

Abstract
Histone acetylation affects many nuclear processes including transcription, chromatin assembly, and DNA damage repair. Acetylation of histone H3 lysine 56 (H3 K56ac) in budding yeast occurs during mitotic S phase and persists during DNA damage repair. Here, we show that H3 K56ac is also present during premeiotic S phase and is conserved in fission yeast. Furthermore, the H3 K56ac modification is not observed in the absence of the histone chaperone Asf1. asf1Δ and H3 K56R mutants exhibit similar sensitivity to DNA damaging agents. Mutational analysis of Asf1 demonstrates that DNA damage sensitivity correlates with (i) decreased levels of H3 K56ac and (ii) a region implicated in histone binding. In contrast, multiple asf1 mutants that are resistant to DNA damage display WT levels of K56ac. These data suggest that maintenance of H3 K56 acetylation is a primary contribution of Asf1 to genome stability in yeast.
Keywords: chromatin assembly, H3 K56ac, DNA damage
Covalent modifications of histone proteins control many aspects of chromatin function. For example, histone acetylation is a dynamic modification that has long been associated with the regulation of gene expression. Acetylation involves the addition of an acetyl moiety to the ε-amino group of target lysine residues, and each of the four core histones is acetylated on multiple lysines. The steady-state balance of histone acetylation is brought about by antagonistic catalytic activities of a family of histone acetyltransferases and histone deacetylases (HDACs) present in large multisubunit complexes that display some substrate specificity for particular N-terminal histone lysines (reviewed in refs. 1 and 2). Although the majority of acetylated lysines in the core histones are localized to the protruding N-terminal tails, recent mass spectrometry approaches have revealed a more complex landscape of modifications throughout the core histone proteins, including novel ones in the histone-fold domains (reviewed in ref. 3). Specifically, two lysines in the globular domain of histones H4 (lysine 91) and H3 (lysine 56) are also acetylated (4–7). In contrast to the majority of acetylated lysines, which are associated with transcription, both H4 K91ac and H3 K56ac have been implicated in chromatin assembly (4, 7). Moreover, recent studies have shown that K56 acetylation occurs during S phase in Saccharomyces cerevisiae and is stimulated by DNA damage (5, 7).
In most eukaryotes, histones deposited during S phase are acetylated on lysine residues 5 and 12 of histone H4 (reviewed in ref. 8); these acetyl groups are incorporated as histones are synthesized or soon thereafter (9). Newly synthesized, acetylated histones are associated with the histone H3/H4 deposition proteins, CAF-I and Asf1 (10–12). H3 K56ac also is bound to CAF-I, which assembles H3 K56ac into nucleosomes during DNA replication (7). H4 K5ac and H4 K12ac are removed after deposition as newly assembled chromatin “matures” (13). This deacetylation is presumably catalyzed by an HDAC activity that remains to be identified. Thus, histone chaperones such as Asf1 and CAF-I may protect acetylated histones from the action of nuclear HDACs before their assembly into chromatin.
Asf1 interacts with both the CAF-I and HIR chromatin assembly complexes, enhancing the replication-dependent histone assembly activity of CAF-I and the replication-independent assembly activity of the HIR complex (refs. 14 and 15; reviewed in refs. 16 and 17). Both CAF-1 and Asf1 are linked to DNA replication, because CAF-I delivers histones H3 and H4 via interaction with PCNA during DNA synthesis (18), whereas Asf1 is required for S-phase progression in the presence of DNA damage and physically interacts with the PCNA-loading complex RFC (19). Notably, previous data showed that Asf1 is particularly important during S phase, because cells lacking this protein are highly sensitive to replication fork-stalling drugs like hydroxyurea (HU) and camptothecin (CPT), phenotypes not shared by cells lacking either the CAF-I or HIR complexes (10, 19, 20).
Here, we report that acetylation of H3 K56 is important for sporulation, indicating a conserved role for this modification in S phase-linked chromatin deposition in both haploid and diploid cells. In addition, we show that this conservation extends to the fission yeast Schizosaccharomyces pombe. Moreover, we show that the highly conserved Asf1 histone chaperone is required for H3 K56ac. Furthermore, mutants that lack either Asf1 or H3 K56ac display similar DNA damage phenotypes, suggesting that Asf1 and K56ac act within the same pathway. In contrast, K56ac is unaffected by the absence of CAF-I or the HIR complex. By examining the presence of K56ac in a variety of asf1 point mutants, we document a correlation between H3 K56ac levels and DNA damage sensitivity. These data suggest that a loss of H3 K56ac is the cause of the DNA damage sensitivity phenotypes of asf1 cells.
