

Short abstract
Tryptophan-pathway genes that exist within an apparent operon-like organization were evaluated. A seven-gene cluster in Xylella fastidiosa exhibits a sharply delineated low-GC content. This strongly implicates lateral gene transfer. In contrast, parametric studies and protein tree phylogenies did not support the origination of a gene block in the Anabaena/Nostoc lineage by lateral gene transfer.
Abstract
Background
Tryptophan-pathway genes that exist within an apparent operon-like organization were evaluated as examples of multi-genic genomic regions that contain phylogenetically incongruous genes and coexist with genes outside the operon that are congruous. A seven-gene cluster in Xylella fastidiosa includes genes encoding the two subunits of anthranilate synthase, an aryl-CoA synthetase, and trpR. A second gene block, present in the Anabaena/Nostoc lineage, but not in other cyanobacteria, contains a near-complete tryptophan operon nested within an apparent supraoperon containing other aromatic-pathway genes.
Results
The gene block in X. fastidiosa exhibits a sharply delineated low-GC content. This, as well as bias of codon usage and 3:1 dinucleotide analysis, strongly implicates lateral gene transfer (LGT). In contrast, parametric studies and protein tree phylogenies did not support the origination of the Anabaena/Nostoc gene block by LGT.
Conclusions
Judging from the apparent minimal amelioration, the low-GC gene block in X. fastidiosa probably originated by LGT at a relatively recent time. The surprising inability to pinpoint a donor lineage still leaves room for alternative, albeit less likely, explanations other than LGT. On the other hand, the large Anabaena/Nostoc gene block does not seem to have arisen by LGT. We suggest that the contemporary Anabaena/Nostoc array of divergent paralogs represents an ancient ancestral state of paralog divergence, with extensive streamlining by gene loss occurring in the lineage of descent representing other (unicellular) cyanobacteria.
Background
Lateral gene transfer
Lateral gene transfer (LGT) has been generally accepted for some time, as exemplified by the endosymbiotic hypothesis of organelle origin [1,2]. Nevertheless, a long-standing background of general conviction has held that LGT is rare, especially between distant organisms. However, the modern era of genomics has been accompanied by increasingly numerous claims that LGT is frequent [3,4,5,6], and there now seems little doubt that LGT exerts a significant influence upon evolutionary histories. Indeed, it has even been asserted that vertical evolutionary patterns of descent might be impossibly masked by rampant events of LGT and that, in fact, instead of bifurcating phylogenetic trees, a reticulate (net-like) pattern exists [7,8,9]. On the other hand, others urge a more balanced perspective, pointing out that alternative explanations for apparent cases of LGT have not always been considered [10,11,12,13,14]. The rationale for explanations other than LGT for genealogical incongruities (such as hidden paralogies and reconstruction artifacts) have been presented in comprehensive detail by Glansdorff [15].
Woese [16] contends that the rRNA tree is a valid representation of organismal genealogy, that LGT was rampant only before the initial bifurcation of the universal phylogenetic tree, and that LGT has become progressively more restricted as a function of elapsed evolutionary time. Using the aminoacyl-tRNA synthases as an example of the modular-type entities asserted to be most amenable to LGT, Woese concludes that the genealogical trace of vertical gene flow is readable, despite a significant jumbling influence of LGT. If correct, this allows the optimistic viewpoint that the complex interplay of vertical gene descent and LGT can be deciphered to yield correct evolutionary histories, provided that sufficiently detailed studies are done.
Approaches for detection of LGT events are either phylogenetic or parametric. Phylogenetic approaches depend on congruence of phylogenetic trees. Aside from technical difficulties of inferring high-quality trees, conflicts between trees under comparison are not necessarily due to LGT, but can arise from coincidental loss of divergent paralogs in different, widely spaced lineages or from convergent evolution. Parametric approaches for detection of LGT include (but are not limited to) the analysis of nucleotide composition, dinucleotide frequencies and codon usage biases. Lawrence and Ochman [17] used such parametric analysis to identify a set of Escherichia coli genes (17.5% of the genome) having putative origin by LGT, and this has stimulated much discussion. High rates of both false positives and false negatives have been asserted by others [18,19], but this is tempered by presentation of a rationale for why phylogenetic and different parametric methods detect different gene subsets [20,21,22]. A consensus seems to be emerging that the most proficient attempts to reconstruct evolutionary events will employ a multifaceted approach that combines tree inference with parametric analysis in a biological context [21,22]. Lawrence and Ochman [21] provide a number of examples of how the context of biological information can assist the analysis, and this approach is implemented herein.
If each member of a linked group of genes is already represented elsewhere in a genome, their origin by LGT is a distinct possibility, as their transfer en bloc as an operon unit would have required only a single evolutionary event. During an ongoing analysis of the genomic distribution of tryptophan-pathway genes, we observed two such cases, that is, where one set of genes was phylogenetically congruent, in contrast to the incongruence of redundant gene copies that were linked to one another. We have evaluated the evidence for the alternative possibilities of LGT or ancient paralogy, as reported here.
A block of Trp-pathway genes in Xylella
The phylogenetic incongruence of trpR, a regulatory gene in Xylella fastidiosa, led to recognition of a low-GC gene block in X. fastidiosa. The tryptophan repressor (TrpR) is quite limited in its phylogenetic distribution, being consistently present only within the enteric lineage, as shown in the protein tree of Figure 1. Here TrpR of Shewanella putrefaciens marks the outlying sequence of the enteric lineage (shown in gray). Outside the boundaries of the enteric lineage, only Coxiella burnetii, X. fastidiosa and two chlamydial species are thus far known to possess trpR. The distribution of trpR in the later three lineages is phylogenetically incongruent because they are widely spaced from one another on the 16S rRNA tree.
