

Abstract
We show here that the African swine fever virus (ASFV) protein pE296R, predicted to be a class II apurinic/apyrimidinic (AP) endonuclease, possesses endonucleolytic activity specific for AP sites. Biochemical characterization of the purified recombinant enzyme indicated that the Km and catalytic efficiency values for the endonucleolytic reaction are in the range of those reported for Escherichia coli endonuclease IV (endo IV) and human Ape1. In addition to endonuclease activity, the ASFV enzyme has a proofreading 3′→5′ exonuclease activity that is considerably more efficient in the elimination of a mismatch than in that of a correctly paired base. The three-dimensional structure predicted for the pE296R protein underscores the structural similarities between endo IV and the viral protein, supporting a common mechanism for the cleavage reaction. During infection, the protein is expressed at early times and accumulates at later times. The early enzyme is localized in the nucleus and the cytoplasm, while the late protein is found only in the cytoplasm. ASFV carries two other proteins, DNA polymerase X and ligase, that, together with the viral AP endonuclease, could act as a viral base excision repair system to protect the virus genome in the highly oxidative environment of the swine macrophage, the virus host cell. Using an ASFV deletion mutant lacking the E296R gene, we have determined that the viral endonuclease is required for virus growth in macrophages but not in Vero cells. This finding supports the existence of a viral reparative system to maintain virus viability in the infected macrophage.
Base excision repair (BER) is the major system for the repair of DNA base lesions such as the products of deamination, oxidation, and alkylation, all of which can arise endogenously (22). As such, BER plays an essential role in the protection of cells against the lethal and mutagenic effects of DNA damage. BER is a multistep process composed of sequential reactions that are proposed to occur in a highly coordinated manner. The mammalian DNA polymerase β (Pol β)-dependent short-patch pathway of BER starts with a monofunctional DNA glycosylase that excises the damaged base, generating an abasic site that is subsequently incised at the 5′ side of the lesion by an apurinic/apyrimidinic (AP) endonuclease, leaving a 5′-terminal deoxyribose phosphate (dRP) (23, 24). This dRP group is eliminated by the dRP lyase activity of Pol β after the gap-filling step (25, 37). Finally, a DNA ligase seals the nick.
A key enzyme of this system is the AP endonuclease, not only because of its endonucleolytic activity, which can resolve mutagenic AP sites, but also because it is able to eliminate 3′ blocking groups, such as phosphates, phosphoglycolates, and α,β-unsaturated aldehydes arising in DNA as products of reactive oxygen species (ROS) attack or generated by enzymatic AP lyase activity, all of which block DNA replication (11). Furthermore, AP endonucleases may interact with and coordinate other enzymes involved in the short-patch route of BER, such as Pol β (2, 44), or in the long-patch subpathway of BER, such as flap endonuclease (Fen) and proliferating cell nuclear antigen (7).
African swine fever virus (ASFV), a complex and enveloped deoxyvirus with icosahedral morphology, is responsible for a highly lethal disease of domestic pigs (33). The virus replicates mainly in the cytoplasm of the infected cell, but an initial phase of viral DNA replication in the nucleus has been described (13, 32). The viral genome is a double-stranded DNA molecule of 170 to 190 kbp that encodes more than 150 polypeptides, including a variety of enzymes involved in gene transcription, protein modification, and DNA replication and repair (46). Among these is the smallest naturally occurring DNA-directed DNA polymerase (174 amino acid residues) described so far (27). This DNA polymerase, designated Pol X, is a highly distributive β-type polymerase lacking a proofreading 3′→5′ exonuclease activity that has been proposed to participate in a BER pathway during ASFV infection (14, 27). In addition, the viral protein pE296R has been annotated as a class II AP endonuclease, while the NP419L gene codes for a DNA ligase (46, 47), which further supports the existence of a BER-like pathway acting in ASFV-infected cells (14, 27). A viral BER process may be crucial for maintaining the integrity of the viral genome in the highly oxidative and potentially mutagenic environment of the swine macrophage (26), the ASFV host cell.
For this work, we purified and characterized the protein codified by the ASFV E296R gene. We show that the recombinant protein possesses an endonuclease activity specific for AP sites, as well as 3′→5′ exonuclease activity. The latter activity may act as a proofreading backup system to increase the fidelity of DNA repair. By using an ASFV deletion mutant lacking the E296R gene, we also show that the viral AP endonuclease is essential for virus growth in swine macrophages but not in Vero cells. These results support a role for the viral AP endonuclease in a reparative BER system to maintain the viability of the virus in its natural target cell.
MATERIALS AND METHODS
Amino acid sequence comparisons.
The multiple sequence alignment of the endonuclease IV (endo IV) subfamily of class II AP endonuclease sequences shown in Fig. 1 was done with Clustal_X (38). All sequences are from the NCBI Protein Database.
FIG. 1.

Multiple amino acid sequence alignment of M. pneumoniae (END4_MYCPN), B. anthracis (1XP3_BANTH), T. maritima (END4_THEMA), E. coli (nfo/END4_ECOL), C. elegans (CeAPN1), S. cerevisiae (APN1_SC), Acanthamoeba polyphaga mimivirus (END4_Mimi), and ASFV (ASFV_E296R) endonuclease IV family proteins, highlighting metal-binding active residues (white letters and open circles at the top of the alignment) and AP-site stabilization residues (yellow letters and yellow circles). Functional enzyme motifs, including the minor groove and phosphate-binding loops, are boxed in navy blue and magenta, respectively. The relative similarity of residues at each position as well as the amino acid background coloring is shown according to Clustal_X codes. Above the sequences, hollow boxes indicate helices and black arrows show strands (taken from the crystal structure of endo IV) (15).
Oligonucleotides and proteins.
The oligonucleotides used in this work are shown in Table 1. They were purchased from Isogen. EndoU, Endo11, and Endo11MM were purified by 7 M urea-20% polyacrylamide gel electrophoresis (PAGE) before use.
TABLE 1.