Results
Histone H3 K56 Mutant Phenotypes.
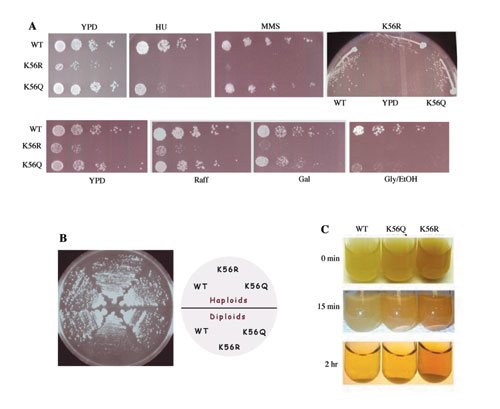
Acetylation of K56 of histone H3 recently has been reported (5, 6, 7, 21), and its function in S. cerevisiae has been addressed by examining the phenotypes of K56 mutant haploid strains. Similarly, we generated K56R and K56Q strains to mimic unacetylated and constitutively acetylated lysine residues, respectively. In addition, haploid and diploid strains containing these mutations were generated to analyze potential mitotic and meiotic growth defects. As previously reported (6, 21), the nonacetylatable K56R haploid was highly sensitive to the DNA damaging agents methyl methane sulfonate (MMS) and HU (see Fig. 5A, which is published as supporting information on the PNAS web site). Additionally, we found that the K56R haploid exhibited a slight growth defect on yeast-rich media (YPD). This defect was more pronounced in a K56R diploid (Fig. 5B). In contrast, a K56Q mutant mimicking acetylation displayed WT growth on YPD but was moderately sensitive to HU (Fig. 5 A and B). These data suggest that formation of the H3 K56ac modification is required for normal growth and that its removal is also important for damage resistance during S phase.
K56R mutants grew slowly on alternative carbon sources, such as galactose, raffinose, or glycerol/ethanol, and became flocculant upon growth to stationary phase (Fig. 5 A and C). Because these K56R growth phenotypes could reflect defects in metabolic gene expression, we asked whether K56ac was required for histone modifications important for transcription, in particular those correlated with transcriptional elongation, H3 K4me2 and H3 K79me2 (22). However, immunoblot analysis of a K56R mutant demonstrated that the global levels of H3 K4me2 or K79me2 were not changed (data not shown), in agreement with an earlier report (6). These results were confirmed by mass spectrometry analyses of H3 purified from the K56R mutant, which showed that the relative stoichiometry of the mono-, di-, and trimethylated forms of K4 and K79 were essentially the same as in a WT strain (data not shown).
H3 K56ac Is Important for Sporulation.
H3 K56ac is present on newly synthesized histone H3 molecules incorporated into chromatin during DNA replication in the mitotic cell cycle (7). We examined whether H3 K56ac also played a role in the S phase of the meiotic pathway. First, we observed that the sporulation efficiency of both K56R and K56Q diploids was greatly reduced compared with the WT strain (Fig. 1A), suggesting that H3 K56ac is important for initiation and/or completion of meiosis. We next asked whether H3 K56ac was observed during premeiotic S phase. Samples were removed from a synchronously sporulating SK1 diploid strain at 1-h intervals, and K56ac was measured by Western blot analysis (Fig. 1B). Consistent with a role in DNA replication-coupled assembly, the levels of K56ac rose dramatically 3–4 h after initiation of meiosis and before the first meiotic division, as judged by the presence of only mononucleated cells until 5–6 h (Fig. 1C). K56ac levels were strongly reduced by 8 h when meiosis was almost complete, when mostly tetranucleated asci were observed (Fig. 1C). The reduction in K56ac preceded phosphorylation of H4 serine 1, a late-stage meiosis modification that persists into spore germination. We conclude that K56ac is a modification associated with DNA replication in both the mitotic and meiotic cell cycles.
Fig. 1.