Figure 1.
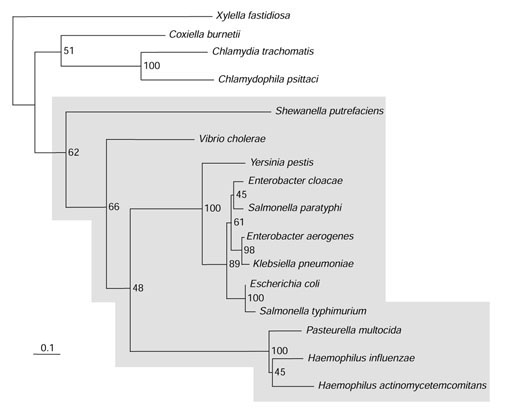
Protein tree for TrpR. Bootstrap values are shown at internal branch positions as percentages (1,000 replicates).
In Chlamydia trachomatis and Chlamydophila psittaci, trpR is positioned near structural genes of tryptophan biosynthesis, but no indication of recent origin by LGT of genes in this region was obtained [23]. X. fastidiosa trpR is separated by three genes from two structural genes of tryptophan biosynthesis. These latter genes do not appear to be essential for the primary task of tryptophan biosynthesis as all seven genes of tryptophan biosynthesis are represented elsewhere in the genome within one of two operons. Thus, in X. fastidiosa the incongruous phylogenetic position of trpR, the redundancy of the trp-linked genes encoding trpAa and trpAb, and the distinct phylogenetic incongruence of the latter gene pair all supported a reasonable possibility of origin by LGT.
The tryptophan supraoperon of Anabaena/Nostoc
All cyanobacteria possess each of the seven Trp-pathway genes at dispersed loci, and individual trees of proteins corresponding to these dispersed genes are phylogenetically congruent. Although this generalization also applies to Anabaena/Nostoc, this latter lineage is unique among cyanobacteria in its possession of an additional set of Trp-pathway genes (lacking only trpC) that coexist within an apparent operon. As shown in Figure 2, both Anabaena and Nostoc exhibit the same relative order of operonic trp genes: trpAa•trpAb → trpD → trpEa → trpEb → trpB. trpAa and trpAb are fused, as indicated in Figure 2 with a filled bar and in the text by the bullet in the notation: trpAa•trpAb. In Anabaena, qor (encoding NADPH: quinone reductase) has been inserted between trpD and trpEa. Another qor paralog is present elsewhere in the genome of Anabaena. Nostoc also has two qor paralogs, but neither resides within the tryptophan operon. Other cyanobacteria lack qor homologs altogether. In Nostoc, tyrP1 (encoding tyrosinase) has been inserted between trpEa and trpEb. All other cyanobacteria, including Anabaena, lack tyrP1. The two trp operons are less compact than frequently observed elsewhere, and relatively large intergenic spacing exists, especially in N. punctiforme. The only instance of translational coupling is between trpAa•trpAb and trpD in N. punctiforme.
Figure 2.
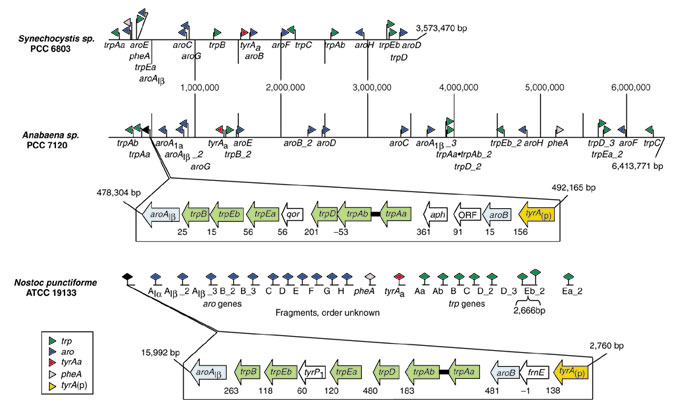
Genomic organization of aromatic-pathway genes in cyanobacteria. Genes relevant to the common pathway segment, the tryptophan branch, the tyrosine branch, and the phenylalanine branch are color-coded, as indicated. A system for uniform genomic naming of Trp-pathway genes or domains has been used as previously implemented [23,57]. Fused catalytic domains are joined by solid black linkers. Gene positions along the entire chromosomes of Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120 are shown. The qualitative presence or absence of genes in Nostoc punctiforme, an unfinished genome, is also indicated. Detailed zoom-in schematics are shown for the gene organizations within the supraoperons of Anabaena and Nostoc, regions spanning 13,000-14,000 bp. In the latter regions, intergenic spacing is shown, with negative values indicating the extent of genic overlap.
