence to reject the uniform distribution for Φw]. Whereas the results for Max χ2 are less striking than those for NSS, the parametric bootstrap fails to account for local patterns of mutation (Hey 2000; McVean 2001; McVean et al. 2002), which are likely to exacerbate the observed bias. These results suggest that there is reason to doubt the validity of the inferences of Max χ2 and NSS concerning the presence of recombination in the Boletales data set.
Figure 6.

Distribution of P-values inferred by the Φw-statistic, the NSS statistic, and the Max χ2-statistic. The results are obtained on the basis of 1000 parametric bootstraps under conditions observed for the Boletales example. None of the replicates contained recombination but the substitution rate autocorrelation was set to ρN = 0.35 and substitution rate heterogeneity was set to α = 1.31.
Conclusion:
We have presented a simple, powerful test for detecting recombination that can be used regardless of sample history. The approach is very general (e.g., does not assume a single population) and aims to determine simply whether there is a recombinant signal present within the sequences. In contrast to two other general tests, Max χ2 and NSS, our test does not falsely infer the presence of recombination because of mutation rate correlation (which is present in some mitochondrial DNA). Interestingly, our approach performs very well even in the presence of population growth, in contrast to methods based on linkage disequilibrium (r2 and |D′|), a coalescent-based likelihood permutation test (from LDHat), Max χ2, and NSS. Our method can be used by itself, or to validate the visual presence of recombination from a phylogenetic network approach, or to independently verify the presence of recombination if a positive estimate of the rate of recombination is obtained. The approach may be particularly useful in distinguishing recurrent mutation from recombination when assumptions such as a single, randomly mating, and constant-size population are not met. The test can be used easily when many sequences and sites are present because of its computational efficiency and indeed is more powerful in such circumstances. A program implementing our test as well as both Max χ2 and NSS is available as a stand-alone program at the following address: http://www.mcb.mcgill.ca/∼trevor. The test is also implemented in SplitsTree 4.2, available at http://www.splitstree.org.
Acknowledgments
T.B. thanks Kirk and Rachel Bevan, Scott Bunnell, Daniel Huson, and Russell Steele, as well as the two anonymous referees for a number of helpful suggestions that greatly improved the manuscript. T.B. is supported by the National Science Engineering and Research Council (NSERC) (postgraduate scholarship B) and by Le Fonds québécois de la recherche sur la nature et les technologies (FQRNT grant 2003-NC-81840). D.B. is supported in part by NSERC (grant 238975-01). H.P. acknowledges Génome Québec.
APPENDIX A
The normal approximation to the permutation test requires calculation of the expectation and variance of the Φw-statistic under permutations of the alignment. This section contains derivations for both the mean and the variance and outlines how to compute both values efficiently. Again, assume that the proportion of informative sites is q and let w be a fixed width (in bases). Throughout this section, let k = wq.
Let M = (Mi,j) be a given n × n refined incompatibility matrix. Note that M is symmetric. Let I = {1, …, n} be an index set. Let σ be any permutation of the index set, and define a permutation of the matrix as σ(M) = (Mσ(i),σ(j)).
Define the sample space Ω by Ω = {σ(M): σ ∈ Sn}. Assume that every permutation σ is equally likely. Define an n × n random matrix 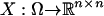 by X = σ(M). Note that X is symmetric, a fact that is used throughout without further mention.
by X = σ(M). Note that X is symmetric, a fact that is used throughout without further mention.
Define for all 1 ≤ i ≤ n: 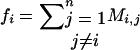 and
and 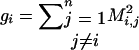 .
.
Also define 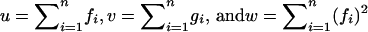 .
.