K56 acetylation state is important for meiosis and sporulation. (A) The K56R and K56Q mutants result in reduced sporulation efficiency as assayed on SPM plates. (B) Meiosis time course analysis of K56ac by Western blot of whole-cell extracts of an SK1 diploid strain. (C) The progress of meiosis was assessed by counting mono- and tetranucleated cells at each time point.
A previous study has shown that K56 acetylation can be detected in other organisms (5), albeit to varying amounts, but the question remains of whether its function is conserved. Interestingly, we found that H3 K56ac also occurs preferentially during S phase in the fission yeast S. pombe, in both mitotic cells (see Fig. 6, which is published as supporting information on the PNAS web site) and cells undergoing meiosis (data not shown). These results suggest that the function of K56ac during S phase is conserved. During growth of asynchronous WT diploid S. cerevisiae cells in sporulation media, which induces arrest in G1 phase, the K56ac signal decreased after a few days, whereas other histone modifications associated with gene expression, such as H3 K4me2, were still present at high levels (data not shown). Thus, high levels of H3 K56ac correlate with the presence of an actively dividing cell population. Taken together, these data suggest that the regulation of the acetylation state of H3 K56ac occurs during both mitotic and premeiotic DNA replication and is crucial for meiotic progression.
Asf1 Is Required for K56ac.
Recently, it has been shown that K56ac-modified H3 physically interacts with the Cac2 subunit of the replication-dependent nucleosome assembly factor CAF-I (7), suggesting that CAF-I deposits H3 K56ac into chromatin during S phase. The histone chaperone Asf1 enhances the histone deposition activity of both the CAF-I and HIR complexes (10, 14, 15, 23), prompting us to test whether Asf1 played a role in the deposition of H3 K56ac into chromatin. We first compared the growth phenotypes of single and double mutants of H3 K56R and asf1Δ. The growth of K56R asf1Δ double mutant cells was similar to either single mutant, suggesting that K56ac and Asf1 act in the same pathway (Fig. 2A). Consistent with this idea, asf1Δ and K56R single and double mutants showed a similar sensitivity to HU and an extended doubling time in liquid YPD (Fig. 2 A and B).
Fig. 2.

K56ac genetic interaction with the chromatin assembly factor Asf1. (A) Growth of the indicated strains plated as 10-fold serial dilutions, 5-μl spots on YPD plates with or without 0.1 M HU. (B) Growth curves of the indicated strains in YPD.
We next examined the presence of K56ac in the absence of Asf1 and Asf1-interacting chromatin assembly factors. As shown by Western blot analysis of strains from two different genetic backgrounds, K56ac is present in cac and hir deletion strains lacking the CAF-1 and HIR complexes, respectively (Fig. 3A). During the course of this work, a histone H3/H4 chaperone was identified in yeast, Rtt106. Rtt106 interacts with CAF-I, and epistasis analysis suggests that it functions in the same pathway with Asf1 for gene silencing (24). We therefore tested whether K56ac would be affected in a rtt106Δ strain. However, K56ac was still present at WT levels in rtt106Δ cells. Notably, these cells were also not HU-sensitive (data not shown). We conclude that Rtt106 is not required for K56ac or HU resistance. In contrast, K56ac was dramatically reduced in asf1Δ mutant cells (Fig. 3A). These data correlate with the HU sensitivity of asf1Δ cells, which is not observed in mutants lacking either the CAF-I or HIR chromatin assembly complexes. Together, these data demonstrate that Asf1, but not other assembly factors, is required for the K56ac modification during S phase histone deposition.
Fig. 3.

K56ac is absent in asf1Δ mutants. (A) Western blot analysis of histone H3 K56ac in chromatin assembly factor mutant strains. Whole-cell extracts of the indicated strains were analyzed by using the K56ac antibody; an anti-H3 C terminus antibody was used to detect total histone H3 levels. Recombinant H3 (rH3) and the K56R mutant strain were analyzed simultaneously to demonstrate the specificity of the K56ac antibody. OB, strains purchased from Open Biosystems in the BY4741 background; HS, strains derived from the histone shuffle strain MSY421. (B) The indicated anti-acetyl histone antibodies were used to analyze the indicated OB-strain WT and asf1Δ extracts.