Sequences of oligonucleotides used in this work
| Name | Sequence (5′→3′) |
|---|---|
| ΔE296R-1 | GAGGGACCGGACCTCTATAG |
| ΔE296R-2 | TCATTCTCTATTGTAATGCGC |
| EndoU | ATATACCGCGGUCGGCCGATCAAGCTTATT |
| EndoG | AATAAGCTTGATCGGCCGGCCGCGGTATAT |
| Endo11 | ATATACCGCGG |
| Endo11MM | ATATACCGCGA |
| Endo19 | CCGGCCGATCAAGCTTATT |
Restriction endonucleases, T4 polynucleotide kinase, Escherichia coli uracil DNA glycosylase (UDG), and E. coli endo IV were purchased from New England Biolabs.
Expression and purification of ASFV protein pE296R.
The E296R open reading frame (ORF) of ASFV cloned in pRSET-A was expressed in E. coli BL21(DE3)/pLys at 30°C in the presence of 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h. Cells were collected by centrifugation for 15 min at 2,000 × g and resuspended in 10 ml of buffer A (50 mM phosphate buffer, pH 8, 1 M NaCl, and 20 mM imidazole). After sonication on ice, an aliquot was withdrawn as total cell extract. The suspension was cleared by centrifugation for 20 min at 20,000 × g, and the soluble supernatant fraction was used for affinity purification.
Ni-nitrilotriacetic acid (Ni-NTA) agarose beads (QIAGEN), previously equilibrated in buffer A, were added to the soluble fraction containing the recombinant protein. After being stirred for 2 h at 4°C, the resin was washed in batches twice with 25 resin volumes of buffer A and then loaded onto a column. Elution was carried out in two steps, with 60 and 250 mM imidazole in buffer B (50 mM phosphate buffer, pH 6, 1 M NaCl). Five 0.5-ml fractions were collected at each step. The fractions were analyzed by sodium dodecyl sulfate-PAGE (SDS-PAGE) followed by staining with Coomassie brilliant blue, and the last four fractions eluted with 250 mM imidazole were pooled, dialyzed against 80 mM Tris-HCl, pH 7.5, 100 mM NaCl, 4 mM dithiothreitol, diluted one-half with 100% glycerol, and stored at −20°C.
Enzymatic assays.
The oligonucleotides used were 5′ labeled with T4 polynucleotide kinase and [γ-32P]ATP (3,000 Ci/mmol; Amersham International Plc.) and purified with a nucleotide removal kit (QIAGEN). After incubation with a 1.5-fold excess of the complementary strand for 10 min at 80°C in 30 mM Tris-HCl [pH 8]-10 mM NaCl, the solution was slowly cooled to room temperature for 1 h. One picomole of the double-stranded, end-labeled substrate was incubated with the indicated enzyme in optimized buffer (25 mM Tris-HCl, pH 8, 10 mM NaCl, and 5 mM MgCl2) in a final volume of 50 μl for AP endonuclease assays. For 3′→5′ exonuclease assays, the final reaction volume was 30 μl, and ZnCl2 was used instead of MgCl2. In all cases, incubations were done for 45 min at 37°C, and the reactions were stopped by adding an equal volume of formamide loading buffer (98% formamide, 20 mM EDTA, 0.5% bromophenol blue, and 0.5% xylene cyanol). The samples were heated for 10 min at 95°C and immediately placed on ice before being loaded into a 20% polyacrylamide-7 M urea-1× Tris-borate-EDTA gel. After electrophoresis, the product bands were detected by autoradiography and measured by densitometry with Quantity One software (Bio-Rad). Kinetic parameters were determined using reaction times, and enzyme concentrations were adjusted to a linear steady state. Km and Vmax values were determined from fitted Lineweaver-Burk plots.
Antibodies.
Antibodies against purified His-tagged pE296R were raised in rabbits. The immune serum obtained recognized the recombinant protein in Western blots. The rat anti-p54 protein, as well as the rabbit anti-pE248R and anti-pB119L proteins, has been described previously (28, 30). Anti-tubulin and anti-β-actin monoclonal antibodies were purchased from Sigma.
Cells and viruses.
Vero cells were obtained from the American Type Culture Collection and grown in Dulbecco modified Eagle medium (DMEM) containing 5% fetal calf serum. Swine alveolar macrophages, obtained as described previously (4), were grown in DMEM containing 10% swine serum. Cell infections with the ASFV BA71V strain and titrations were carried out as previously described (8). Highly purified ASFV was prepared as described before (3).
The E296R deletion virus was obtained by insertion of the gusA gene of E. coli into the E296R ORF of the Vero-cell-adapted ASFV strain BA71V. A 1,318-bp DNA fragment containing the E296R gene was generated by PCR using the primers ΔE296R-1 and ΔE296R-2 and Vent DNA polymerase (New England Biolabs). The PCR product was cloned into EcoRV-digested pZErO (Invitrogen), generating the plasmid pZErO-E296R. Plasmid p72Gus10T (12) was digested with SmaI/HindIII and treated with Klenow fragment. The fragment obtained was cloned into AvrII-digested pZErO-E296R, which was previously treated with Klenow fragment to obtain the transfer vector pΔE296R-Gus. The recombinant BA71V-ΔE296R (vΔE296R) virus was generated in Vero cells and purified by sequential rounds of plaque purification in the presence of 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid (X-Gluc) as previously described (29). The disruption of the E296R gene was confirmed by DNA hybridization and Western blot analysis of vΔE296R-infected Vero cells (data not shown).
Analysis of pE296R expression in ASFV-infected Vero cells.
Vero cells in DMEM containing 2% fetal calf serum were either mock infected or infected with ASFV BA71V at a multiplicity of infection (MOI) of 10 PFU per cell, and at different times postinfection, the cells were lysed in electrophoresis sample buffer at a density of 3 × 106 cells per ml. Equivalent amounts of the cell lysates were electrophoresed in 12% SDS-polyacrylamide gels and subsequently transferred to nitrocellulose membranes for Western blot analysis. The membranes were incubated with the antibody against the pE296R protein (1:2,000 dilution) and then with a 1:10,000 dilution of peroxidase-labeled anti-rabbit serum (Amersham International Plc.). The proteins were detected with the ECL system (Amersham International Plc.) according to the manufacturer's recommendations.
Immunofluorescence.