The tryptophan operons appear to be nested within what could be a larger unit of transcription that is reminiscent of what has been called a supraoperon in Bacillus subtilis [24]. The genes comprising the supraoperon of B. subtilis are aroG → aroB → aroH → trpAaBDCEbEa → hisHb → tyrAp → aroF. A hierarchy of internal promoters and terminators exists for differential control of the B. subtilis supraoperon. The Anabaena/Nostoc linkage group is additionally reminiscent of the B. subtilis supraoperon in the presence of aroB and tyrA. Although B. subtilis does not have aroAIβ represented in its supraoperon (as do Anabaena and Nostoc), aroAIβ is the homology class (of three possible DAHP synthase homologs distributed in nature [25,26,27]) that is utilized by B. subtilis. A number of supraoperon gene insertions have occurred outside of the trp operon as well. These differ for Anabaena and Nostoc as depicted in Figure 2. Anabaena has genes encoding aph and a hypothetical gene (open reading frame (ORF)) inserted between aroB and the trpAa•trpAb fusion. The aph gene encodes an uncharacterized protein of the defined alkaline phosphatases (metalloenzyme superfamily) (group COG1524 in the COGS database). Among cyanobacteria, only Nostoc has homologs of these two Anabaena genes, although they are not inserted in the Nostoc supraoperon. Nostoc has frnE (encoding a thiol-disulfide isomerase) inserted between tyrA(p) and aroB. Four subclasses of tyrA are defined according to the substrate specificities of the TyrA gene product: tyrAp, specific for prephenate; tyrAa, specific for arogenate; tyrAc, accepts either prephenate or arogenate; and tyrA(p), has broad specificity but exhibits a distinct preference for prephenate. Among all cyanobacteria, only Nostoc possesses frnE.
In their genomes outside the supraoperon boundaries, Nostoc and Anabaena possess a full complement of genes for biosynthesis of tryptophan, tyrosine and phenylalanine. Even these extra-supraoperonic genes of the Anabaena/Nostoc lineage are represented by multiple paralogs in many cases (Figure 2). If one considers the single-copy assemblage of aromatic-pathway genes present in the Synechocystis/Synechococcus/Prochlorococcus lineage as a fundamental complement of genes common to all cyanobacteria, the Anabaena/Nostoc genomic repertoire contains substantial redundancy. Thus, Anabaena has two additional extra-operonic paralogs of aroAIβ and trpD. In addition to extra-operonic, free-standing copies of trpAa and trpAb, a second fused gene (trpAa•trpAb_2) encoding the two domains of anthranilate synthase is present in Anabaena. Nostoc has two extra-operonic copies of aroAIβ, aroB and trpD. All cyanobacteria possess AroA of the Iβ class (aroAIβ). While this is also true of the Anabaena/Nostoc lineage (in fact, having multiple copies), both Anabaena and Nostoc possess an additional gene encoding AroA of the Iα class (aroAIα). All cyanobacteria possess a tyrA gene of the arogenate-specificity class (tyrAa), but the Anabaena/Nostoc supraoperons also possess a tyrA gene deemed to be a cyclohexadienyl dehydrogenase [28] with a favored specificity for prephenate (tyrA(p)) (C.A.B., R.A.J., N.K. and McNally A., unpublished observation).
Figure 3 shows an evolutionary scenario, using a Fitch diagram [29], that depicts the suggested origin of trpD paralogs via two gene duplication events (Dp1 and Dp2) that preceded the node of speciation divergence (Sp4) to Nostoc (Npu) and Anabaena (Asp). Consistent with the latter conclusion, Npu TrpD_1 exhibits greater identity with its ortholog Asp TrpD_1 than with its paralogs Npu TrpD_2 and Npu TrpD_3. Likewise, Npu TrpD_2 and Npu TrpD_3 exhibit greater identity with their Asp orthologs than with their Npu paralogs.
Figure 3.
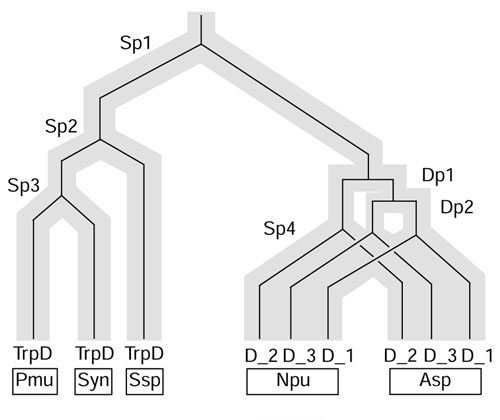
Fitch diagram [29] illustrating the origin and distribution of ortholog and paralogs of trpD in cyanobacteria. Paralogs, originating by gene duplication events (Dp1 and Dp2), track back to a horizontal line, whereas orthologs, originating by speciation (Sp1, Sp2, Sp3 and Sp4), track back to an inverted Y. The six trpD genes of Nostoc (Npu) and Anabaena (Asp) comprise a paralog set, and each of those comprises a four-member ortholog set with respect to the trpD genes from P. marinus (Pmu), Synechococcus sp. (Syn), and Synechocystis sp. (Ssp).
Since the basic single-copy repertoire of dispersed aromatic-pathway genes shown in Figure 2 for Synechocystis (Ssp) is representative of other cyanobacteria such as Synechococcus (Syn) and Prochlorococcus (Pmu) and is also present at dispersed extra-operonic loci of Anabaena and Nostoc, an obvious possibility would seem to be that the genes of the supraoperon originated by LGT in a common ancestor of Anabaena and Nostoc. If so, speciation was followed by different species-specific gene-insertion events. Because the divergence of Anabaena and Nostoc was relatively recent, evidence for LGT by analysis of GC content, codon usage, or dinucleotide frequency might be forthcoming. A number of distinctive properties of the supraoperon gene block represent items of biological context (as discussed by Lawrence and Ochman [21]) that potentially could provide excellent tracking clues about the identity of the putative donor in LGT. These include the overall gene organization of the trp operon, for which many microbial patterns are known; the extremely rare gene order of trpEa trpEb instead of the typical order trpEb trpEa; the fusion of genes encoding the alpha (trpAa) and beta (trpAb) subunits of anthranilate synthase, a fusion that exists in only a limited number of other taxa, and the presence of operonic genes exhibiting distinctive homology subtypes (aroAIβ and tyrA(p)).