The N-terminal 155 residues of Asf1 (Asf1N) make up the most evolutionarily conserved region of this protein and form an Ig fold domain in both yeast and human cells (25, 26). The C termini of Asf1 proteins are less well conserved in evolution; in fungi these tails contain large polyacidic tracts (25, 27). Budding yeast Asf1N is sufficient for binding to histones H3/H4 and to the checkpoint kinase Rad53, and also for stimulation of CAF-I-dependent nucleosome assembly (25). We generated a strain in which the sole source of Asf1 was Asf1N, which is WT for growth on YPD and also for HU sensitivity as previously reported (25) (data not shown). We therefore predicted that K56ac would still be present in this strain. Western blot analysis of Asf1N extracts showed that this is indeed the case (Fig. 3A), indicating that K56ac does not require the C-terminal polyacidic stretch of Asf1. We conclude that the region of Asf1 sufficient for DNA-damage resistance also maintains H3 K56 acetylation.
Because Asf1 interacts with newly synthesized, acetylated histones, we tested whether acetylation of lysines on the histone H3 or H4 tails was also regulated by this protein. As shown in Fig. 3B, H3 K18ac and the deposition-associated H4 K12ac were unaffected in an asf1Δ mutant. In a previous immunoblotting study, H3 K9ac was reported to be absent in an asf1Δ strain (28), which prompted us to perform mass spectrometry analyses of H3 and H4 isolated from log-phase cultures of WT and asf1Δ strains (see Table 1, which is published as supporting information on the PNAS web site). The results revealed that although acetylation of most residues in the histone H4 N terminus was unaffected in the asf1Δ strain, some modest differences were observed. However, in agreement with our antibody results, the most striking difference was that K56ac was undetectable in asf1Δ cells (Table 1). Thus, Asf1 plays a major role in maintaining the levels of H3 K56 acetylation but is not required for many other well known modifications, including H3 K9ac.
The inability of both asf1Δ and H3 K56R mutants to grow in the presence of MMS and HU suggested a common role in genome stability. To further explore histone modifications from cells experiencing genotoxic stress, we extracted histones from an asynchronous culture in parallel with an HU-treated culture arrested in S phase. Mass spectrometry analysis of these samples revealed that the amount of K56ac histone H3 was 5-fold higher in the HU-treated sample relative to the asynchronous population, whereas other, N-terminal H3 acetylations were not significantly altered (Table 1). Because ≈20% of histone H3 is in the K56ac form in asynchronous cells (7), the 5-fold increase in HU suggests that the great majority of H3 synthesized in S phase is in the K56ac form, consistent with other data linking this mark to S phase (5, 7). In addition, the levels of acetylation at four lysines in the histone H4 tail (K5, K8, K12, and K16) were increased upon HU treatment, probably because of an increase in the proportion of nascent, newly synthesized molecules with H4 K5ac and H4 K12ac modifications. We conclude that the most dramatic alteration in histone modifications induced by HU is the increased level of H3K56ac.
A Surface of Asf1 Related to Damage Resistance Is Required for H3 K56ac.
We sought to determine whether mutations that affect protein interactions of Asf1 alter H3 K56 acetylation. For example, residues H36/D37 in the Asf1 N-terminal globular domain are required for interaction with Hir proteins in both yeast and human cells (25, 29). Additionally, residues D54 and V94 in the Asf1 N terminus interact with the C terminus of histone H3 (26). We therefore determined whether asf1 mutations that affected H3 K56 acetylation correlated with these interactions or with DNA damage sensitivity. We first generated a series of mutations that altered Asf1 surface residues and tested for effects on sensitivity to drugs that affect S-phase progression: HU, which depletes dNTP pools, and CPT, which traps covalent topoisomerase I–DNA reaction intermediates. As noted before, asf1Δ mutants are highly sensitive to these compounds (10, 19). For each mutant, the degree of sensitivity to these drugs (Fig. 4A; and see Fig. 7, which is published as supporting information on the PNAS web site) and to the alkylating agent MMS and the double-strand breaking reagent bleomycin was correlated, such that mutations that caused severe sensitivity to one compound caused sensitivity to all. As observed previously, mutation of the Hir binding site (H36A, D37A) did not alter drug sensitivity (25). Mutations in the histone binding region also displayed phenotypes consistent with published results. Specifically, in previous studies a V94R mutation caused drug sensitivity, and a D54R mutation did not (26); we observed that the double mutation V94D, L96D caused severe drug sensitivity, whereas H53A, D54A resulted in mild sensitivity to CPT but not HU at the levels tested. We also identified Y112E (but not Y112F) as a mutation that causes severe DNA damage sensitivity and R145A, T147A as a mutation that causes CPT and HU sensitivity nearly as great as that observed for Y112E and V94D, L96D. None of the mutations generated altered steady-state levels of Asf1 protein significantly (Figs. 4B and 8, which is published as supporting information on the PNAS web site).
