

Abstract
Thrombospondin (TSP)-1, a multidomain glycoprotein, is secreted from astrocytes and promotes synaptogenesis. However, little is known about the mechanisms regulating its expression and release. In this article, we report that purinergic signaling participates in the production and secretion of TSP-1. Treatment of primary cultures of rat cortical astrocytes with extracellular ATP caused an increase in TSP-1 expression in a time- and concentration-dependent manner and was inhibited by antagonists of P2 and P1 purinergic receptors. Agonist studies revealed that UTP, but not 2′,3′-O-(4-benzoyl)benzoyl-ATP, 2-methylthio-ADP, adenosine, or 5′-N-ethyl-carboxamidoadenosine, caused a significant increase in TSP-1 expression. In addition, release of TSP-1 was stimulated by ATP and UTP but not by 2-methylthio-ADP or adenosine. Additional studies indicated that P2Y4 receptors stimulate both TSP-1 expression and release. P2Y receptors are coupled to protein kinase cascades, and signaling studies demonstrated that blockade of mitogen-activated protein kinases or Akt inhibited ATP- and UTP-induced TSP-1 expression. Using an in vitro model of CNS trauma that stimulates release of ATP, we found that TSP-1 expression increased after mechanical strain and was completely blocked by a P2 receptor antagonist and by inhibition of p38/mitogen-activated protein kinase and Akt, thereby indicating a major role for P2 receptor/protein kinase signaling in TSP-1 expression induced by trauma. We conclude that TSP-1 expression can be regulated by activation of P2Y receptors, particularly P2Y4, coupled to protein kinase signaling pathways and suggest that purinergic signaling may be an important factor in TSP-1-mediated cell-matrix and cell–cell interactions such as those occurring during development and repair.
Keywords: neuron–glia interaction, traumatic injury, nucleotide receptors, gliosis, protein kinase
Recent evidence has shifted the focus on astrocytes from primarily a passive role involved in homeostasis to a more active role in a number of key physiological and pathological interactions with neurons (1–4). One of the ways in which astrocytes can interact with neurons is by the expression or release of recognition molecules. Thrombospondins (TSPs) are large multimeric, multidomain glycoproteins that participate in cell–cell and cell–matrix interactions (5). TSPs are expressed in many tissues, but their importance in the CNS is just beginning to be understood. For instance, two recent studies have focused attention on the role of TSPs in CNS development and repair. Christopherson et al. (6) showed that application of purified TSP-1, TSP-2, or astrocyte-conditioned medium was sufficient to increase the number of synapses in retinal ganglion cells and that these synapses were presynaptically active. In addition, Lin et al. (7) reported that after ischemia, increased levels of TSP-1 were localized in astrocytes and endothelial cells near blood vessels. These studies indicate the importance of TSPs in synapse formation during development and in remodeling after CNS injury, but little is known about factors that induce expression and secretion of TSPs from astrocytes.
Nucleotides and nucleosides are released by a variety of cells (8–10) and have both short-term effects as neurotransmitters and long-term effects as trophic factors (11). The actions of extracellular ATP are a result of stimulation of P2-type purinergic receptors, which are categorized into ligand-gated ion channels (P2X1–7) or metabotropic heptahelical G protein-coupled receptors (P2Y1,2,4,6,11–14) (12). Astrocytes express both P2Y and P2X receptors (13–18), and these receptors are coupled to protein kinase cascades, including mitogen-activated protein kinases (MAPKs) and protein kinase B/Akt (14, 15, 19, 20), that mediate gene expression (21, 22).
In smooth muscle and mesangial cells, TSP-1 gene expression is rapidly induced by serum and growth factors such as platelet-derived growth factor and fibroblast growth factor-2 (23–27), but little is known about TSP-1 regulation in the brain. Platelet-derived growth factor stimulated synthesis of TSP in a human glial cell line (28) and transforming growth factor-β1 induced TSP-1 mRNA in astrocytes (29), but the potential role of extracellular ATP and P2 receptor signaling in TSP-1 expression and release in astrocytes has not been investigated. In this study, we show that extracellular ATP, through the activation of P2Y4 receptors, stimulates TSP-1 expression and release in astrocytes and that this nucleotide-induced increase is mediated by protein kinase signaling pathways. Because ATP is released after trauma (30, 31) and other types of tissue injury (10, 32), we also tested the effect of mechanical strain on TSP-1 expression and found an increase in TSP-1 that depended on activation of P2 receptors and protein kinase signaling.
Results
Activation of Astrocytic Purinergic Receptors Stimulates Expression and Release of TSP-1.
To determine whether TSP-1 expression could be regulated by purinergic signaling, we conducted time-course and concentration–response experiments using extracellular ATP. Immunoblots and densitometric analyses (Fig. 1A) showed that extracellular ATP significantly increased TSP-1 expression by 19- and 30-fold at 6 and 12 h, respectively. Immunoblot analysis also showed that TSP-1 expression depended on the concentration of extracellular ATP; treating cultures with ≥100 μM extracellular ATP resulted in a significant increase in TSP-1 level (Fig. 1B).
Fig. 1.