Lemma 1. Let X be a random matrix. Then for any arbitrary but distinct {i, j, k, l}
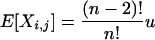 |
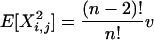 |
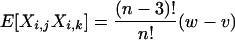 |
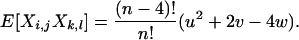 |
Proof. Note that a permutation σ of I can be viewed as mapping to  . Denote the value of σ(i) by σi. The total number of permutations is then n!. The number of permutations that have m distinct elements fixed in some mapping is (n − m)! (e.g., σ(a1) = b1, σ(a2) = b2,…, σ(am) = bm). Since every permutation is equally likely the probability of such a permutation is
. Denote the value of σ(i) by σi. The total number of permutations is then n!. The number of permutations that have m distinct elements fixed in some mapping is (n − m)! (e.g., σ(a1) = b1, σ(a2) = b2,…, σ(am) = bm). Since every permutation is equally likely the probability of such a permutation is
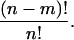 |
Note that every distinct pair (i, j), i ≠ j can be mapped to any distinct pair (a, b), a ≠ b, by some σ. Note also that Pr[Xi,j = Ma,b] = Pr[σa = i ∧ σb = j]. Finally, for notational convenience the summation 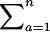 is written as
is written as 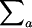 . Hence,
. Hence,
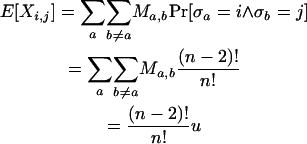 |
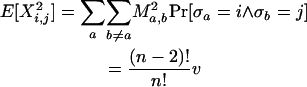 |
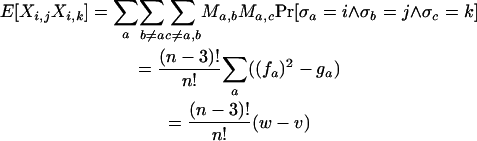 |
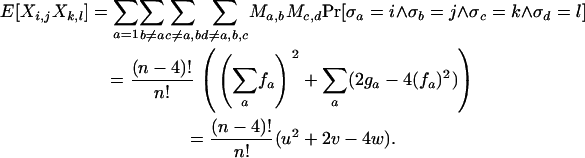 |
▪
Consider the statistic Φw defined on a random matrix X as
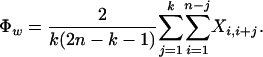 |
Define (for 1 ≤ a, b ≤ n)
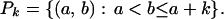 |
Note that
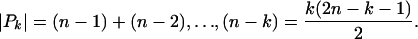 |
Then
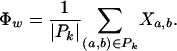 |
Theorem 1. The expectation and variance of Φw can be written as
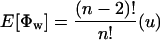 |
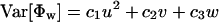 |
(for n ≥ 2k), where
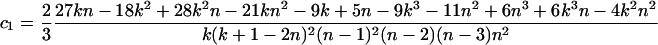 |
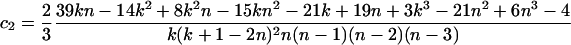 |
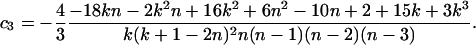 |
Moreover, both E[Φw] and Var[Φw] can be calculated in O(n2) time.
Proof. The expectation is straightforward:
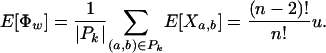 |
The variance is a little more involved,
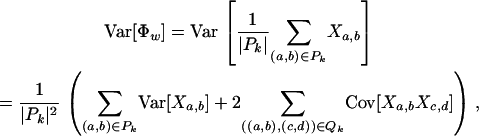 |
where
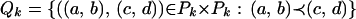 |
and ≺ denotes standard lexicographical ordering.
Note that Qk can be partitioned into two disjoint sets Qk,0 and Qk,1, where Qk,m = {((a, b), (c, d)) ∈ Qk : |{a, b} ∩ {c, d}| = m} [by definition Qk does not contain pairs of the type ((a, b), (a, b))]. One way to determine Qk,1 is to set up a recurrence.
Note that
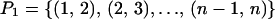 |
so that
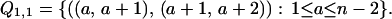 |
Hence |Q1,1| = (n − 2).
Next let ((a1, a2), (a3, a4)) ∈ Qk − Qk−1. Then at least one (a1, a2) = (a, a + k) or (a3, a4) = (a, a + k) must be true. Consider the four subcases:
Case 1: ((a, b), (a, a + k)), where 1 ≤ a ≤ n − k and a < b < a + k. There are precisely (n − k)(k − 1) terms of this type.