Fig. 4.

The loss of K56ac correlates with DNA damage sensitivity of asf1 alleles. (A) A subset of Asf1 mutations affects DNA damage resistance in vivo. Yeast strains carrying the indicated asf1 alleles were grown to logarithmic phase, and 10-fold serial dilutions were spotted onto nonselective rich media (YPD) to score total cell numbers or onto media containing 0.1 M HU or 8 μM CPT as indicated. Plates were photographed after 2–3 days at 30°C. (B) Whole-cell extracts of cells with the indicated asf1 alleles were analyzed by immunoblotting with the indicated antibodies. Recombinant yeast histone H3 was loaded in the last lane, demonstrating the specificity of the histone antibodies. Total protein on the blot was detected by Ponceau S staining. Mutations that cause severe reduction in H3 K56ac are indicated in red; mild effects are indicated in blue. The negative charge imparted by the V94D, L96D double mutation causes a slight reduction in gel mobility. (C) Mutations that affect H3 K56ac cluster on the Asf1 surface. The structure of yeast Asf1 (PDB entry 1ROC) was rendered in pymol (DeLano Scientific). Side chains are color-coded as above. R108 (colored gray) contacts the histone H3 C terminus in addition to D54 and V94 (25).
Western blot analysis of cells expressing these asf1 mutants (Fig. 4B) showed that the levels of H3 K56ac generally correlated with the degree of DNA damage sensitivity. For example, there was no acetylation defect in H36A, D37A cells or in other mutants that are not drug-sensitive. These data indicate that interaction with the HIR complex is not important for H3 K56 acetylation. Because heterochromatic silencing is also compromised by the H36A, D37A mutations, we conclude that the silencing function of Asf1 is not required for K56ac. In contrast, the highly damage-sensitive Y112E and V94D, L96D mutants lacked H3 K56 acetylation, and mutants that displayed intermediate drug resistance (H53A, D54A; V45D) displayed intermediate H3 K56ac levels. The one exception to this correlation is the R145A, T147A mutant, which is highly damage-sensitive but only moderately defective in H3 K56ac levels. The R145A, T147A data suggest that K56ac may be necessary but not sufficient for efficient resistance to DNA damaging agents. Together, our data support the idea that H3 K56ac is the primary underlying molecular event by which Asf1 contributes to DNA damage resistance.
To further test this idea, we examined additional genetic interactions between Asf1 and H3 K56. The nucleosomal DNA–histone interactions in H3 K56Q mutant cells appear more “open” (7), as detected by more rapid nuclease digestion of chromatin and reduced supercoiling of endogenous plasmids in these cells. These effects are thought to occur because H3 K56 is a critical electrostatic DNA interaction site that is neutralized by the K56Q mutation. We therefore propose that the H3 K56–DNA interaction is constitutively strong in asf1 cells, because histone H3 remains unacetylated at K56. A prediction of this K56 “charge” model is that the growth and DNA damage sensitivity defects of asf1Δ cells should be suppressed by a histone H3 K56Q mutation. As shown in Fig. 9 (which is published as supporting information on the PNAS web site), we did indeed observe suppression of growth and damage sensitivity phenotypes. In particular, the HU sensitivity of asf1Δ cells is nearly completely suppressed by the H3 K56Q mutation, and growth rates on rich media are substantially suppressed. We conclude that the growth and damage sensitivity phenotypes of asf1 cells are largely the result of defects in H3 K56 acetylation. Weakening the electrostatic interaction between H3 K56 and DNA during replication is critical for genome stability and is enforced by Asf1 via maintenance of acetylation of this residue.