Extracellular ATP increases TSP-1 protein expression. (A and B) Immunoblot analyses of TSP-1 expression. After extracellular ATP treatment, TSP-1 expression increased in a time- (A; n = 3) and concentration-dependent (B; n = 4) manner. Purified TSP-1 from platelets was used as a positive control (Pos) for the TSP-1 antibody. β-Actin is a loading control. Line and bar graphs show fold stimulation of TSP-1 protein levels compared with vehicle-treated controls after ATP treatment (∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001). CON, control. (C) Immunocytochemistry of cultured rat astrocytes. ATP-treated cells (100 μM; 6 h) exhibited strong TSP-1 expression (d and f) compared with vehicle-treated controls (b and e). GFAP was also increased in ATP-treated cells (c) compared with vehicle-treated controls (a). Subcellular analysis showed that ATP-induced TSP-1 increase is mainly localized in the cytoplasm (f). Images a and b, and c and d, are from the same fields, respectively. Red, TSP-1; green, GFAP; blue, DAPI. (Scale bars: a–d, 10 μm; e and f, 50 μm.)
Immunocytochemical experiments confirmed the effect of extracellular ATP on TSP-1 expression. Vehicle-treated astrocytes show very weak TSP-1 immunoreactivity (Fig. 1C b and e). Addition of extracellular ATP for 6 h led to a pronounced increase in TSP-1 immunoreactivity (Fig. 1Cd) with expression concentrated mainly in the cytoplasm (Fig. 1Cf). Consistent with previous reports (33, 34), extracellular ATP treatment also led to an increase in glial fibrillary acidic protein (GFAP) expression when compared with vehicle-treated controls (Fig. 1C a and c).
Extracellular ATP exerts its effects by activating P2 purinergic receptors. Because of the presence of ectonucleotidases on astrocytes (35, 36), ATP can be degraded to ADP, AMP, and adenosine, which could lead to activation of P1 purinergic receptors. As one approach to assess the involvement of P2 and P1 receptors in TSP-1 expression, astrocyte cultures were treated with P2 or P1 receptor antagonists before addition of extracellular ATP. We found that antagonists of P2 receptors, reactive blue 2 (RB2) and pyridoxalphosphate-6-azophenyl-2′-4′-disulfonic acid (PPADS), or the P1 receptor antagonist 8-(p-sulfophenyl)-theophylline caused an 80% or 50% decrease in TSP-1 expression, respectively (Fig. 2A). As another approach, we used apyrase, an ATP diphosphohydrolase that metabolizes ATP to AMP. Cultures treated with apyrase before ATP treatment showed weak TSP-1 immunoreactivity (Fig. 2Bd) when compared with ATP-treated cultures in the absence of apyrase (Fig. 2Bb). This finding indicates that, although P1 receptors may be coupled to TSP-1 production, degradation of ATP to AMP or adenosine is not required to induce expression of TSP-1. In addition, GFAP immunoreactivity was less intense in cultures treated with apyrase and ATP (Fig. 2Bc) when compared with ATP-treated cultures without apyrase (Fig. 2Ba).
Fig. 2.

ATP-induced TSP-1 expression is mediated by purinergic receptors. Astrocytes were treated with P2 antagonists (RB2 or PPADS; 50 μM; 15 min) or a P1 antagonist [8-(p-sulfophenyl)-theophylline; (8-PSPT); 10 μM; 15 min] before addition of ATP (100 μM; 6 h). (A) Immunoblotting shows that RB2, PPADS, or 8-(p-sulfophenyl)-theophylline decreased the expression of TSP-1 induced by ATP. GRB-2 is a loading control. The bar graph shows fold stimulation of TSP-1 protein levels compared with ATP-treated cultures after antagonist treatment (∗, P < 0.05; ∗∗∗, P < 0.001; n = 8). (B) Immunocytochemistry of cultured rat astrocytes after apyrase and ATP treatment. Astrocytes were treated with apyrase (90 units; 15 min), an ATP diphosphohydrolase that metabolizes ATP to AMP, before addition of 100 μM ATP for 6 h. Apyrase-treated cultures exhibited weak TSP-1 immunoreactivity (d) when compared with ATP-treated cultures (b). In addition, apyrase-treated cultures also showed weak GFAP immunoreactivity (c) when compared with ATP-treated cultures (a). Corresponding images in left and right panels are from the same fields. Red, TSP-1; green, GFAP; blue, DAPI. (Scale bar: 50 μm.)