Case 2: ((a, a + k), (b, a + k)), where 1 ≤ a ≤ n − k and a < b < a + k. Again, there are precisely (n − k)(k − 1) terms of this type.
Case 3: ((a, a + k), (a + k, b)), where 1 ≤ a ≤ n − k and a + k < b ≤ min(a + 2k, n). For n ≥ 2k there are (k)((n − k) − k) + (k)(k − 1)/2 such terms.
Case 4: ((b, a), (a, a + k)), where 1 ≤ a ≤ n − k and max(1, a − k) ≤ b < a. For n ≥ 2k there are again (k)((n − k) − k) + (k)(k − 1)/2 such terms.
Cases 3 and 4 can coincide for n ≥ 2k when |a − b| = k. All other combinations of cases are disjoint. There are precisely (n − k) − k such coincidences. This gives the following recurrence for Qk,1:
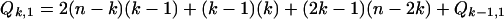 |
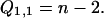 |
The recurrence can be solved by standard techniques resulting in
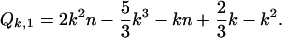 |
Note that 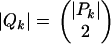 . Since Qk is the disjoint union of Qk,0 and Qk,1, then
. Since Qk is the disjoint union of Qk,0 and Qk,1, then
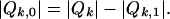 |
The variance of Φw can then be written as
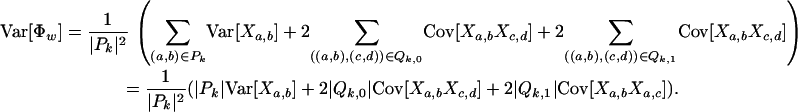 |
Noting that Cov[Xa,bXc,d] = E[Xa,bXc,d] − E[Xa,b]E[Xc,d] and Var[Xa,b] = E[Xa,b2] − E[Xa,b]2, the constants c1, c2, and c3 can be solved for using the relations from the previous lemma. Since the quantities u, v, and w can be computed in O(n2) time, so can the variance and expectation. ▪
APPENDIX B
The rate of recombination is here referred to as ρ = 4Nrt, where r is the per base recombination rate and t is the sequence length. Here N was set to 1000 (diploid population), t was set to 1000 as well, and r solved for accordingly.
For population growth ρ† was obtained so that the expected number of recombinations was equal under scenarios (i.e., Eβ=5000[R(m)] = Eβ=0[R(m)]), where R(m) is the number of recombinations for a sample of size m (Wiuf et al. 2001), and β = Nb, where b is the population growth rate per generation (Wiuf et al. 2001). The expected number of recombinations for β = 0 can be found by the following formula (Hudson and Kaplan 1985):
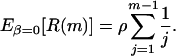 |
Table B1 shows the values used for ρ = 1 (when β = 0). For values of ρ > 1 (e.g., ρ = 2) one can simply double the values in the table.
TABLE B1.
Conversion of the rate of recombination ρ between β = 0 and β = 5000
| ρ
|
|||
|---|---|---|---|
| Sample size | E[R(m)] | β = 0 | β = 5000 |
| m = 5 | 2.08 | 1 | 550 |
| m = 10 | 2.83 | 1 | 400 |
| m = 15 | 3.25 | 1 | 325 |
| m = 25 | 3.78 | 1 | 250 |
| m = 50 | 4.48 | 1 | 175 |
Similarly, the rate of mutation is here referred to as θ = 4Nμt, where μ is the per base mutation rate and t is the sequence length. Under a Jukes–Cantor model if β = 0 then
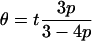 |
(Wiuf et al. 2001). This allows θ to be found for a fixed amount of sequence diversity p. For β = 5000 the appropriate value of θ was found by simulation. The values used are shown in Table B2.
TABLE B2.