Discussion
Together, the data presented here support a model in which the DNA damage sensitivity phenotypes of asf1Δ mutant cells can be explained by the absence of H3K56ac, an idea that affects the interpretation of several published studies of asf1 cells. For example, a role for H3 K56ac has recently been shown in the recruitment of the Swi/Snf chromatin remodeling complex to the genes encoding histone H2A during S phase, and also to the SUC2 gene under inducing conditions (5). The absence of Swi/Snf-dependent remodeling would alter transcription factor access at these and other genes and may explain some of the growth defects observed in asf1 and H3 K56 mutants (10) (Figs. 2 and5).
Additionally, the DNA damage sensitivity of asf1 cells has been linked to the stability of replisomes in the presence of DNA damage (19). Because histone H3 K56ac occurs in S phase during both mitosis and meiosis (5, 7) (Fig. 1), we hypothesize that this modification is fundamentally important to the stability of replisomes and is normally delivered to replication forks via Asf1. However, the contribution of Asf1 to heterochromatic gene silencing is separable from H3 K56ac, because mutation of the HIR binding site does not affect this modification. In contrast, H3 K56ac appears to depend on the Asf1–histone interaction, because mapping of the data from our studies onto the structure of Asf1 (25) shows that residues that affect H3 K56ac cluster in a region overlapping the area implicated in histone H3 binding (Fig. 4C) (26).
Our results reveal a role for H3 K56ac in DNA damage repair through the action of the H3/H4 histone chaperone Asf1. The DNA damaging agents to which both H3 K56R and asf1Δ mutants are most sensitive are those that most significantly affect S phase (MMS, CPT, HU), suggesting that the Asf1-mediated deposition of histone H3 acetylated on K56 during S phase has a crucial role in genome stability during DNA replication. We suggest that K56ac creates a more “open” chromatin environment in which the newly assembled DNA strands are more accessible to remodeling/repair factors. Once completed, we speculate that a yet unidentified HDAC activity would be activated to result in the more “closed” chromatin conformation as observed in G2/M or in the absence of K56ac (7). Two alternative scenarios for the role of Asf1 in H3 K56ac metabolism can be envisioned: (i) Asf1 is essential for targeting the H3 K56ac histone acetyltransferase to histone H3 in S phase, and (ii) Asf1 may protect H3K56ac from a nuclear HDAC activity before and during chromatin deposition. These proposed roles may not be mutually exclusive.
Materials and Methods
Yeast Strains and Plasmids.
Strain MSY421 from M. M. Smith (University of Virginia) [MATa, Δ(hht1-hhf1) Δ(hht2-hhf2) leu2-3, 112, ura3-62, trp1, his3, pMS329 (HHT1-HHF1, URA3, CEN)] was used to shuffle in plasmids containing histone H3 K56 mutations (TRP1, CEN, hht2-HHF2) using 5-FOA as a counterselecting agent for the URA3 plasmid. The mutant plasmids were generated by PCR mutagenesis (hht2 K56R or K56Q) and confirmed by sequencing. The final yeast strain was confirmed by PCR amplification of the HHT2-HHF2 locus and DNA sequencing. Gene disruptions of the ASF1 coding region and the ASF1-N truncation (Asf1 1–155) in these strains were generated by transformation with a DNA fragment containing ≈50 bp of flanking sequences containing a kan-MX cassette as described in ref. 30 . These strains are referred to as “histone shuffle” background (HS) as opposed to deletion mutants in the BY4741 background, which were purchased from Open Biosystems. Yeast growth, plasmid and DNA fragment transformation of yeast cells, and tetrad dissection and mating type testing were done according to standard yeast protocols (31). Growth curves were an average of three independent cultures for each strain, diluted in YPD to an OD600 of 0.1 from saturated cultures, and measured each hour during growth at 30°C shaking at 200 rpm. DNA damage plates were YPD with or without 0.03% MMS or 0.1 M HU, photographed after 3 days of growth at 30°C. Sporulation medium (SPM) was 0.3% potassium acetate with 0.02% raffinose. Percent sporulation was calculated by counting the number of asci in SPM containing four spores versus no spores. For each strain, sporulation efficiency was assayed in triplicate, and ≈800–1,000 asci were counted after 3–6 days in SPM. For the experiments in, yeast strain PKY952 (MATα, asf1Δ::HIS3, lys2–128δ, URA3-VIIL, in the W303 genetic background) was transformed with low-copy plasmids carrying the natural ASF1 promoter driving expression of the indicated asf1 alleles (25). Mutant asf1 alleles were generated by standard site-directed mutagenesis protocols and were sequenced before use. For the damage sensitivity assays, cells were grown 3 days at 30°C before photographing.