Vero cells grown in chamber slides were either mock infected or infected with BA71V or vΔE296R at an MOI of 10 PFU per cell and then fixed at different times postinfection with 4% paraformaldehyde-2% sucrose in 1× phosphate-buffered saline (PBS) for 15 min at room temperature, followed by 5 min with methanol at −20°C. Fixed cells were washed twice with 1× PBS and blocked for 30 min with blocking buffer (3% bovine serum albumin, 0.1% Triton X-100, 1× PBS). The cells were then sequentially incubated for 1 h with primary antibodies and the corresponding secondary antibodies diluted with 1% bovine serum albumin in 1× PBS. The primary antibodies were used at the following dilutions: anti-tubulin, 1:200; anti-p54, 1:200; and anti-pE296R, 1:500. The secondary antibodies used were goat anti-mouse, goat anti-rat, and goat anti-rabbit immunoglobulin G coupled to Alexa 594, Alexa 697, and Alexa 488, respectively (1:500 dilution) (Molecular Probes). After incubation with the antibodies, the cells were washed three times for 15 min each in 1× PBS. Finally, the coverslips were mounted with Mowiol/Dabco on glass slides. Preparations were examined with a Bio-Rad Radiance 2000 confocal laser scanning microscope. Images were processed using Adobe Photoshop software.
RESULTS
ASFV protein pE296R contains the conserved catalytic center of E. coli endonuclease IV.
By complete DNA sequencing of the ASFV BA71V strain and subsequent database searches for amino acid sequence similarities, it had been reported that the ASFV E296R ORF potentially encoded a class II AP endonuclease (46). Figure 1 shows a multiple amino acid sequence alignment of ASFV protein pE296R and selected members of the E. coli endo IV subgroup of the class II AP endonuclease family. A dendrogram obtained from this alignment (not shown) indicates that the ASFV protein pE296R more closely resembles endo IV from Acanthamoeba polyphaga mimivirus, which has been reported to be related to ASFV (20). All of the residues that are involved in catalytic Zn atom coordination (15) are fully conserved (residues in white below open circles in Fig. 1), except for E261 of endo IV, which is not conserved in the ASFV protein. Amino acids that stabilize the AP site by stacking interactions with base pairs at the 3′ and 5′ ends are also highly conserved (residues in yellow below yellow circles). The sugar-phosphate backbone interaction residues are clearly conserved in the enzymes of E. coli, Bacillus anthracis, Mycoplasma pneumoniae, Candida elegans, and Saccharomyces cerevisiae, whereas the Thermotoga maritima, mimivirus, and ASFV proteins show more sequence diversity (magenta boxes). Nevertheless, this diversity does not essentially modify the DNA recognition loop (R loop) predictions (see below), and therefore the proposed mechanism (15) for interaction with productively cleaved AP site-containing DNA could be applied to all the enzymes in the group.
Expression and purification of recombinant protein pE296R.
In order to characterize the ASFV pE296R protein, the E296R ORF was cloned into the pRSET-A vector and expressed in E. coli. As shown in Fig. 2, a protein of about 34 kDa, the size expected for the recombinant protein, was detected in total lysates from cells induced with IPTG. Based on the His tag present at its N-terminal end, the recombinant protein could be purified from the soluble fractions of these lysates in a single step by Ni-NTA affinity chromatography (see Materials and Methods). After purification, a major band corresponding to the recombinant pE296R protein could be observed (Fig. 2).
FIG. 2.
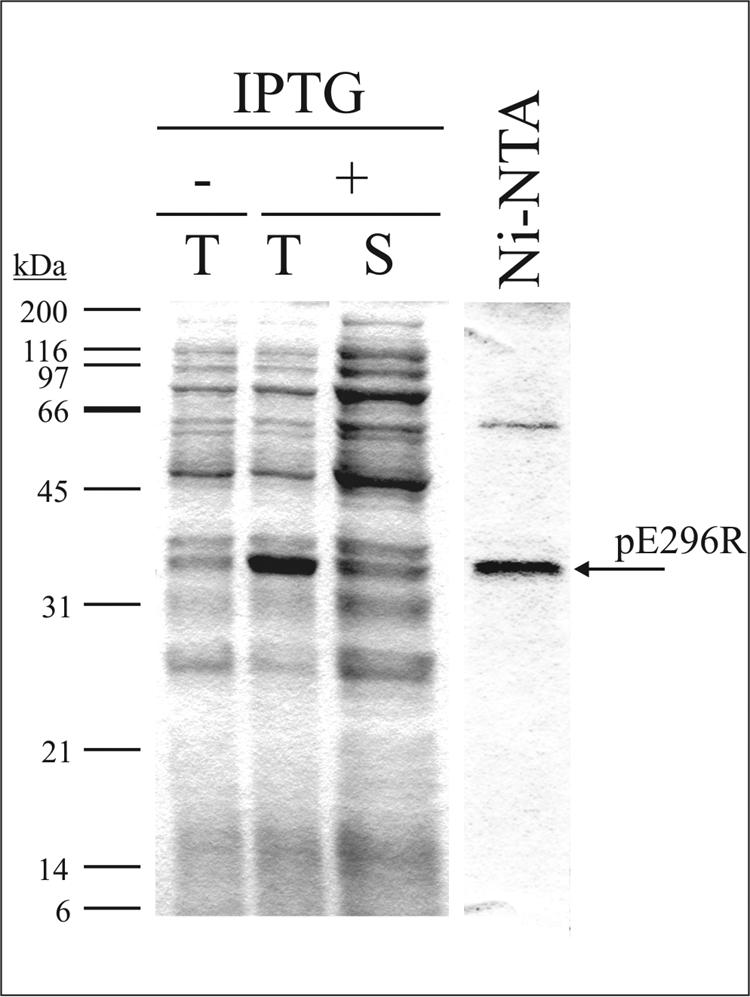
Expression and purification of recombinant ASFV pE296R protein. A Coomassie blue-stained gel after 12% SDS-PAGE separation of total extracts from control noninduced (−T) and IPTG-induced (+T) E. coli BL21(DE3)/pLysS cells transformed with the recombinant plasmid pRSET-E296R is shown. The soluble (S) fraction of the latter extract and the purified fraction (Ni-NTA) obtained after Ni-NTA affinity chromatography of the soluble fraction are also shown. The electrophoretic migration of molecular size markers is indicated on the left. The arrow shows the expected position of the recombinant protein.