Conversion of the rate of mutation θ between β = 0 and β = 5000
| θ
|
||
|---|---|---|
| Diversity (%) | β = 0 | β = 5000 |
| p = 1 | 10.1 | 6,600 |
| p = 5 | 53.6 | 33,000 |
| p = 10 | 115.4 | 68,000 |
| p = 15 | 187.5 | 106,000 |
| p = 25 | 375 | 193,600 |
References
- Anderson, J. B., C. Wickens, M. Khan, L. E. Cowen, N. Federspiel et al., 2001. Infrequent genetic exchange and recombination in the mitochondrial genome of Candida albicans. J. Bacteriol. 183(3): 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla, P., 2003. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4(1): 50–60. [DOI] [PubMed] [Google Scholar]
- Awadalla, P., A. Eyre-Walker and J. M. Smith, 1999. Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286(5449): 2524–2525. [DOI] [PubMed] [Google Scholar]
- Brown, C. J., E. C. Garner, A. Keith Dunker and P. Joyce, 2001. The power to detect recombination using the coalescent. Mol. Biol. Evol. 18(7): 1421–1424. [DOI] [PubMed] [Google Scholar]
- Bruen, T., and D. Bryant, 2006. A subdivision approach to maximum parsimony. Ann. Combinator. (in press).
- Camin, J. H., and R. R. Sokal, 1965. A method for deducing branching sequences in phylogeny. Evolution 19(3): 311–326. [Google Scholar]
- Casella, G., and R. L. Berger, 2001. Statistical Inference. Duxbury Press, Belmont, CA.
- Crandall, K. A., and A. R. Templeton, 1999. Statistical approaches to detecting recombination, pp. 153–176 in The Evolution of HIV, edited by K. A. Crandall. Johns Hopkins University Press, Baltimore.
- Drouin, G., F. Prat, M. Ell and G. D. Clarke, 1999. Detecting and characterizing gene conversions between multigene family members. Mol. Biol. Evol. 16(10): 1369–1390. [DOI] [PubMed] [Google Scholar]
- Fearnhead, P., and P. Donnelly, 2001. Estimating recombination rates from population genetic data. Genetics 159: 1299–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 2004. Inferring Phylogenies. Sinauer Associates, Sunderland, MA.
- Grassly, N. C., and E. C. Holmes, 1997. A likelihood method for the detection of selection and recombination using nucleotide sequences. Mol. Biol. Evol. 14(3): 239–247. [DOI] [PubMed] [Google Scholar]
- Grassly, N. C., P. H. Harvey and E. C. Holmes, 1999. Population dynamics of HIV-1 inferred from gene sequences. Genetics 151: 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R. C., and P. Marjoram, 1996. Ancestral inference from samples of DNA sequences with recombination. J. Comput. Biol. 3(4): 479–502. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1998. The age of a mutation in a general coalescent tree. Stoch. Models 14: 273–295. [Google Scholar]
- Hagenblad, J., and M. Nordborg, 2002. Sequence variation and haplotype structure surrounding the flowering time locus FRI in Arabidopsis thaliana. Genetics 161: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon, D. T., A. D. S. Bastos and P. Awadalla, 2004. Low linkage disequilibrium indicative of recombination in foot-and-mouth disease virus gene sequence alignments. J. Gen. Virol. 85: 1095–1100. [DOI] [PubMed] [Google Scholar]
- Hein, J., 1990. Reconstructing evolution of sequences subject to recombination using parsimony. Math. Biosci. 98(2): 185–200. [DOI] [PubMed] [Google Scholar]
- Hein, J., 1993. A heuristic method to reconstruct the history of sequences subject to recombination. J. Mol. Evol. 36(4): 396–405. [Google Scholar]
- Hein, J., M. H. Schierup and C. Wiuf, 2005. Gene Genealogies, Variation and Evolution. Oxford University Press, London/New York/Oxford.