Results and discussion
Lateral gene transfer of a block of genes in Xylella
The trpR gene in X. fastidiosa was previously noted [23] to have anomalously low GC content, relative to that of the genome. Low-GC blocks of genes have been attributed to LGT before, for example, argF (present in E. coli K-12 but not in other strains) is bracketed with unidentified high-GC (59%) genes that together comprise a distinctive block of LGT genes [30]. The flanking genes of trpR were accordingly analyzed for GC content. Figure 4 shows that trpR in X. fastidiosa is at one end of a block of seven genes, all of which have a distinctively low GC content (highlighted in green), compared to the flanking genes (highlighted in yellow).
Figure 4.
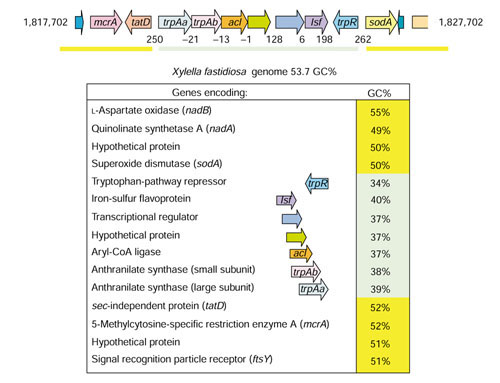
Block of genes acquired by lateral gene transfer (LGT) in Xylella fastidiosa. The gene map at the top shows the LGT block of genes with a green bar. The gene block begins with trpAa on the left and ends with trpR on the right. Intergenic spacing is given. The vertical pale green bar in the lower panel shows the corresponding genes from bottom to top. The GC% for each gene is shown, and the gene products are named. The hypothetical protein belongs to pfam00583, the acetyltransferase (GNAT) family. The low-GC gene block of the X. fastidiosa genome corresponds to gene numbers XF1914 (trpAa)-XF1920 (trpR).
If the block of low-GC genes in Xylella really reflects an alien origin, differences in dinucleotide frequencies might be expected, as such context biases differ from organism to organism. A 3:1 dinucleotide bias (third nucleotide position in a codon analysis algorithm followed by the first nucleotide position in the succeeding codon) was utilized, as it is the dinucleotide that is least restricted by amino-acid preference and codon usage in individual genes [31]. The 3:1 dinucleotide frequencies were calculated for the entire block of low-GC genes, as well as for the immediately flanking genes. These results presented in Figure 5 with a set of four selected dinucleotides shows that dinucleotides frequencies of the flanking genes were within a variance of about 4% from genomic frequencies, whereas the low-GC block of genes exhibited recognizably greater variances from the genomic dinucleotides frequencies of X. fastidiosa.
Figure 5.
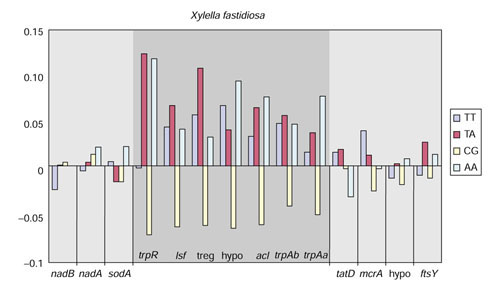
Three-to-one dinucleotide analysis of the putative LGT-block of X. fastidiosa genes shown in Figure 4. For easier viewing, four of the 16 dinucleotide combinations have been selected. The frequency variation of each gene is shown as positive variation (upward-pointing bars) or negative variation (downward-pointing bars) with respect to the average genomic frequencies (set to a value of zero at the midline), the absolute values of which can be seen in Table 1. treg, transcriptional regulator; hypo, hypothetical gene.
The co-variation of 3:1 dinucleotide frequencies of genes in the low-GC gene block of Xylella with the corresponding genomic frequencies was also evaluated using the Spearman rank correlation coefficient. Table 1 illustrates the data used to compare the Xylella trpR gene and the Xylella genome. A p-value of 0.730 indicated that the 3:1 dinucleotide frequencies of trpR from Xylella did not exhibit significant co-variation with the frequencies of the Xylella genome. In contrast, the 3:1 dinucleotide frequencies of trpR from Chlamydia trachomatis did exhibit significant co-variation with the frequencies characteristic of the C. trachomatis genome (p-value = 0.031). These analyses are consistent with occurrence of recent LGT in X. fastidiosa.
Table 1.
Statistical test of co-variation of 3:1 dinucleotide frequencies of trpR and its cognate genome
| Xylella fastidiosa | Chlamydia trachomatis | |||
| 3:1 Dinucleotide frequencies | Genome | trpR | Genome | trpR |
| TT | 4.5 | 4.3 | 9.3 | 9.6 |
| TC | 5.0 | 6.5 | 8.4 | 9.6 |
| TA | 4.2 | 16.3 | 8.6 | 9.6 |
| TG | 11.4 | 7.6 | 10.2 | 3.2 |
| CT | 4.5 | 4.3 | 4.7 | 4.3 |
| CC | 6.1 | 2.2 | 3.4 | 2.1 |
| CA | 8.9 | 8.7 | 4.5 | 4.3 |
| CG | 9.6 | 2.2 | 4.0 | 4.3 |
| AT | 3.2 | 1.1 | 4.9 | 6.4 |
| AC | 5.2 | 6.5 | 4.4 | 4.3 |
| AA | 3.7 | 15.2 | 7.5 | 7.4 |
| AG | 5.5 | 9.8 | 12.2 | 18.1 |
| GT | 4.6 | 3.3 | 3.3 | 5.3 |
| GC | 8.1 | 2.2 | 4.1 | 4.3 |
| GA | 6.4 | 5.4 | 5.6 | 5.3 |
| GG | 9.0 | 4.3 | 5.0 | 2.1 |
Xfa genome/trpR p-value = 0.031; Ctr genome/trpR p-value = 0.730.