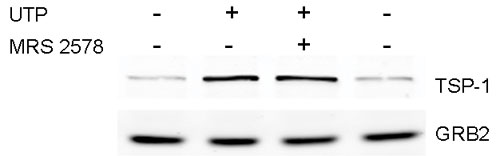
Rat cortical astrocytes express both P2Y and P2X receptors. To determine whether one or both types of P2 receptors are involved in the ATP-mediated increase of TSP-1 expression, various P2 agonists were used. We found that UTP, an agonist of P2Y2, P2Y4, and P2Y6 receptors, caused a robust (92-fold) increase in TSP-1 expression, whereas 2-methylthio-ADP (2MeSADP), an agonist of P2Y1 receptors, and 2′,3′-O-(4-benzoyl)benzoyl-ATP (BzATP), an agonist of P2X receptors, were weak or ineffective (Fig. 3A). In addition, neither adenosine nor N-ethylcarboxamidoadenosine, agonists of P1 receptors, significantly increased TSP-1 protein expression (Fig. 3A). We also found that ATP and UTP significantly increased TSP-1 release by an average of 2.3- and 3.5-fold, respectively (Fig. 3B). However, the P2Y1 agonist 2MeSADP and the P1 agonist, adenosine, did not stimulate release of TSP-1. UTP can activate three P2 receptors, P2Y2, P2Y4, and P2Y6. Several observations shed light on the identity of the P2Y receptor subtype(s) activated by UTP. First, RB2 is an antagonist of rat P2Y4 receptors, but not P2Y2 receptors (37), and we found that RB2 reduced the expression and release of TSP-1 induced by UTP (see Fig. 7 A and B, which is published as supporting information on the PNAS web site). Secondly, BzATP is an agonist for P2Y2 receptors but not for P2Y4 receptors (37); BzATP did not significantly induce TSP-1 expression (Fig. 3). Thirdly, an antagonist of P2Y6 receptors, MRS2578 (38), did not significantly inhibit expression of TSP-1 induced by UTP (P = 0.37; n = 4; see Fig. 8, which is published as supporting information on the PNAS web site). These findings point to a role for P2Y4 receptors in the expression of TSP-1, although other P2 receptors may be involved (see Discussion).
Fig. 3.

P2Y receptors mediate ATP-induced expression and secretion of TSP-1. (A) Astrocytes were treated with UTP (100 μM), BzATP (100 μM), 2MeSADP (10 μM), adenosine (100 μM), or N-ethylcarboxamidoadenosine (NECA; 10 μM) for 6 h, and cell lysates were collected. Immunoblotting and quantitation of data in the bar graph show that UTP caused a robust increase in TSP-1 expression (≈90-fold; ∗∗∗, P < 0.001; n = 7), whereas significant differences were not observed for other agonists tested. (B) Astrocytes were treated with 100 μM ATP, 100 μM UTP, 10 μM 2MeSADP, or 100 μM adenosine for 6 h, after which time media were collected and concentrated. Immunoblots and quantitation of data in bar graph show that ATP and UTP, but not 2MeSADP or adenosine, caused a significant increase in TSP-1 release (∗∗∗, P < 0.001; n = 3). Equal loading for media was confirmed with Coomassie blue staining (data not shown).
Signaling from P2 Receptors to Protein Kinases Mediates Expression of TSP-1.
Protein kinase cascades are key regulators of gene expression (21, 22), but little is known about their involvement in TSP-1 expression. Because P2 receptors in astrocytes are coupled to extracellular signal-regulated protein kinase (ERK) and p38/MAPK, both members of the MAPK family, as well as protein kinase B/Akt (14, 15, 19, 20), we examined the role of P2 receptor/protein kinase signaling in the expression of TSP-1. Time-course studies confirmed that P2 receptors stimulated ERK, Akt, and p38/MAPK and demonstrated that P2 receptors are also coupled to the stress-activated protein kinase/c-Jun N-terminal protein kinase (SAPK/JNK) (see Fig. 9, which is published as supporting information on the PNAS web site). To assess the role of P2 receptor/protein kinase signaling in TSP-1 expression, we conducted a series of experiments with protein kinase inhibitors. We used UO126, an inhibitor of mitogen-activated protein kinase kinase, the upstream activator of ERK (39), wortmannin, an inhibitor of phosphatidylinositol 3-kinase, the upstream activator of Akt (40), SB202190, an inhibitor of p38/MAPK (41), or SP600125, an inhibitor of SAPK/JNK (42). First, we tested the selectivity of these inhibitors in our system. UO126, wortmannin, and SP600125 selectively inhibit ERK, Akt, and SAPK/JNK signaling, respectively, whereas SB202190 can inhibit both p38/MAPK and Akt signaling, but not ERK and SAPK/JNK (see Fig. 10, which is published as supporting information on the PNAS web site).
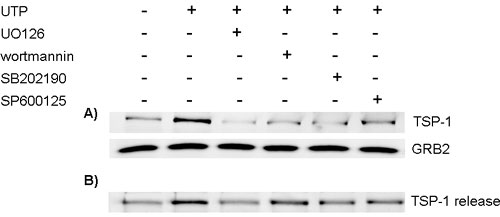
To test the role of these protein kinase signaling pathways in the ATP-induced TSP-1 expression, cells were treated with UO126, wortmannin, SB202190, or SP600125 before treating with extracellular ATP. Blocking ERK, Akt, p38/MAPK, and SAPK/JNK pathways significantly decreased the ATP-mediated increase of TSP-1 by 53.8%, 74.6%, 100%, and 67.4%, respectively (Fig. 4), thereby suggesting that the regulation of TSP-1 expression by P2 receptors is mediated by multiple protein kinase cascades, particularly p38/MAPK and Akt. The drugs by themselves did not affect TSP-1 levels (Fig. 4A). Similar results were obtained when cells were treated with UTP, although ERK may play a more prominent role in the induction of TSP-1 by UTP than by the broad-spectrum agonist ATP (see Fig. 11A, which is published as supporting information on the PNAS web site). In addition to reducing TSP-1 expression, UO126, wortmannin, SB202190, and SP600125 reduced the release of TSP-1 (see Fig. 11B).