SK1 Meiosis.
Singles colonies from a derivative of the diploid S. cerevisiae SK1 strain NKY1551 (ho::LYS2 lys2 ura3 leu2::hisG) were inoculated separately into 10 ml of YPD. The saturated cultures were used to inoculate 200 ml of YPA (1% Bacto yeast extract/2% Bacto peptone/1% potassium acetate) to an OD600 of 0.2 and grown for 13.5 h at 30°C shaking at 200 rpm. The cells were washed with sterile water and resuspended into 200 ml of SPM (0.3% potassium acetate/0.02% raffinose). The cultures were shaken at 200 rpm at 30°C for 10 h, 15-ml samples were taken at 0, 1, 2, 3, 4, 5, 6, 7, 8, and 10 h, and trichloroacetic acid extracts were prepared and used for Western blot analysis. Additional 0.5-ml samples were taken at each time point, fixed in ethanol 50%, and DAPI-stained for quantification of mono-, di-, and tetranucleated cells. A total of three diploids were analyzed.
Western Blot Analysis.
Trichloroacetic acid whole-cell extracts were prepared from 12 ml of cells grown to an OD600 of 0.8 in YPD as described in ref. 32. The final volume of the extracts was 150 μl, from which 4 μl was loaded onto 15% SDS/PAGE for Western blot analysis. All histone H3-modification antibodies were from Upstate Biotechnology. The polyclonal anti-Asf1 antibody was described in ref. 25. The following antibody dilutions were used: K56ac (1:5,000); K18ac (H3), K12ac, and S1phos (H4) (1:3,000); anti-H3 (Abcam) (1:10,000); and anti-Asf1 (1:5,000). All primary and secondary antibodies were diluted in TBS containing 0.1% Tween 20 and 5% milk. An ECL Plus kit (Amersham Pharmacia) was used according to manufacturer's instructions.
RP-HPLC Separation of Histones and Mass Spectrometry Analysis.
See Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank Upstate Biotechnology for their role in developing the K56ac antibody, Mary Ann Osley for critical reading of the manuscript and helpful discussions throughout the course of this work, and Fred Winston and Vanessa Cheung for helpful suggestions, especially regarding Fig. 9. This work was supported by National Institutes of Health Method to Extend Research in Time (MERIT) Grant GM53512 (to C.D.A.), National Science Foundation Grant MCB-0549131 (to P.D.K.), and National Institutes of Health National Center for Research Resources Biomedical Research Technology Program Grant RR01614 (to A.L.B.).
Abbreviations
- CPT
camptothecin
- HDAC
histone deacetylase
- HU
hydroxyurea
- MMS
methyl methane sulfonate
- SPM
sporulation medium
- YPD
yeast-rich media.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Roth S. Y., Denu J. M., Allis C. D. Annu. Rev. Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 2.Kurdistani S., Grunstein M. Nat. Rev. Mol. Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- 3.Freitas M. A., Sklenar A. R., Parthun M. R. J. Cell. Biochem. 2004;92:691–700. doi: 10.1002/jcb.20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye J., Ai X., Eugei E. E., Zhang L., Carpenter L. R., Jelinek M. A., Freitas M. A., Parthun M. R. Mol. Cell. 2005;18:122–130. doi: 10.1016/j.molcel.2005.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu F., Zhang K., Grunstein M. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Ozdemir A., Spicuglia S., Lasonder E., Vermeulen M., Campsteijn C., Stunnenberg H.G., Logie C. J. Biol. Chem. 2005;280:25949–25952. doi: 10.1074/jbc.C500181200. [DOI] [PubMed] [Google Scholar]
- 7.Masumoto H., Hawke D., Kobayashi R., Verreault A. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 8.Verreault A. Genes Dev. 2000;14:1430–1438. [PubMed] [Google Scholar]
- 9.Ruiz-Carrillo A., Wangh L. J., Allfrey V. G. Science. 1975;190:117–128. doi: 10.1126/science.1166303. [DOI] [PubMed] [Google Scholar]