Enzymatic properties of ASFV AP endonuclease.
The predicted AP endonuclease activity of the recombinant pE296R protein was examined using a 5′-32P-labeled 30-mer double-stranded oligonucleotide containing a uracil (U) residue at position 12 (Fig. 3A). This substrate was incubated with E. coli UDG to remove the U residue and generate an AP site and with various amounts of the purified recombinant pE296R protein. AP endonucleolytic activity would release a 32P-labeled cleavage product of 11 nucleotides. As shown in Fig. 3A, the pE296R protein exhibited AP endonuclease activity in a dose-dependent manner (lanes 5 to 9). As shown, no activity was detected in the absence of UDG (lane 3), indicating the requirement of an AP site for endonuclease activity. As also shown in lane 3 of Fig. 3A, no contaminating nuclease activity was detected in the purified recombinant protein preparation used.
FIG. 3.

(A) AP endonuclease activity of ASFV pE296R protein. The 5′-32P-labeled 30-mer double-stranded oligonucleotide (EndoU/EndoG) substrate (1 pmol) containing a U-G pair at position 12 was treated simultaneously with 0.5 units of UDG to create an AP site and the following amounts of purified pE296R protein: 30, 60, 90, 120, and 200 ng (lanes 5, 6, 7, 8, and 9, respectively). E. coli endo IV (0.5 units) was used as a control (lane 4). Results for mixtures containing only UDG (0.5 units), endo IV (0.5 units), and pE295R (90 ng) are shown in lanes 1, 2, and 3, respectively. The reaction was monitored by autoradiography after resolution in a 20% polyacrylamide-7 M urea gel. The position of the 11-mer product is indicated. (B) Optimal pH (B1), salt (B2), and magnesium (B3) conditions for AP endonuclease activity of pE296R. To study pH dependence, the following buffers were used: 20 mM sodium acetate for pHs 5.5 and 6, 20 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] for pHs 6, 6.5, and 7, and 20 mM Tris-HCl for pHs 7, 7.5, 8, and 8.5. Each point is the mean of three independent experiments.
Figure 3B shows the pH, ionic strength, and divalent cation dependence of ASFV AP endonuclease activity. As can be seen in this figure, the AP endonuclease was active over the range from pH 6.0 to pH 8.5, with the highest activity obtained at pH 8.0. The AP endonuclease showed maximal activity in the presence of 10 to 20 mM NaCl, with concentrations of 100 mM and higher being strongly inhibitory. MgCl2 stimulated the activity, with maximal stimulation in the 4 to 10 mM range.
The class II AP endonucleases have also been described to possess a 3′→5′ proofreading exonuclease activity specific for double-stranded DNA that may improve the fidelity of BER by removing mispaired bases (6, 18, 39, 40, 42). To see whether the viral enzyme possesses this activity, we used a 30-mer duplex substrate with a 19-base recessed 3′ end or an internal nick, with or without a mismatch at the 3′ end of the recessed oligonucleotide or at the 3′ side of the nick (Fig. 4). As shown in Fig. 4, the viral AP endonuclease exhibited dose-dependent 3′→5′ exonuclease activity with all substrates tested. Interestingly, this activity was essentially limited to the removal of the last nucleotide at the 3′ end of the 5′-32P-labeled oligonucleotide and more efficiently eliminated a mismatched than a correctly paired base. It is also noteworthy that the best substrate for the enzyme was the nicked oligonucleotide with a mismatch, which resembles DNA intermediates arising in the BER route after misinsertion of a nucleotide by reparative DNA polymerases. These properties would make the enzyme well fitted for the correction of possible misinsertions during a viral BER pathway. It can also be seen in Fig. 4 that E. coli endo IV, which was carried as a control in these experiments, shows, like the viral AP endonuclease, a strong preference for mismatched substrates (compare lanes 2 and 7 with lanes 12 and 17).
FIG. 4.

3′→5′ exonuclease activity of ASFV pE296R protein. The 5′-32P-labeled double-stranded oligonucleotide substrate (1 pmol) was treated with the following amounts of purified pE296R protein: 35 ng (lanes 3, 8, 13, and 18), 100 ng (lanes 4, 9, 14, and 19), and 200 ng (lanes 5, 10, 15, and 20). Lanes 2, 7, 12, and 17 show control reactions with 5 units of endo IV. Lanes 1, 6, 11, and 16 show control reactions with no enzyme. The reaction products were monitored by autoradiography after resolution in a 20% polyacrylamide-7 M urea gel.
The Km and catalytic efficiency values obtained for the endonucleolytic activity of the viral AP endonuclease were 53.4 nM and 1.07 min−1 nM−1, respectively. These values are comparable to those reported for endo IV (17) and human Ape1 (43). On the other hand, the Km and catalytic efficiency values for the 3′→5′ exonuclease activity of the viral enzyme on the nicked substrate with a mismatch were 180 nM and 0.45 min−1 nM−1, respectively, which are in the range of those found for the endonucleolytic activity.
Expression and immunolocalization of ASFV AP endonuclease in infected cells.
In order to explore the role of the viral AP endonuclease during infection, we first analyzed the expression of this enzyme in Vero cells infected with the BA71V strain of ASFV. As shown in Fig. 5, Western blot analysis of extracts from infected cells using an antibody prepared against the purified recombinant pE296R protein revealed the presence of a specific band migrating at the expected position for the pE296R protein. This band was first observed at 6 h postinfection (hpi) and accumulated for up to 20 hpi. The protein was expressed in the presence of cytosine arabinoside (AraC), an inhibitor of viral DNA replication and late transcription, indicating that it is an early protein, although it was also present at late times postinfection. On the other hand, as also shown in this figure, the protein is not a structural component of the virus particle.
FIG. 5.

Western blot analysis of cell extracts from ASFV-infected cells with the anti-pE296R antibody. Immunoblotting was carried out for mock-infected (mock) and ASFV-infected cells harvested at the indicated times postinfection. Results obtained with cells infected for 16 h in the presence of 40 μg/ml of cytosine arabinoside (AraC) are also shown. A sample of 2 μg of highly purified ASFV (lane ASFV) was also analyzed. The band corresponding to the pE296R protein is indicated by an arrow. The levels of β-actin are also shown as a loading control.