Fig. 4.

ATP-induced expression of TSP-1 is mediated by protein kinase signaling. Astrocytes were treated with UO126 (10 μM), wortmannin (100 nM), SB202190 (10 μM), or SP600125 (10 μM) for 30 min before the addition of ATP (100 μM; 6 h). (A) Immunoblotting shows involvement of ERK, Akt, p38//MAPK, and SAPK/JNK in the ATP-induced TSP-1 expression. GRB-2 is a loading control. (B) Quantitation of data in the bar graph shows that UO126 (∗, P < 0.05; n = 9), wortmannin (∗∗, P < 0.01; n = 9), SB202190 (∗∗∗, P < 0.001; n = 5), and SP600125 (∗, P < 0.05; n = 5) significantly decreased ATP-induced expression of TSP-1.
Mechanical Strain Increases TSP-1 Protein Expression via P2 Receptor Activation and Protein Kinase Signaling.
Tissue injury, such as caused by trauma, leads to the release of ATP (30, 31). To examine the potential significance of the induction of TSP-1 by P2 receptor/protein kinase signaling, we used an in vitro cell culture model of traumatic CNS injury developed by Ellis et al. (43). In previous studies with this model, we demonstrated that biaxial mechanical strain (stretch) led to the rapid release of ATP from astrocytes, which in turn stimulated P2 receptors coupled to ERK and Akt (31, 44). Here, we found that, when cultured astrocytes grown on a deformable membrane were subjected to a rapid and reversible stretch injury, TSP-1 levels were significantly increased after 6 and 12 h (Fig. 5 A and B). Consistent with this finding, immunocytochemical experiments demonstrated that unstretched controls exhibited very weak TSP-1 immunoreactivity (Fig. 5D), whereas stretched astrocytes exhibited robust TSP-1 staining, which was concentrated in the cytoplasm (Fig. 5F). In addition, after mechanical strain astrocytes became reactive as shown by their stellate morphology, longer processes, and stronger GFAP immunoreactivity (compare Fig. 5 C and E).
Fig. 5.

Mechanical strain increases TSP-1 expression. Astrocytes grown on deformable SILASTIC membranes were subjected to a rapid and reversible stretch (50-ms, 7.5-mm membrane displacement), and at various times later, TSP-1 expression was analyzed by immunoblotting (A and B) or immunocytochemistry (C–F). (A) Immunoblotting shows a time-dependent increase in TSP-1 expression after injury. β-Actin is a loading control. (B) Quantitation of data shows TSP-1 expression significantly increased 6 and 12 h (∗∗∗, P < 0.001; n = 3) after injury and returned to noninjured levels by 24 h. (C–F) Immunocytochemical experiments of uninjured cells or 6 h after injury show that mechanical strain increased TSP-1 expression (F) when compared with uninjured controls (D). In addition, after mechanical strain cells were more stellate and had longer processes and more intense GFAP immunoreactivity (E) than uninjured controls (C). Corresponding images in Left and Right are from the same fields. Red, TSP-1; green, GFAP; blue, DAPI. (Scale bars: 10 μm.)
To examine the involvement of P2 receptors or protein kinase signaling in the strain-induced expression of TSP-1, astrocytes were treated with the P2 receptor antagonist PPADS or the p38/MAPK and Akt inhibitor SB202190 before trauma. When P2 receptors were blocked, TSP-1 expression was significantly reduced compared with that in stretched cells, TSP-1 levels being similar to those in unstretched cells (Fig. 6A). In addition, SB202190 markedly inhibited stretch-induced expression of TSP-1 (Fig. 6B), thereby indicating that signaling from P2 receptors and protein kinase cascades regulates TSP-1 expression after trauma.
Fig. 6.

Mechanical strain-induced TSP-1 expression is mediated by P2 receptors and protein kinase signaling. Astrocytes grown on deformable silicone membranes were treated with the P2 receptor antagonist PPADS (50 μM; 15 min) or the p38/MAPK/Akt inhibitor SB202190 (10 μM; 30 min) before being subjected to mechanical strain (50-ms, 7.5-mm membrane displacement). After 6 h, cell lysates were collected. Immunoblottings and quantitation of data as bar graphs show that PPADS (A) and SB202190 (B) significantly blocked mechanical strain-induced expression of TSP-1 (∗, P < 0.05; ∗∗, P < 0.01; n = 3), indicating involvement of P2 receptor and protein kinase signaling in the expression of TSP-1 induced by trauma.
Discussion
Our findings demonstrated that extracellular ATP and mechanical strain stimulate astrocyte TSP-1 protein expression and release by activation of P2 receptors coupled to protein kinase signaling pathways, particularly p38/MAPK and Akt. In view of the emerging evidence for the participation of thrombospondin in synaptogenesis (6) and ischemia (7), our data suggest an important role for purinergic signaling in cell–cell and cell–matrix interactions during CNS development and repair.