- 10.Tyler J. K., Adams C. R., Shaw-Ree C., Kobayashi R., Kamakaka R., Kadonaga J. T. Nature. 1999;402:555–560. doi: 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman P. D., Kobayashi R., Kessler N., Stillman B. Cell. 1995;81:1105–1114. doi: 10.1016/s0092-8674(05)80015-7. [DOI] [PubMed] [Google Scholar]
- 12.Verreault A., Kaufman P. D., Kobayashi R., Stillman B. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 13.Annunziato A. T., Seale R. L. J. Biol. Chem. 1983;258:12675–12684. [PubMed] [Google Scholar]
- 14.Prochasson P., Florens L., Swanson S. K., Washburn M. P., Workman J. L. Genes Dev. 2005;19:2534–2539. doi: 10.1101/gad.1341105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green E. M., Antczak A. J., Bailey A. O., Franco A. A., Wu K. J., Yates J. R., III, Kaufman P. D. Curr. Biol. 2005;15:2044–2049. doi: 10.1016/j.cub.2005.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loyola A., Almouzni G. Biochim. Biophys. Acta. 2004;1677:3–11. doi: 10.1016/j.bbaexp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Franco A. A., Kaufman P. D. Cold Spring Harbor Symp. Quant. Biol. 2004;69:1–8. doi: 10.1101/sqb.2004.69.201. [DOI] [PubMed] [Google Scholar]
- 18.Shibahara K., Stillman B. Cell. 1999;96:575–585. doi: 10.1016/s0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- 19.Franco A. A., Lam W. M., Burgers P. M., Kaufman P. D. Genes Dev. 2005;19:1–11. doi: 10.1101/gad.1305005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramey C. J., Howar S., Adkins M., Linger J., Spicer J., Tyler J. K. Mol. Cell. Biol. 2004;24:10313–10327. doi: 10.1128/MCB.24.23.10313-10327.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyland E. M., Cosgrove M. S., Molina H., Wang D., Pandey A., Cottee R. J., Boeke J. D. Mol. Cell. Biol. 2005;25:10060–10070. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao T., Kao C. F., Krogan N. J., Sun Z. W., Greenblatt J. F., Osley M. A., Strahl B. D. Mol. Cell. Biol. 2005;25:637–651. doi: 10.1128/MCB.25.2.637-651.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp J. A., Fouts E. T., Krawitz D. C., Kaufman P. D. Curr. Biol. 2001;11:463–473. doi: 10.1016/s0960-9822(01)00140-3. [DOI] [PubMed] [Google Scholar]
- 24.Huang S., Zhou H., Katzmann D., Hochstrasser M., Atasanova E., Zhang Z. Proc. Natl. Acad. Sci. USA. 2005;102:13410–13415. doi: 10.1073/pnas.0506176102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daganzo S. M., Erzberger J. P., Lam W. M., Skordalakes E., Zhang R., Franco A. A., Brill S. J., Adams P. D., Berger J. M., Kaufman P. D. Curr. Biol. 2003;13:2148–2158. doi: 10.1016/j.cub.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 26.Mousson F., Lautrette A., Thuret J. Y., Agez M., Courbeyrette R., Amigues B., Becker E., Neumann J. M., Guerois R., Mann C., Ochsenbein F. Proc. Natl. Acad. Sci. USA. 2005;102:5975–5980. doi: 10.1073/pnas.0500149102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umehara T., Chimura T., Ichikawa N., Horikoshi M. Genes Cells. 2002;7:59–73. doi: 10.1046/j.1356-9597.2001.00493.x. [DOI] [PubMed] [Google Scholar]
- 28.Adkins M. W., Tyler J. K. J. Biol. Chem. 2004;279:52069–52074. doi: 10.1074/jbc.M406113200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang R., Poustovoitov M. V., Ye X., Santos H. A., Chen W., Daganzo S. M., Erzberger J. P., Serebriiskii I. G., Canutescu A. A., Dunbrack R. L., et al. Dev. Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 30.Longtine M. S., McKenzie A., III, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 31.Rose M. D., Winston F., Hieter P. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1990. [Google Scholar]
- 32.Kao C. F., Osley M. A. Methods. 2003;31:59–66. doi: 10.1016/s1046-2023(03)00088-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}