We next determined the localization of the pE296R protein by immunofluorescence analysis with the anti-pE296R antibody. The cells were also stained with an anti-tubulin antibody to identify the cytoplasm and with an antibody against the viral late protein p54 as a marker of viral factories (30). Confocal images of mock-infected and ASFV-infected cells are shown in Fig. 6. Essentially no background signal was detected in mock-infected cells with the anti-pE296R antibody. At early times postinfection (6 and 8 hpi), the viral AP endonuclease was found in both the nucleus and the cytoplasm, while at later times (12 and 16 hpi) the protein was found only in the cytoplasm, where it appeared dispersed throughout but also localized in the viral factories. Interestingly, in the presence of AraC, the viral AP endonuclease was retained in the nucleus, possibly as a consequence of the blockage in virus replication. The strong signal detected in the nucleus under these conditions suggests that the disappearance of the AP endonuclease from the nucleus at late times in the absence of AraC is not due to degradation of the protein, but rather to its exit from the nucleus at these times. It can also be seen in this figure that no signal was detected with the anti-pE296R antibody in cells infected with a deletion mutant lacking this protein (see below), indicating that the signal observed was not due to a cellular protein that might be induced during infection.
FIG. 6.

Subcellular localization of pE296R protein in ASFV-infected Vero cells by immunofluorescence microscopy analysis. Mock-infected (mock) or ASFV-infected Vero cells were fixed at the indicated times postinfection (hpi) and labeled with anti-tubulin, anti-p54, and anti-pE296R. Antigens were visualized with secondary antibodies coupled to Alexa 697 (tubulin, blue channel), 594 (p54, green channel), and 488 (pE296R, red channel). Cells infected in the presence of AraC were examined at 8, 12, and 16 hpi. A cell infected with the ASFV deletion mutant vΔE296R is also shown. Samples were examined with a Bio-Rad Radiance 2000 confocal laser scanning microscope.
We also tested whether leptomycin B, an inhibitor of the CMR1-dependent pathway of nuclear export (19), blocks the release of the viral AP endonuclease from the nucleus. We found that in contrast with the case for AraC, treatment of the infected cells with 20 ng/ml of leptomycin B did not cause a significant retention of the viral enzyme in the nucleus, suggesting that the CRM1-dependent route does not play a major role in the exit of the viral enzyme from the nucleus at late times postinfection (not shown).
Replication of the deletion mutant vΔE296R in Vero cells and swine macrophages.
To further examine the function of the ASFV AP endonuclease in infection, we constructed a recombinant E296R gene deletion mutant (vΔE296R) as described in Materials and Methods. The vΔE296R deletion mutant was generated by homologous recombination in cultured Vero cells, which indicates that the viral AP endonuclease is not an essential protein for virus replication in this cell type. However, the viral endonuclease, if it is indeed involved in a reparative BER pathway, might be required for virus replication in its natural host cell, the swine macrophage, which produces and releases ROS in response to stimulation by various agents (10). This oxidative and potentially genotoxic environment might cause lesions in the viral DNA whose repair via a BER system could be essential to prevent a deleterious effect on the virus. We therefore compared the growth of the parental BA71V virus with that of the deletion mutant vΔE296R in both Vero cells and swine macrophages. As shown in Fig. 7A, the mutant and parental viruses replicated with the same kinetics and essentially to the same extent in Vero cells, confirming that deletion of the AP endonuclease gene has no effect on ASFV replication in this cell type. In contrast, the total yield of mutant virus in cultured porcine macrophages was reduced to 2 to 4% that of the parental virus (Fig. 7B). This finding indicates that the viral AP endonuclease plays an essential role in virus growth in its natural target cell.
FIG. 7.

(A and B) Growth curves of parental BA71V and mutant vΔE296R viruses in Vero cells (A) and swine macrophage cultures (B). Vero cells or swine macrophages were infected with BA71V or vΔE296R at an MOI of 2 PFU per cell. At different times postinfection, samples were collected and titrated by plaque assay on fresh Vero cells as previously described (8). (C) Viral DNA synthesis in macrophages infected with parental BA71V or mutant vΔE296R virus. Swine macrophages infected with the parental or mutant virus as described for panel B, as well as mock-infected cultures, were labeled with 5 mCi/ml of [3H]thymidine (80 Ci/mmol) for a 12-h period. At the indicated times postinfection, samples were collected, and the acid-insoluble radioactivity was measured. (D) Expression of late viral proteins in macrophages infected with parental or mutant virus. Macrophage cultures were mock infected or infected as described for panel B, and at the indicated times, the expression of the late viral proteins pE248R and pB119L was examined by Western blotting using specific antibodies against these proteins (28). The positions of the pE248R and pB119L proteins, as well as β-actin as a loading control, are indicated by arrows. Molecular size markers are indicated on the left.
To investigate the stage at which the growth of the ASFV deletion mutant in macrophages is blocked, we first compared the extents of virus DNA synthesis in cells infected with the parental and mutant viruses. For this comparison, the infected macrophages were labeled with [3H]thymidine for a 12-h period, and the acid-insoluble radioactivity was measured at different times postinfection. Figure 7C shows that although there was a slight delay in the onset of viral DNA replication in cells infected with the deletion mutant, the amount of viral DNA synthesized at 10 to 12 hpi was about 70% of that obtained with the parental virus. This difference probably does not account for the strong decrease in the production of infectious progeny virus.
We then analyzed the expression of late viral proteins in macrophages infected with the parental and mutant viruses by determining by Western blotting the levels of the structural protein pE248R and the nonstructural protein pB119L (28) at different times postinfection. As shown in Fig. 7D, the expression of these two proteins was considerably reduced in macrophages infected with the deletion mutant virus compared with the parental BA71V virus. This defect in late protein expression observed with the ASFV deletion mutant may significantly contribute to the reduction in virus yield.