What is the origin of the LGT gene block?
Gene organization is subject to constant change. For precisely this reason, the overall gene organization within the low-GC gene block might implicate a donor organism because the LGT event is inferred to be recent. Because the enteric lineage is a reasonable source of the LGT gene block, it is pertinent that the gene organization around trpR is highly conserved in the enteric lineage. Without exception, trpR in the enteric lineage is preceded upstream by a gene encoding soluble lytic murein transglycosylase (slt). hemK is usually positioned directly downstream, except for the Haemophilus actinomycetemcomitans/H. influenzae/Pasteurella multocida grouping (where the downstream gene encodes a monofunctional biosynthetic peptidoglycan transglycosylase (mtgA)). No genomes of the enteric lineage were found to possess trpR in a context of flanking genes that resembled the X. fastidiosa gene organization.
The LGT-block of Xylella genes conceivably could have originated from a donor similar to a common ancestor of the chlamydiae before the massive gene reduction associated with the chlamydial lifestyle. This would be consistent with the low GC content of both the chlamydial genome and the LGT-block of genes, as well as with the observation that chlamydiae and Xylella are the only two known taxa where trpR is positioned near structural genes of the tryptophan pathway. Direct comparison of chlamydial trpAa and trpAb genes with those of the Xylella operon is not possible because all chlamydial genomes thus far mapped lack trpAa and trpAb [23]. In this context, sequencing of genomes from closely related free-living relatives of the chlamydiae could be informative. The currently available chlamydial genomes also lack other genes of the low-GC block.
C. burnetii was also considered as a possible source of the low-GC gene block in X. fastidiosa because it possesses trpR. This potential LGT event seems ruled out because trpR is not near any structural genes encoding TrpAa and TrpAb in C. burnetti; C. burnetii TrpAa and TrpAb are not close to the corresponding X. fastidiosa enzymes on phylogenetic trees; and C. burnetii lacks the remaining genes in the low-GC gene block of X.fastidiosa.
If LGT accounts for the low-GC gene block in X. fastidiosa, how recent was this event? Presumably, it was sufficiently recent that significant amelioration to the genomic GC content has not yet occurred. The closest sequenced genome to Xylella is Xanthomonas. Genomes representing two species of the latter genus have been sequenced, and both lack the low-GC gene block. Therefore, the putative LGT event occurred some time after lineage divergence of Xylella and Xanthomonas. On the other hand, LGT presumably has predated speciation in the Xylella genus as all three strains of Xylella in the National Center for Biotechnology Information (NCBI) database possess the low-GC gene block. The T2 score of Hooper and Berg [31] measures the covariance of 3:1 dinucleotide signatures, and is designed to recognize very recent imports of alien genes by LGT. T2 scores calculated for the low-GC gene block of X. fastidiosa were not above the required threshold for very recent gene imports.
What is the function of the low-GC block of genes in Xyella?
Within the low-GC block, trpR is separated by four ORFs from genes encoding the two subunits of anthranilate synthase (trpAa and trpAb). These probably do not function for general tryptophan biosynthesis since paralogs of these genes, which exhibit a phylogenetically congruent context of gene organization, exist elsewhere in the genome (Figure 6). The latter genes are located within either of two separate operon clusters (Figure 6) with the GC content characteristic of X. fastidiosa. The GC-content values for the latter genes: trpAa, trpAb, trpB, trpC, trpD, trpEa, and trpEb are 52%, 49%, 54%, 55%, 51%, 59% and 55%, respectively. Furthermore, Figure 6 shows that the organization of the full complement of trp-pathway genes into two operons in X. fastidiosa is similar or identical to that of some of its nearest neighbors on the 16S rRNA tree, although the Xylella operons exhibit atypically large intergenic spacings. None of these neighbors possesses the low-GC block of Xylella genes illustrated in Figure 4. Hence, the two operons shown in Figure 6 can be inferred to be responsible for primary tryptophan biosynthesis throughout this clade.
Figure 6.