- Hey, J., 2000. Human mitochondrial DNA recombination: Can it be true? Trends Ecol. Evol. 15(5): 181–182. [DOI] [PubMed] [Google Scholar]
- Hey, J., and J. Wakeley, 1997. A coalescent estimator of the population recombination rate. Genetics 145: 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 33: 54–78. [DOI] [PubMed] [Google Scholar]
- Hudson, R., 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23: 183–201. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H., and D. Bryant, 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Innan, H., and M. Nordborg, 2002. Recombination or mutational hot spots in human mtDNA? Mol. Biol. Evol. 19(7): 1122–1127. [DOI] [PubMed] [Google Scholar]
- Jakobsen, I. B., and S. Easteal, 1996. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput. Appl. Biosci. 12(4): 291–295. [DOI] [PubMed] [Google Scholar]
- Jiggins, F. M., 2002. The rate of recombination in Wolbachia bacteria. Mol. Biol. Evol. 19(9): 1640–1643. [DOI] [PubMed] [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969. Mammalian Protein Metabolism, Vol. III, pp. 21–132. Academic Press, New York/London.
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J., 1982. The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kretzer, A. M., and T. D. Bruns, 1999. Use of atp6 in fungal phylogenetics: an example from the boletales. Mol. Phylogenet. Evol. 13(3): 483–492. [DOI] [PubMed] [Google Scholar]
- Kuhner, M. K., J. Yamato and J. Felsenstein, 2000. Maximum likelihood estimation of recombination rates from population data. Genetics 156: 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoukakis, E. D., and E. Zouros, 2001. Recombination in animal mitochondrial DNA: evidence from published sequences. Mol. Biol. Evol. 18(11): 2127–2131. [DOI] [PubMed] [Google Scholar]
- Le Quesne, W. J., 1969. A method of selection of characters in numerical taxonomy. Syst. Zool. 18(2): 201–205. [Google Scholar]
- Lewontin, R., 1964. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D., and E. Rybicki, 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16(6): 562–563. [DOI] [PubMed] [Google Scholar]
- Martin, Y., G. Gerlach, C. Schlotterer and A. Meyer, 2000. Molecular phylogeny of European muroid rodents based on complete cytochrome b sequences. Mol. Phylogenet. Evol. 16(1): 37–47. [DOI] [PubMed] [Google Scholar]
- Massey, F. J., 1951. The Kolmogorov-Smirnov test for goodness of fit. J. Am. Stat. Assoc. 46(253): 68–78. [Google Scholar]
- Maynard Smith, J., 1992. Analyzing the mosaic structure of genes. J. Mol. Evol. 34(2): 126–129. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and N. H. Smith, 2002. Recombination in animal mitochondrial DNA. Mol. Biol. Evol. 19(12): 2330–2332. [DOI] [PubMed] [Google Scholar]
- McGuire, G., and F. Wright, 2000. TOPAL 2.0: improved detection of mosaic sequences within multiple alignments. Bioinformatics 16: 130–134. [DOI] [PubMed] [Google Scholar]
- McVean, G., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G. A., 2001. What do patterns of genetic variability reveal about mitochondrial recombination? Heredity 87: 613–620. [DOI] [PubMed] [Google Scholar]
- McVean, G. A. T., 2002. A genealogical interpretation of linkage disequilibrium. Genetics 162: 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin, V. N., K. S. Dorman, F. Fang and M. A. Suchard, 2005. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics 21: 3034–3042. [DOI] [PubMed] [Google Scholar]
- Miyashita, N., and C. H. Langley, 1988. Molecular and phenotypic variation of the white locus region in Drosophila melanogaster. Genetics 120: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, S. R., and R. C. Griffiths, 2003. Bounds on the minimum number of recombination events in a sample history. Genetics 163: 375–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 1997. Site-by-site estimation of the rate of substitution and the correlation of rates in mitochondrial DNA. Syst. Biol. 46(2): 346–353. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., 2000. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154: 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny, D., and M. Hendy, 1986. Estimating the reliability of evolutionary trees. Mol. Biol. Evol. 3(5): 403–417. [DOI] [PubMed] [Google Scholar]
- Piganeau, G., M. Gardner and A. Eyre-Walker, 2004. A broad survey of recombination in animal mitochondria. Mol. Biol. Evol. 21(12): 2319–2325. [DOI] [PubMed] [Google Scholar]