DISCUSSION
Our results show that ASFV protein pE296R, predicted to be a class II AP endonuclease, possesses endonucleolytic activity specific for abasic sites. Biochemical characterization of the purified recombinant enzyme, with determination of the kinetic parameters for the endonucleolytic reaction on AP substrates, has shown that the Km and catalytic efficiency values for the viral AP endonuclease are in the range of those reported for E. coli endo IV (17) and human Ape1 (43). In addition to the endonuclease activity, the viral enzyme has, as do other class II AP endonucleases, a 3′→5′ exonuclease activity, with Km and catalytic efficiency values comparable to those found for the endonucleolytic activity. Several features of the 3′→5′ exonuclease activity of the viral enzyme are of special interest. The activity is essentially restricted to the last nucleotide at the 3′ end and is considerably more efficient at eliminating a mismatch than a correctly paired base. These properties make the enzyme most suitable for performing a proofreading function during viral DNA repair by a BER system.
The resolution of the E. coli endo IV three-dimensional structure (15) demonstrated that it is a single-domain αβ protein with secondary structure elements arranged as a β-barrel, with eight parallel β-strands surrounded by eight peripheral α-helices (Fig. 8A). The alignment shown in Fig. 1 and the ASFV pE296R three-dimensional structure prediction made by homology modeling using endo IV allowed us to deduce that the viral protein maintains the α8β8 folding (Fig. 8B). The main differences are due to an insertion of two amino acids in the β1α1 loop and to a gap in the α3 helix and β4α4 loop of the pE296R protein (Fig. 1). This loop, as well as the β5α5 loop, is not well conserved in the ASFV protein, which leads to the loss of βA and βB strands. The β1α1 loop, as can be seen in Fig. 8B, could interfere to some extent with the binding of the pE296R protein to DNA, which is shown superimposed in the figure. However, a similar loop is also found in the three-dimensional structure of B. anthracis endo IV, which has recently been resolved (accession no. 1XP3 in the RCSB Protein Data Bank [M. J. Fogg, V. M. Levdikov, E. V. Blagova, J. A. Brannigan, A. J. Wilkinson, and K. S. Wilson, unpublished data]). In this regard, it should be mentioned that a model of the pE296R protein that we built using the endonuclease of B. anthracis as a template (not shown) also maintains folding similar to that of the E. coli protein-based model.
FIG. 8.

Three-dimensional structure prediction for ASFV pE296R protein by homology modeling inferred from the structure of E. coli endo IV. The structure of endo IV bonded to AP site-containing DNA (15) (A and C) was used as a template (accession no. 1QUM in the RCSB Protein Data Bank), and the structure was predicted for the ASFV pE296R protein (B and D), with DNA strands shown as blue lines, α helices shown as red coils, and β strands shown as yellow arrows. Zn atoms are indicated by dots (A and B). The AP site-containing DNA from the 1QUM structure has been superimposed on the pE296R protein model in panels B and D. The ASFV protein pE296R model was obtained with the SwissModel server (34), using the alignment shown in Fig. 1. Panels C and D show the high similarity between catalytic center residue dispositions in the 1QUM template (C) and the model obtained for the ASFV pE296R protein (D).
Endo IV contains a trinuclear Zn2+ cluster at its active site, and because the phosphodiester bond of the sugar-phosphate backbone is always cleaved by a three-metal-ion mechanism, all of the repair activities of endo IV have been explained by the cleavage activity of this active site (15). In endo IV, the AP site is stabilized by stacking interactions of the enzyme with both 5′ and 3′ base pairs, with residues Tyr72 and Leu73 at the 5′ side and Arg37 at the 3′ side filling up the AP site and bracing DNA in a dramatically bent conformation. Again, in the modeled viral protein, the conserved residues in the catalytic center are located in the same positions as those in endo IV (compare Fig. 8C and D). Because of this catalytic center disposition, the E. coli enzyme does not make specific contacts with the base opposite the AP site, which is consistent with the finding in biochemical studies that E. coli endo IV shows no preference for the base opposite the AP site (16, 17). However, we have found that the 3′→5′ exonuclease activities of both endo IV of E. coli and ASFV protein pE296R show a strong preference for mismatched base pairs. This may suggest the existence in the two enzymes of recognition mechanisms which discriminate between correctly and incorrectly paired bases. In summary, the three-dimensional prediction for ASFV protein pE296R underscores the structural similarities between E. coli endo IV and the viral enzyme, which support a common mechanism for the cleavage reaction.
The ASFV AP endonuclease, together with the viral Pol X and ligase, could act in a short-patch BER-like system to maintain the integrity of the virus genome in the potentially genotoxic environment of the macrophage, the main target cell in natural infections. Macrophages are known to generate ROS in response to a variety of stimuli (10). These free radicals could cause lesions in the bases of the viral DNA that, if uncorrected, would have a deleterious effect on the virus. The efficiency of a BER system in preventing DNA damage will rely on the accuracy of the repair reaction. In relation to this, a previous biochemical characterization of ASFV Pol X in our laboratory (14) showed that its nucleotide insertion fidelity is comparable to that of mammalian Pol β (1, 5). The present finding that the viral AP endonuclease has a 3′→5′ exonuclease activity raises the possibility that the enzyme could also provide a proofreading activity to correct possible errors made by the viral reparative polymerase during the gap-filling step, thus increasing the fidelity of the viral BER system.
Since the ASFV reparative polymerase is expressed at late times postinfection (27), a Pol X-dependent short-patch BER would act only at these times, possibly to correct misincorporations during viral DNA replication or to protect already replicated genomes. The viral AP endonuclease, which is present in the infected cell at these late times and is in part localized in the virus factory, could participate in such a late viral BER process. It should also be taken into account that, unexpectedly, the viral endonuclease was found to be expressed at early times of infection. In contrast with the exclusively cytoplasmic localization of the protein at late times postinfection, the early enzyme is present in both the cytoplasm and the nucleus. The function of the nuclear early enzyme is intriguing. In relation to this, it should be mentioned that human Ape1 also functions in reductively activating certain transcription factors, such as c-Jun and p53 (9, 45). However, the viral enzyme lacks the Cys65 residue involved in the redox function of human Ape1 (41). On the other hand, the possibility that the ASFV AP endonuclease could play, in addition to a role in DNA repair, an essential function in virus replication is unlikely, since the enzyme is not required for virus growth in Vero cells. It is thus possible that the early endonuclease could perform a repair role related to the nuclear phase of viral DNA replication. The finding that the enzyme disappears from the nucleus at the times when the nuclear stage of replication is completed (13, 32), together with the observation that it is retained in this compartment under conditions that block viral DNA replication, is in line with this possibility. A repair system that might function to protect the viral DNA in the nucleus would, however, be Pol X independent and might involve the participation of the viral replicative DNA polymerase, which is expressed at early times postinfection (31). Further work will be needed to clarify this question.