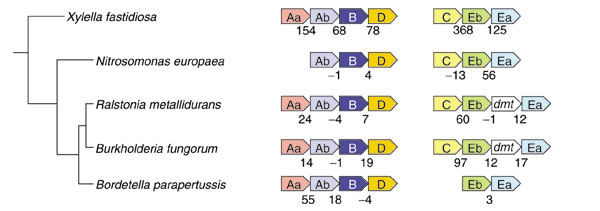
Organization of trp-pathway genes in X. fastidiosa and its nearest phylogenetic neighbors. The position of the organisms indicated on a 16S rRNA subtree is shown at the far left. To enhance the presentation, the trp-gene acronyms are shortened. Thus, trpAa is shown as Aa, etc. Intergenic spacing is indicated. dmt refers to a putative DNA methyltransferase. TrpAa in Nitrosomonas europeae and trpC in Bordetella parapertussis are located in other chromosomal positions, unlinked to other trp-pathway genes. X. fastidiosa and N. europeae, but not the other organisms shown in the figure, possess truA (encoding tRNA pseudouridine synthase A) upstream of trpC. truA and trpC are translationally coupled with 31-bp and 105-bp overlaps in X. fastidiosa and N. europeae, respectively. The gene organizations shown for a given organism is identical to the other organisms shown in parentheses as follows: Ralstonia metallidurans (R. solanacearum), Burkholderia fungorum (B. pseudomallei, B. mallei), and B. parapertussis (B. pertussis, B. bronchiseptica). R. solanacearum, in addition to the genes shown, has adjacent paralogs of trpB and trpD located on a large plasmid. The trpAaAbBD and trpCEbEa operons of the X. fastidiosa 9a5c genome correspond to gene numbers XF0210-XF0213 and XF1374-XF1376, respectively.
Genes encoding the two anthranilate synthase subunits (trpAa and trpAb) and aryl-CoA ligase (acl) surely belong to an operon, as translational coupling is evident from the overlap of start and stop codons (Figure 4). Acl exhibits strong similarity to coenzyme F390 synthetase of methanogenic archaea, as well as to phenylacetate-CoA ligase of E. coli. As Xylella does not appear to make the F420 cofactor that is the substrate of F390 synthetase, the function of Acl is likely to be closer to phenylacetate-CoA ligase. The aromatic ring is highly stable, and CoA thioesterification can provide chemical activation, allowing cleavage of the aromatic ring, as exemplified by catabolism of benzoate, 4-hydroxybenzoate, and anthranilate [32]. Because acl is tightly organized with trpAa and trpAb, it seems feasible that anthranilate might be the substrate of acl. An anthranilate-CoA ligase has been described recently in Azoarcus evansii by Schuhle et al. [33]. The Xylella Acl exhibited greater identity with phenylacetate-CoA ligase of E. coli than with anthranilate-CoA ligase of A. evansii, but a given substrate specificity within homology groups often can be associated with different subgroupings [25,34].
If anthranilate is indeed the substrate of Acl in Xylella, it would be a futile cycle if anthranilate were formed biosynthetically, only to be subsequently catabolized. Therefore, it seems more likely that the activation of anthranilate could be a step in the formation of a siderophore or antibiotic compound that is assembled by a nonribosomal peptide synthetase mechanism (see Quadri et al. [35] and references therein for numerous examples). Pyochelin from Pseudomonas aeruginosa exemplifies an iron siderophore whose peptide-based synthesis depends on CoA-activated salicylate (closely related to anthranilate) as a starter unit [36].
While it appears likely that trpR, aryl-CoA ligase, trpAa and trpAb belong to a common functional unit, the possible roles of the remaining three genes downstream of acl are problematic at the present time.
The Anabaena/Nostoc gene blocks
The large gene blocks in Anabaena and Nostoc that begin with aroAIβ and end with tyrAr(p) exhibited GC ratios that were similar to that of the host genome (Table 2). This is not necessarily inconsistent with their possible origin by LGT because the GC ratio of a putative donor genome could have been coincidentally similar to that of Anabaena and Nostoc.
Table 2.
Did operonic genes originate by LGT?
| First BLAST hit | Second BLAST hit | |||||
| Gene product | % GC | Organism | % Identity | Organism | % Identity | |
| Anabaena | AroAIβ | 46 | Nostoc punctiforme | 75 | Nostoc punctiforme | 71 |
| TrpB | 47 | Nostoc punctiforme | 76 | Anabaena sp. | 60 | |
| TrpEb | 46 | Nostoc punctiforme | 88 | Anabaena sp. | 88 | |
| TrpEa | 46 | Nostoc punctiforme | 85 | Anabaena sp. | 74 | |
| Qor | 45 | Enterococcus faecalis | 40 | Streptomyces coelicolor | 37 | |
| TrpD | 41 | Nostoc punctiforme | 72 | Anabaena sp. | 56 | |
| TrpAa•TrpAb | 41 | Nostoc punctiforme | 81 | Anabaena sp. | 77 | |
| Gpm1 | 41 | Nostoc punctiforme | 70 | Streptomyces coelicolor | 51 | |
| ORF | 41 | Nostoc punctiforme | 58 | Streptomyces coelicolor | 30 | |
| AroB | 41 | Nostoc punctiforme | 68 | Nostoc punctiforme | 62 | |
| TyrA(p) | 38 | Nostoc punctiforme | 72 | Yersinia pseudotuberculosis | 43 | |
| Nostoc† | AroAIβ | 46 | Anabaena sp. | 80 | Nostoc punctiforme | 79 |
| TrpB | 47 | Anabaena sp. | 77 | Nostoc punctiforme | 64 | |
| TrpEb | 46 | Anabaena sp. | 88 | Nostoc punctiforme | 88 | |
| TyrP_1 | 44 | Nostoc punctiforme | 56 | Nitrosomonas europeae | 30 | |
| TrpEa | 46 | Anabaena sp. | 85 | Anabaena sp. | 74 | |
| TrpD | 42 | Anabaena sp. | 72 | Anabaena sp. | 68 | |
| TrpAa•TrpAb | 42 | Anabaena sp. | 81 | Anabaena sp. | 76 | |
| AroB | 43 | Anabaena sp. | 68 | Nostoc punctiforme | 68 | |
| FrnE | 40 | Deinococcus radiodurans | 30 | Rhodobacter capsulatus | 29 | |
| TyrA(p) | 39 | Anabaena sp. | 72 | Yersinia pseudotuberculosis | 38 | |
*Anabaena sp. PCC 7120 has a genomic GC ratio of 42.82%. †Nostoc punctiforme has a genomic GC ratio of 43.90%.