TSP-1 is produced in a variety of cell types (5), but little is known about the mechanisms that regulate synthesis and release of TSP-1 by brain cells. Our work demonstrates that stimulation of astrocyte P2Y purinergic receptors coupled to protein kinases markedly increases TSP-1 expression and release. Agonist and antagonist studies point to a role for P2Y4 receptors. However, other P2 receptors might be involved because RB2 and PPADS did not completely block expression of TSP-1 induced by nucleotides (Fig. 2 and see Fig. 7). Future studies with short interfering RNAs and/or mice deficient in specific P2 receptor subtypes will help to further define the P2 receptors that stimulate TSP production and release. Involvement of P2Y11 receptors can be excluded because P2Y11 receptors do not appear to be functionally expressed in cortical astrocytes cultured from 1-day-old rat neonates (13). In addition, P2X receptors do not appear to stimulate TSP-1 expression. However, in other studies we observed that both P2Y and P2X receptors regulate N-cadherin expression in astrocytes (unpublished observations), thereby exemplifying the diversity of P2 receptor signaling.
P2 receptors are coupled to MAPKs and Akt (14, 15, 19, 20). Consistent with previous studies, we found that extracellular ATP was able to stimulate ERK, Akt, and p38//MAPK in astrocytes. We also demonstrated that extracellular ATP induces phosphorylation of SAPK/JNK in astrocytes. Experiments with pathway inhibitors suggest that multiple protein kinase pathways can regulate TSP-1 expression. Interestingly, SB202190 treatment resulted in complete inhibition of the ATP-induced expression of TSP-1. Because SB202190 affects both Akt and p38/MAPK phosphorylation, the complete reduction in TSP-1 induced by ATP could be a result of the combinatorial effect of blocking both Akt and p38/MAPK signaling. We have considered the possibility that MAPK pathway inhibitors may affect functions of P2Y receptors such as receptor activation or G protein coupling because it has been reported that SB202190 can directly interact with the G protein-coupled receptor subtype CCK1, although not with the CCK2 subtype (45). However, the other kinase inhibitors we have used, U0126, wortmannin, and SP600125, are structurally unrelated to SB202190. Although the selectivity of these inhibitors compared with a large number of protein kinases is very high (46), it still remains possible that they could also have other effects not yet determined. Nonetheless, because the effect of SB202190 is limited to one particular subtype of a G protein-coupled receptor and because of other considerations such as the distinct structures of the other inhibitors, the high selectivity of these inhibitors, as well as our demonstration that the inhibitors we have used to block ERK, SAPK, and Akt signaling are selective in P2 receptor signaling in astrocytes (see Fig. 10), it is reasonable to suggest that the inhibition of TSP-1 expression by these agents reflects their actions at specific protein kinases. The observation that MAPKs are involved in TSP-1 expression is in agreement with a report by Nakagawa et al. (47), who showed that TSP-1 in rat kidney proximal tubular cells and mouse fibroblasts is regulated by both ERK1/2 and p38/MAPK in response to transforming growth factor-β1.
It has recently been demonstrated that TSP-1 and TSP-2 promote synaptogenesis (6), thereby suggesting a role for TSPs in CNS development. Consistent with this finding, TSPs are expressed in the developing nervous system when synaptogenesis occurs and decline with maturation (6, 48). P2 receptors are also expressed in embryonic and postnatal brains and have been implicated in the development of the nervous system (49, 50). This finding suggests that ATP receptor signaling could be involved in synaptogenesis by inducing the synthesis and secretion of TSPs during CNS development.
Injury leads to the release of ATP from many cell types (9, 10), including astrocytes (30, 31). Our data with a well characterized in vitro model of traumatic CNS injury (43) demonstrated that TSP-1 expression is induced by mechanical strain in a manner mediated by P2 receptor/protein kinase signaling. Damage to the CNS evokes a process in which astrocytes shift into a reactive state by undergoing hypertrophic and hyperplastic changes. These characteristics of astrogliosis, as well as increased levels of GFAP, can be induced in vitro and in vivo by ATP and nucleotide analogs (33, 34, 51–54). Astrocytes, when activated, can release and/or express a variety of factors, including recognition and extracellular matrix molecules (55, 56). Because TSP-1 can enhance CNS synaptogenesis (6), an increase in TSP-1 after injury has occurred would support the idea that TSP-1 is actively involved in forming new synapses, provided the lesioned neuron can survive and regenerate through the glial scar. The timing of the increase in TSP expression would be critical. It has been reported that TSP-1 protein increased 4 and 72 h after focal cerebral ischemia/reperfusion (7). We found that TSP-1 was maximal from 6 to 12 h after trauma and returned to near basal levels 24 h after injury. An early increase in TSP-1 may be related to the enhanced plasticity often observed in areas surrounding the injured site. For example, traumatic or ischemic injuries result in changes in synapse number and structures and also dendritic growth and neurogenesis (57–61). Because TSP-1 is involved in cell–cell interactions, cell adhesion, and synaptogenesis, the early increase in TSP-1 expression after injury could support the concept that a component of the early phase of reactive astrogliosis involves an attempt to stabilize neurons and their synapses in areas surrounding the injury (62, 63). In this view, TSP-1 released by astrocytes might be one of the molecules that support neurons in areas surrounding an injury to go through periods of dendritic growth, axonal sprouting, and synaptogenesis. During a later phase of recovery, the use of hydrolysis-resistant ATP analogs may offer a new therapeutic approach to stimulate repair and remodeling.