The existence of a viral reparative system in infected cells is supported by the results obtained with the ASFV deletion mutant lacking the AP endonuclease gene. The finding that the viral endonuclease is needed for virus growth in swine macrophages but not in Vero cells suggests that this requirement is related to the induction of more extensive damage in the viral DNA due to the highly oxidative environment of the macrophage. In the absence of the viral AP endonuclease, the oxidative lesions caused in the virus DNA might not be repaired, which would lead to a decrease in the production of infectious progeny virus. The block in the growth of the deletion mutant may be due, at least in part, to a defect in late protein expression, possibly originating by alterations in viral gene transcription as a consequence of accumulated DNA lesions. However, the packaging of mutated genomes in progeny viruses might also contribute to the reduction in virus yield. In relation to these observations, it is interesting that endo IV has been reported as a DNA damage-inducible essential factor for oxidative injury resistance in E. coli (21, 26).
Our observations with the ASFV deletion mutant lacking the AP endonuclease are not in agreement with the proposal made by others that the ASFV BER system is a mutagenic system that would account for virus variability (35, 36). Instead, our findings support the existence of an ASFV reparative BER system to maintain the viability of the virus in the infected macrophage.
Acknowledgments
We thank P. Gómez-Puertas for helpful comments on three-dimensional structure prediction for the ASFV AP endonuclease.
This study was supported by grants from the Spanish Ministerio de Educación y Ciencia (BFU2004-00298) and the European Commission (QLK2-CT-2001-02216) and by an institutional grant from Fundación Ramón Areces. M. Redrejo-Rodríguez was a predoctoral fellow of the Ministerio de Educación y Ciencia.
REFERENCES
- 1.Ahn, J., V. S. Kraynov, X. Zhong, B. G. Werneburg, and M. D. Tsai. 1998. DNA polymerase beta: effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem. J. 331:79-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett, R. A., D. M. Wilson III, D. Wong, and B. Demple. 1997. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc. Natl. Acad. Sci. USA 94:7166-7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrascosa, A. L., M. del Val, J. F. Santarén, and E. Viñuela. 1985. Purification and properties of African swine fever virus. J. Virol. 54:337-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrascosa, A. L., J. F. Santarén, and E. Viñuela. 1982. Production and titration of African swine fever virus in porcine alveolar macrophages. J. Virol. Methods 3:303-310. [DOI] [PubMed] [Google Scholar]
- 5.Chagovetz, A. M., J. B. Sweasy, and B. D. Preston. 1997. Increased activity and fidelity of DNA polymerase beta on single-nucleotide gapped DNA. J. Biol. Chem. 272:27501-27504. [DOI] [PubMed] [Google Scholar]
- 6.Chou, K. M., and Y. C. Cheng. 2002. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature 415:655-659. [DOI] [PubMed] [Google Scholar]
- 7.Dianova, I. I., V. A. Bohr, and G. L. Dianov. 2001. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry 40:12639-12644. [DOI] [PubMed] [Google Scholar]
- 8.Enjuanes, L., A. L. Carrascosa, M. A. Moreno, and E. Viñuela. 1976. Titration of African swine fever (ASF) virus. J. Gen. Virol. 32:471-477. [DOI] [PubMed] [Google Scholar]
- 9.Evans, A. R., M. Limp-Foster, and M. R. Kelley. 2000. Going APE over ref-1. Mutat. Res. 461:83-108. [DOI] [PubMed] [Google Scholar]
- 10.Forman, H. J., and M. Torres. 2001. Redox signaling in macrophages. Mol. Aspects Med. 22:189-216. [DOI] [PubMed] [Google Scholar]
- 11.Friedberg, E. C., and F. D. Wood. 1996. DNA excision repair pathways, p. 249-266. In M. DePamphilis (ed.), DNA replication in eukaryotic cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 12.García, R., F. Almazán, J. M. Rodríguez, M. Alonso, E. Viñuela, and J. F. Rodríguez. 1995. Vectors for the genetic manipulation of African swine fever virus. J. Biotechnol. 40:121-131. [DOI] [PubMed] [Google Scholar]
- 13.García-Beato, R., M. L. Salas, E. Viñuela, and J. Salas. 1992. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 188:637-649. [DOI] [PubMed] [Google Scholar]
- 14.García-Escudero, R., M. García-Díaz, M. L. Salas, L. Blanco, and J. Salas. 2003. DNA polymerase X of African swine fever virus: insertion fidelity on gapped DNA substrates and AP lyase activity support a role in base excision repair of viral DNA. J. Mol. Biol. 326:1403-1412. [DOI] [PubMed] [Google Scholar]
- 15.Hosfield, D. J., Y. Guan, B. J. Haas, R. P. Cunningham, and J. A. Tainer. 1999. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell 98:397-408. [DOI] [PubMed] [Google Scholar]
- 16.Ide, H., K. Tedzuka, H. Shimzu, Y. Kimura, A. A. Purmal, S. S. Wallace, and Y. W. Kow. 1994. Alpha-deoxyadenosine, a major anoxic radiolysis product of adenine in DNA, is a substrate for Escherichia coli endonuclease IV. Biochemistry 33:7842-7847. [DOI] [PubMed] [Google Scholar]
- 17.Ishchenko, A. A., H. Ide, D. Ramotar, G. Nevinsky, and M. Saparbaev. 2004. Alpha-anomeric deoxynucleotides, anoxic products of ionizing radiation, are substrates for the endonuclease IV-type AP endonucleases. Biochemistry 43:15210-15216. [DOI] [PubMed] [Google Scholar]
- 18.Kerins, S. M., R. Collins, and T. V. McCarthy. 2003. Characterization of an endonuclease IV 3′-5′ exonuclease activity. J. Biol. Chem. 278:3048-3054. [DOI] [PubMed] [Google Scholar]