Accordingly, the 3:1 dinucleotide frequencies of the aroAIβ-tyrAc gene block and the immediately flanking genes were analyzed, but these dinucleotide frequencies also did not suggest LGT. Figure 7 shows that dinucleotide frequencies did not deviate more than 5% from the genomic frequencies across the aroAIβ-tyrAc gene block. This contrasts with the distinctly greater deviation of 3:1 dinucleotide frequencies within the low-GC gene block of Xylella, which is shown on the same scale as the Anabaena data.
Figure 7.
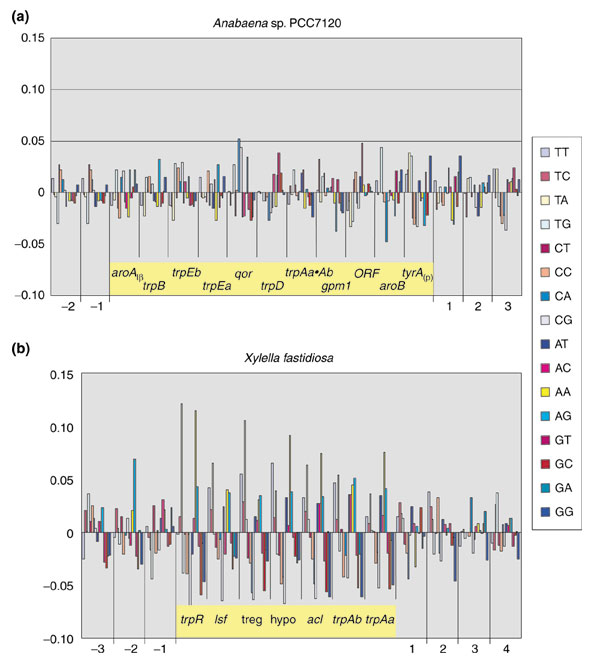
Three-to-one dinucleotide analysis. (a) The aroAIβ-tyrAc gene block in Anabaena. Deviations from genomic frequencies are expressed as positive (upward-pointing bars) or negative (downward-pointing bars) percentages. (b) For comparison, the results obtained for the low-GC gene block of X. fastidiosa (of which Figure 4 is a subset). The gene blocks of interest are highlighted in yellow, and the flanking genes are indicated by numbers.
Codon usage was analyzed throughout the gene block and also failed to implicate LGT. Figure 8 exemplifies this with a comparison of the pair of TrpAa domains in Anabaena, one encoded from within the gene block and the other outside. As Xylella also possesses a TrpAa pair, one encoded from within the low-GC gene block and the other outside, the analyses of these are also included in Figure 8 as a kind of positive control. In Anabaena (two bars on the right of each panel) the codon usage for leucine, serine, arginine, glycine, valine and proline was very similar for the TrpAa domain of TrpAa•TrpAb in the aroAIβ-tyrA(p) gene block and for the stand-alone TrpAa protein. This contrasts with the results obtained for the two Xylella TrpAa proteins, one in the low-GC gene block (on the far left) and the other (second bar in each panel) encoded by the gene in the trpAaAbBD operon (Figure 6). Thus, in contrast to the Anabaena TrpAa pair, the Xylella TrpAaAb pair exhibited distinctly different codon usage. Although this result is certainly consistent with an explanation of LGT in Xylella, one cannot be certain that the different functional roles of TrpAa domains might be associated with differing intra-genomic patterns of codon usage that are not yet well characterized [37].
Figure 8.
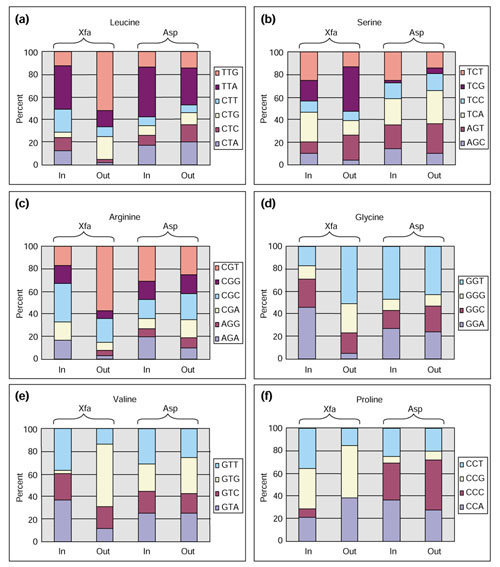
Codon usage for the pairs of TrpAa domains in the genomes of Anabaena sp. (Asp) and Xylella fastidiosa (Xfa). (a) Leucine; (b) serine; (c) arginine; (d) glycine; (e) valine and (f) proline. From left-to-right, Xfa TrpAa_1 is encoded from the low-GC gene block (In) and Xfa TrpAa_2 is encoded from outside (Out) the gene block; Asp TrpAa_1 is encoded from within the aroAIβtyrA(p) gene block (In) and Asp TrpAa_2 is encoded from outside (Out) the latter gene block. Synonymous codons are shown at the right of each amino acid set and color-coded to match the percent usage indicated by the bars.