In conclusion, TSP-1 protein expression and release is stimulated by extracellular ATP through activation of P2 receptors coupled to protein kinase signaling pathways. In addition, stretch-induced traumatic injury of brain cells leads to TSP-1 expression via activation of P2 receptors. Our findings point to a trophic role of purinergic signaling for interactions that occur between glial cells and neurons during CNS development and repair.
Materials and Methods
Cell Culture and Treatment.
Primary cultures of astrocytes were obtained from cerebral cortices of neonatal rats (Fischer), as described in ref. 34. More information is available in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Mechanical Strain-Induced Trauma.
Confluent cultures of astrocytes grown in flex plates were subjected to mechanical strain by means of a Cell Injury Controller (model 94A; Custom Design and Fabrication, Department of Radiology, Virginia Commonwealth University, Richmond), a device that regulates a pulse of compressed gas to rapidly and transiently deform the silicone membrane and adherent cells in a manner such that the magnitude and duration of the injury can be controlled (43). Before each experiment, the injury controller device was calibrated as described by the manufacturer. Cells were injured by a 50-ms, 7.5-mm displacement. These parameters have been correlated with severe trauma, as defined by studies with this in vitro stretch injury model (30). Membrane displacement generated with this model corresponds to biaxial strain, or stretch, similar to those that occur in humans after rotational acceleration–deceleration injury, as indicated by studies with gel-filled human skulls (64). Care was taken to avoid excessive handling of the flex plates to minimize release of ATP caused by fluid flow and perturbation of the SILASTIC membranes.
Immunoblotting.
Proteins from cultured rat astrocytes were processed for immunoblotting by standard procedures (see Supporting Materials and Methods).
Immunocytochemistry.
Immunocytochemistry of cultured rat astrocytes was performed 6 h after ATP treatment or mechanical strain. Cells were washed twice with Dulbecco’s PBS and processed for immunocytochemistry following standard procedures (see Supporting Materials and Methods).
Statistical Analysis.
The number of experimental replications is given in the figure legends; experiments were conducted with cultures from different seedings. Data were analyzed by Student’s t test for two groups or ANOVA for multiple groups followed by post hoc multiple comparisons (Bonferroni test) using an instat software package (GraphPad, San Diego).
Supplementary Material
Acknowledgments
We thank Dr. Brian King (University College London, London) for advice on the pharmacology of P2Y receptors and Yuan Kang and You-Fang Shi for technical assistance. This work was supported by the Department of Veteran Affairs (J.T.N.), National Institutes of Health Grants NS046651 (to J.T.N.) and NS045470 (to M.D.T.), and a Lois Pope Life Fellowship (to M.D.T.).
Abbreviations
- TSPs
thrombospondins
- MAPKs
mitogen-activated protein kinases
- GFAP
glial fibrillary acidic protein
- RB2
reactive blue 2
- PPADS
pyridoxalphosphate-6-azophenyl-2′-4′-disulfonic acid
- 2MeSADP
2-methylthio-ADP
- BzATP
2′,3′-O-(4-benzoyl)benzoyl-ATP
- ERK
extracellular signal-regulated protein kinase
- SAPK/JNK
stress-activated protein kinase/c-Jun N-terminal kinase.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Haydon P. G. Nat. Rev. Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 2.Newman E. A. Trends Neurosci. 2003;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- 3.Tekkok S. B., Ransom B. R. In: Glial–Neuornal Signaling. Hatton G. I., Parpura V., editors. Boston: Kluwer; 2004. pp. 1–20. [Google Scholar]
- 4.Auld D. S., Robitaille R. Neuron. 2003;40:389–400. doi: 10.1016/s0896-6273(03)00607-x. [DOI] [PubMed] [Google Scholar]
- 5.Adams J. C., Lawler J. Int. J. Biochem. Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christopherson K. S., Ullian E. M., Stokes C. C., Mullowney C. E., Hell J. W., Agah A., Lawler J., Mosher D. F., Bornstein P., Barres B. A. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 7.Lin T. N., Kim G. M., Chen J. J., Cheung W. M., He Y. Y., Hsu C. Y. Stroke. 2003;34:177–186. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- 8.Schwiebert E. M., Zsembery A., Geibel J. P. Curr. Top. Membr. 2003;34:31–58. [Google Scholar]