- 19.Kudo, N., B. Wolff, T. Sekimoto, E. P. Schreiner, Y. Yoneda, M. Yanagida, S. Horinouchi, and M. Yoshida. 1998. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242:540-547. [DOI] [PubMed] [Google Scholar]
- 20.La Scola, B., S. Audic, C. Robert, L. Jungang, X. de Lamballerie, M. Drancourt, R. Birtles, J. M. Claverie, and D. Raoult. 2003. A giant virus in amoebae. Science 299:2033. [DOI] [PubMed] [Google Scholar]
- 21.Levin, J. D., and B. Demple. 1996. In vitro detection of endonuclease IV-specific DNA damage formed by bleomycin in vivo. Nucleic Acids Res. 24:885-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindahl, T. 1993. Instability and decay of the primary structure of DNA. Nature 362:709-715. [DOI] [PubMed] [Google Scholar]
- 23.Lindahl, T. 1990. Repair of intrinsic DNA lesions. Mutat. Res. 238:305-311. [DOI] [PubMed] [Google Scholar]
- 24.Lindahl, T., and R. D. Wood. 1999. Quality control by DNA repair. Science 286:1897-1905. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto, Y., and K. Kim. 1995. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 269:699-702. [DOI] [PubMed] [Google Scholar]
- 26.Nunoshiba, T., T. DeRojas-Walker, J. S. Wishnok, S. R. Tannenbaum, and B. Demple. 1993. Activation by nitric oxide of an oxidative-stress response that defends Escherichia coli against activated macrophages. Proc. Natl. Acad. Sci. USA 90:9993-9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliveros, M., R. J. Yáñez, M. L. Salas, J. Salas, E. Viñuela, and L. Blanco. 1997. Characterization of an African swine fever virus 20-kDa DNA polymerase involved in DNA repair. J. Biol. Chem. 272:30899-30910. [DOI] [PubMed] [Google Scholar]
- 28.Rodríguez, I., M. Redrejo-Rodríguez, J. M. Rodríguez, A. Alejo, J. Salas, and M. L. Salas. 2006. African swine fever virus pB119L protein is a FAD-linked sulfhydryl oxidase. J. Virol. 80:3157-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodríguez, J. M., F. Almazán, E. Viñuela, and J. F. Rodríguez. 1992. Genetic manipulation of African swine fever virus: construction of recombinant viruses expressing the beta-galactosidase gene. Virology 188:67-76. [DOI] [PubMed] [Google Scholar]
- 30.Rodríguez, J. M., R. García-Escudero, M. L. Salas, and G. Andrés. 2004. African swine fever virus structural protein p54 is essential for the recruitment of envelope precursors to assembly sites. J. Virol. 78:4299-4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rojo, G. 1999. Ph.D. thesis. Universidad Autónoma de Madrid, Madrid, Spain.
- 32.Rojo, G., R. García-Beato, E. Viñuela, M. L. Salas, and J. Salas. 1999. Replication of African swine fever virus DNA in infected cells. Virology 257:524-536. [DOI] [PubMed] [Google Scholar]
- 33.Salas, M. L. 1999. African swine fever virus (Asfarviridae), p. 30-38. In R. Webster and A. Granof (ed.), Encyclopedia of virology, 2nd ed. Academic Press, London, United Kingdom.
- 34.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Showalter, A. K., I. J. Byeon, M. I. Su, and M. D. Tsai. 2001. Solution structure of a viral DNA polymerase X and evidence for a mutagenic function. Nat. Struct. Biol. 8:942-946. [DOI] [PubMed] [Google Scholar]
- 36.Showalter, A. K., and M. D. Tsai. 2001. A DNA polymerase with specificity for five base pairs. J. Am. Chem. Soc. 123:1776-1777. [DOI] [PubMed] [Google Scholar]
- 37.Sobol, R. W., R. Prasad, A. Evenski, A. Baker, X. P. Yang, J. K. Horton, and S. H. Wilson. 2000. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature 405:807-810. [DOI] [PubMed] [Google Scholar]
- 38.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unk, I., L. Haracska, R. E. Johnson, S. Prakash, and L. Prakash. 2000. Apurinic endonuclease activity of yeast Apn2 protein. J. Biol. Chem. 275:22427-22434. [DOI] [PubMed] [Google Scholar]
- 40.Unk, I., L. Haracska, S. Prakash, and L. Prakash. 2001. 3′-Phosphodiesterase and 3′→5′ exonuclease activities of yeast Apn2 protein and requirement of these activities for repair of oxidative DNA damage. Mol. Cell. Biol. 21:1656-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker, L. J., C. N. Robson, E. Black, D. Gillespie, and I. D. Hickson. 1993. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol. 13:5370-5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallace, F. 1997. Oxidative damage to DNA and its repair, p. 49-90. In J. Scandalios (ed.), Oxidative stress and the molecular biology of antioxidant defenses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 43.Wilson, D. M., III. 2005. Ape1 abasic endonuclease activity is regulated by magnesium and potassium concentrations and is robust on alternative DNA structures. J. Mol. Biol. 345:1003-1014. [DOI] [PubMed] [Google Scholar]
- 44.Wong, D., and B. Demple. 2004. Modulation of the 5′-deoxyribose-5-phosphate lyase and DNA synthesis activities of mammalian DNA polymerase beta by apurinic/apyrimidinic endonuclease 1. J. Biol. Chem. 279:25268-25275. [DOI] [PubMed] [Google Scholar]
- 45.Xanthoudakis, S., and T. Curran. 1992. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 11:653-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yáñez, R. J., J. M. Rodríguez, M. L. Nogal, L. Yuste, C. Enríquez, J. F. Rodríguez, and E. Viñuela. 1995. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 208:249-278. [DOI] [PubMed] [Google Scholar]
- 47.Yáñez, R. J., and E. Viñuela. 1993. African swine fever virus encodes a DNA ligase. Virology 193:531-536. [DOI] [PubMed] [Google Scholar]