- Posada, D., 2001. Unveiling the molecular clock in the presence of recombination. Mol. Biol. Evol. 18(10): 1976–1978. [DOI] [PubMed] [Google Scholar]
- Posada, D., 2002. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol. Biol. Evol. 19(5): 708–717. [DOI] [PubMed] [Google Scholar]
- Posada, D., and K. A. Crandall, 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. USA 98(24): 13757–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D., and K. A. Crandall, 2002. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 54(3): 396–402. [DOI] [PubMed] [Google Scholar]
- Rohayem, J., J. Munch and A. Rethwilm, 2005. Evidence of recombination in the norovirus capsid gene. J. Virol. 79(8): 4977–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4(4): 406–425. [DOI] [PubMed] [Google Scholar]
- Sawyer, S., 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6(5): 526–538. [DOI] [PubMed] [Google Scholar]
- Schaeffer, S. W., and E. L. Miller, 1993. Estimates of linkage disequilibrium and the recombination parameter determined from segregating nucleotide sites in the alcohol dehydrogenase region of Drosophila pseudoobscura. Genetics 135: 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H., and J. Hein, 2000. a Consequences of recombination on traditional phylogenetic analysis. Genetics 156: 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H., and J. Hein, 2000. b Recombination and the molecular clock. Mol. Biol. Evol. 17(10): 1578–1579. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1994. Linkage disequilibrium in growing and stable populations. Genetics 137: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., and R. R. Hudson, 1991. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129: 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneath, P., M. Sackin and R. Ambler, 1975. Detecting evolutionary incompatibilities from protein sequences. Syst. Zool. 24(3): 311–332. [Google Scholar]
- Song, Y. S., and J. Hein, 1999. On the minimum number of recombination events in the evolutionary history of DNA sequences. J. Math. Biol. 48(2): 160–186. [DOI] [PubMed] [Google Scholar]
- Suerbaum, S., J. M. Smith, K. Bapumia, G. Morelli, N. H. Smith et al., 1998. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA 95(21): 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida, M., M. Ogata and M. Nishioka, 2000. Molecular phylogenetic relationships of pond frogs distributed in the Palearctic region inferred from DNA sequences of mitochondrial 12S ribosomal RNA and cytochrome b genes. Mol. Phylogenet. Evol. 16(2): 278–285. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L., 1998. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sinauer Associates, Sunderland, MA.
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaousis, A. D., D. P. Martin, E. D. Ladoukakis, D. Posada and E. Zouros, 2005. Widespread recombination in published animal mtDNA sequences. Mol. Biol. Evol. 22(4): 925–933. [DOI] [PubMed] [Google Scholar]
- Uzzell, T., and K. W. Corbin, 1971. Fitting discrete probability distributions to evolutionary events. Science 172: 1089–1096. [DOI] [PubMed] [Google Scholar]
- Wall, J. D., 2000. A comparison of estimators of the population recombination rate. Mol. Biol. Evol. 17(1): 156–163. [DOI] [PubMed] [Google Scholar]
- Weiller, G. F., 1998. Phylogenetic profiles: a graphical method for detecting genetic recombinations in homologous sequences. Mol. Biol. Evol. 15(3): 326–335. [DOI] [PubMed] [Google Scholar]
- Weir, B., and W. Hill, 1986. Nonuniform recombination within the human beta-globin gene cluster. Am. J. Hum. Genet. 38(5): 776–781. [PMC free article] [PubMed] [Google Scholar]
- Wiuf, C., and J. Hein, 2000. The coalescent with gene conversion. Genetics 155: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiuf, C., T. Christensen and J. Hein, 2001. A simulation study of the reliability of recombination detection methods. Mol. Biol. Evol. 18(10): 1929–1939. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1993. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 10(6): 1396–1401. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1995. A space-time process model for the evolution of DNA sequences. Genetics 139: 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13(5): 555–556. [DOI] [PubMed] [Google Scholar]