- 9.Bergfeld G. R., Forrester T. Cardiovasc. Res. 1992;26:40–47. doi: 10.1093/cvr/26.1.40. [DOI] [PubMed] [Google Scholar]
- 10.Bodin P., Burnstock G. Experientia. 1995;51:256–259. doi: 10.1007/BF01931108. [DOI] [PubMed] [Google Scholar]
- 11.Burnstock G., Williams M. J. Pharmacol. Exp. Ther. 2000;295:862–869. [PubMed] [Google Scholar]
- 12.Ralevic V., Burnstock G. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 13.Lenz G., Gottfried C., Luo Z., Avruch J., Rodnight R., Nie W. J., Kang Y., Neary J. T. Br. J. Pharmacol. 2000;129:927–936. doi: 10.1038/sj.bjp.0703138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panenka W., Jijon H., Herx L. M., Armstrong J. N., Feighan D., Wei T., Yong V. W., Ransohoff R. M., MacVicar B. A. J. Neurosci. 2001;21:7135–7142. doi: 10.1523/JNEUROSCI.21-18-07135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacques-Silva M. C., Rodnight R., Lenz G., Liao Z., Kong Q., Tran M., Kang Y., Gonzalez F. A., Weisman G. A., Neary J. T. Br. J. Pharmacol. 2004;141:1106–1117. doi: 10.1038/sj.bjp.0705685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon S. J., Yu R., Panupinthu N., Wilson J. X. Glia. 2004;47:367–376. doi: 10.1002/glia.20048. [DOI] [PubMed] [Google Scholar]
- 17.Yamakuni H., Kawaguchi N., Ohtani Y., Nakamura J., Katayama T., Nakagawa T., Minami M., Satoh M. J. Neuroimmunol. 2002;129:43–50. doi: 10.1016/s0165-5728(02)00179-0. [DOI] [PubMed] [Google Scholar]
- 18.Fumagalli M., Brambilla R., D’Ambrosi N., Volonte C., Matteoli M., Verderio C., Abbracchio M. P. Glia. 2003;43:218–230. doi: 10.1002/glia.10248. [DOI] [PubMed] [Google Scholar]
- 19.Neary J. T., Kang Y., Bu Y., Yu E., Akong K., Peters C. M. J. Neurosci. 1999;19:4211–4220. doi: 10.1523/JNEUROSCI.19-11-04211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neary J. T., Zhu Q. NeuroReport. 1994;5:1617–1620. doi: 10.1097/00001756-199408150-00019. [DOI] [PubMed] [Google Scholar]
- 21.Chang L., Karin M. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 22.Brazil D. P., Yang Z. Z., Hemmings B. A. Trends Biochem. Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Majack R. A., Cook S. C., Bornstein P. J. Cell Biol. 1985;101:1059–1070. doi: 10.1083/jcb.101.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donoviel D. B., Amacher S. L., Judge K. W., Bornstein P. J. Cell. Physiol. 1990;145:16–23. doi: 10.1002/jcp.1041450104. [DOI] [PubMed] [Google Scholar]
- 25.Bornstein P. FASEB J. 1992;6:3290–3299. doi: 10.1096/fasebj.6.14.1426766. [DOI] [PubMed] [Google Scholar]
- 26.Hugo C., Pichler R., Meek R., Gordon K., Kyriakides T., Floege J., Bornstein P., Couser W. G., Johnson R. J. Kidney Int. 1995;48:1846–1856. doi: 10.1038/ki.1995.483. [DOI] [PubMed] [Google Scholar]
- 27.Horiguchi H., Jin L., Ruebel K. H., Scheithauer B. W., Lloyd R. V. Endocrine. 2004;24:141–146. doi: 10.1385/ENDO:24:2:141. [DOI] [PubMed] [Google Scholar]
- 28.Asch A. S., Leung L. L., Shapiro J., Nachman R. L. Proc. Natl. Acad. Sci. USA. 1986;83:2904–2908. doi: 10.1073/pnas.83.9.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott-Drew S., ffrench-Constant C. J. Neurosci. Res. 1997;50:202–214. doi: 10.1002/(SICI)1097-4547(19971015)50:2<202::AID-JNR9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed S. M., Rzigalinski B. A., Willoughby K. A., Sitterding H. A., Ellis E. F. J. Neurochem. 2000;74:1951–1960. [PubMed] [Google Scholar]
- 31.Neary J. T., Kang Y., Tran M., Feld J. J. Neurotrauma. 2005;22:491–500. doi: 10.1089/neu.2005.22.491. [DOI] [PubMed] [Google Scholar]
- 32.Gourine A. V., Llaudet E., Dale N., Spyer K. M. J. Neurosci. 2005;25:1211–1218. doi: 10.1523/JNEUROSCI.3763-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abbracchio M. P., Saffrey M. J., Hopker V., Burnstock G. Neuroscience. 1994;59:67–76. doi: 10.1016/0306-4522(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 34.Neary J. T., Baker L., Jorgensen S. L., Norenberg M. D. Acta Neuropathol. (Berl.) 1994;87:8–13. doi: 10.1007/BF00386249. [DOI] [PubMed] [Google Scholar]
- 35.Lai K. M., Wong P. C. J. Neurochem. 1991;57:1510–1515. doi: 10.1111/j.1471-4159.1991.tb06345.x. [DOI] [PubMed] [Google Scholar]
- 36.Zimmermann H., Braun N. Prog. Brain Res. 1999;120:371–385. [PubMed] [Google Scholar]
- 37.Wildman S. S., Unwin R. J., King B. F. Br. J. Pharmacol. 2003;140:1177–1186. doi: 10.1038/sj.bjp.0705544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mamedova L. K., Joshi B. V., Gao Z. G., von Kugelgen I., Jacobson K. A. Biochem. Pharmacol. 2004;67:1763–1770. doi: 10.1016/j.bcp.2004.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Favata M. F., Horiuchi K. Y., Manos E. J., Daulerio A. J., Stradley D. A., Feeser W. S., Van Dyk D. E., Pitts W. J., Earl R. A., Hobbs F., et al. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 40.Hazeki O., Hazeki K., Katada T., Ui M. J. Lipid Mediat. Cell. Signal. 1996;14:259–261. doi: 10.1016/0929-7855(96)00534-2. [DOI] [PubMed] [Google Scholar]
- 41.Lee J. C., Laydon J. T., McDonnell P. C., Gallagher T. F., Kumar S., Green D., McNulty D., Blumenthal M. J., Heys J. R., Landvatter S. W. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 42.Bennett B. L., Sasaki D. T., Murray B. W., O’Leary E. C., Sakata S. T., Xu W., Leisten J. C., Motiwala A., Pierce S., Satoh Y., et al. Proc. Natl. Acad. Sci. USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellis E. F., McKinney J. S., Willoughby K. A., Liang S., Povlishock J. T. J. Neurotrauma. 1995;12:325–339. doi: 10.1089/neu.1995.12.325. [DOI] [PubMed] [Google Scholar]
- 44.Neary J. T., Kang Y., Willoughby K. A., Ellis E. F. J. Neurosci. 2003;23:2348–2356. doi: 10.1523/JNEUROSCI.23-06-02348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morel C., Ibarz G., Oiry C., Carnazzi E., Berge G., Gagne D., Galleyrand J. C., Martinez J. J. Biol. Chem. 2005;280:21384–21393. doi: 10.1074/jbc.M408851200. [DOI] [PubMed] [Google Scholar]
- 46.Davies S. P., Reddy H., Caivano M., Cohen P. Biochem. J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakagawa T., Lan H. Y., Glushakova O., Zhu H. J., Kang D. H., Schreiner G. F., Bottinger E. P., Johnson R. J., Sautin Y. Y. J. Am. Soc. Nephrol. 2005;16:899–904. doi: 10.1681/ASN.2004080689. [DOI] [PubMed] [Google Scholar]
- 48.Adams J. C., Tucker R. P. Dev. Dyn. 2000;218:280–299. doi: 10.1002/(SICI)1097-0177(200006)218:2<280::AID-DVDY4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 49.Bogdanov Y. D., Dale L., King B. F., Whittock N., Burnstock G. J. Biol. Chem. 1997;272:12583–12590. doi: 10.1074/jbc.272.19.12583. [DOI] [PubMed] [Google Scholar]
- 50.Meyer M. P., Clarke J. D., Patel K., Townsend-Nicholson A., Burnstock G. Dev. Dyn. 1999;214:152–158. doi: 10.1002/(SICI)1097-0177(199902)214:2<152::AID-AJA5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 51.Franke H., Krugel U., Grosche J., Heine C., Hartig W., Allgaier C., Illes P. Neuroscience. 2004;127:431–441. doi: 10.1016/j.neuroscience.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Franke H., Krugel U., Illes P. Glia. 1999;28:190–200. [PubMed] [Google Scholar]
- 53.Franke H., Krugel U., Schmidt R., Grosche J., Reichenbach A., Illes P. Br. J. Pharmacol. 2001;134:1180–1189. doi: 10.1038/sj.bjp.0704353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rathbone M. P., Middlemiss P. J., Kim J. K., Gysbers J. W., DeForge S. P., Smith R. W., Hughes D. W. Neurosci. Res. 1992;13:1–17. doi: 10.1016/0168-0102(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 55.Eddleston M., Mucke L. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ridet J. L., Malhotra S. K., Privat A., Gage F. H. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 57.Arvidsson A., Collin T., Kirik D., Kokaia Z., Lindvall O. Nat. Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 58.Biernaskie J., Corbett D. J. Neurosci. 2001;21:5272–5280. doi: 10.1523/JNEUROSCI.21-14-05272.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones T. A., Kleim J. A., Greenough W. T. Brain Res. 1996;733:142–148. doi: 10.1016/0006-8993(96)00792-5. [DOI] [PubMed] [Google Scholar]
- 60.Liu J., Solway K., Messing R. O., Sharp F. R. J. Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheff S. W., Price D. A., Hicks R. R., Baldwin S. A., Robinson S., Brackney C. J. Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- 62.Fitch M. T., Silver J. Cell Tissue Res. 1997;290:379–384. doi: 10.1007/s004410050944. [DOI] [PubMed] [Google Scholar]
- 63.Faulkner J. R., Herrmann J. E., Woo M. J., Tansey K. E., Doan N. B., Sofroniew M. V. J. Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shreiber D. I., Gennarelli T. A., Meaney D. F. Proceedings of the 1995 International Conference on Biomechanics of Impact; Switzerland: Brunnen; 1995. pp. 233–244. Int. Res. Conf. on Biomechanics of Impact, Bron, France. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}