Analysis of protein trees
We evaluated whether the closest BLAST hits, using as queries the amino-acid sequences corresponding to the operonic genes of Anabaena or Nostoc, would be with other cyanobacteria (and therefore consistent with origin by gene duplication) or with another taxon grouping (consistent with LGT). In either case, one would expect that the sequences encoded by the operonic genes of Anabaena would be the best matches for the operonic genes of Nostoc, as was indeed the case. For all of the operonic Anabaena/Nostoc Trp-pathway proteins used as queries, homolog sequences from other cyanobacteria (Synechocystis, Synechococcus, Prochlorococcus) were the remaining top hits returned in the BLAST queue. As BLAST hits must be considered imperfect indicators of nearest-neighbor homologs [38], the conclusion that the operonic trp-pathway genes are of cyanobacterial lineage origin was confirmed more rigorously by examination of extensive trees (available upon request) constructed for each trp protein of Anabaena and Nostoc. For the Trp-pathway proteins, all the cyanobacterial proteins clustered together, regardless of whether they were Anabaena or Nostoc paralogs or whether they were the singly represented proteins of Synechocystis, Synechococcus, or Prochlorococcus. The same result was obtained for AroAIβ protein trees. All the redundant genes exhibited identity relationships that suggested their origin by one or more gene-duplication events in the common ancestor of Anabaena and Nostoc; that is, exactly as diagrammed in Figure 3.
A different result was obtained for genes encoding AroB and TyrA. AroB sequences in nature are rather divergent. All of the cyanobacterial AroB proteins form a compact cluster in the AroB tree (including the non-operonic Anabaena/Nostoc aroB genes), except for those encoded by the Anabaena/ Nostoc supraoperons. The supraoperonic AroB proteins occupy a tree position that is not particularly close to other AroB proteins (the closest matches being on the order of 30-35% identity with some enteric bacteria). A similar situation applies to TyrA(p). All cyanobacteria possess the arogenate dehydrogenase specificity class (denoted TyrAa) of the TyrA superfamily. The additional TyrA(p) present only in Anabaena and Nostoc and located as the carboxy-terminal gene of the supraoperon exhibits identities of 39-43% with the TyrA(p) proteins of some enteric bacteria. These results for supraoperonic aroB and tyrA(p) could be consistent with LGT, but with no clear donor candidates available. On the other hand, origin as ancient paralogs is also a possibility.
The trpAa•trpAb fusion
A particularly fortuitous gene that could favor or disfavor the hypothesis of LGT of the aroAIβ-tyrA(p) gene block in Anabaena/Nostoc is trpAa•trpAb, a fusion corresponding to two genes that are usually separate (free-standing). As only a limited number of trpAa•trpAb fusions are known, possible LGT donors can be evaluated. Organisms known to possess the trpAa•trpAb fusion are listed at the top of Table 3. Another small group of trpAa•trpAb fusions are known, which are dedicated to phenazine biosynthesis and which form a distinct cluster. These are denoted trpAa•trpAb_phz in Table 3. Thus far, the trpAa•trpAb_phz, fusions are limited to species of Pseudomonas and Streptomyces. pabAa and pabAb are homologs of trpAa and trpAb, and the distribution of fusions involving these domains are also listed in Table 3 to give a general sense of the frequencies of such gene fusions. A variety of data (G.X. and R.A.J., unpublished observation) indicates that equivalent fusions often arise independently of one another in widely spaced lineages.
Table 3.
Gene fusions involving trpAa and or trpAb homologs
| trpAa•trpAb | Brucella melitensis; Sinorhizobium meliloti; Agrobacterium tumefaciens; Azospirillum brasilense; Nostoc punctiforme; Thermomonospora fusca; Rhodopseudomonas palustris; Rhizobium loti; Legionella pneumophila; Anabaena sp._1; Anabaena sp._2 |
| trpAa•trpAb_phz* | Pseudomonas aureofaciens; Pseudomonas aeruginosa; Pseudomonas chlororaphis; Pseudomonas fluorescens; Streptomyces venezuelae; Streptomyces coelicolor |
| trpAb•trpB | Escherichia coli; Salmonella typhi; Campylobacter jejuni; Thermotoga maritima |
| pabAa•pabAb† | Deinococcus radiodurans |
| pabAa•pabAc† | Neisseria meningitides; Neisseria gonorrhoeae; Chlorobium tepidum; Helicobacter pylori; Campylobacter jejuni; Streptococcus pneumoniae; Streptococcus pyogenes; Streptococcus equi; Streptococcus gordonii; Listeria innocua; Listeria monocytogenes; Geobacter sulfurreducens; Ralstonia solanacearum; Burkholderia fungorum; Novosphingobium aromaticivorans; Chlorobium tepidum; Ralstonia metallidurans; Lactococcus lactis; Burkholderia pseudomallei; Magnetococcus sp. |
| pabAb•pabAa† | Streptomyces griseus; Streptomyces venezuelae; Streptomyces pristinaespiralis; Thermomonospora fusca; Anabaena sp.; Nostoc punctiforme; Corynebacterium glutamicum; Saccharomyces cerevisiae; Aspergillus fumigatus; Plasmodium falciparum; Coprinus cinereus; Schizosaccharomyces pombe |
*Also known as phzE. †pabAa, pabAb and pabAc are also known as pabB, pabA and pabC.
Figure 9 shows a segment of the 16S rRNA tree that contains all of the trpAa•trpAb fusions which are known so far. Cyanobacteria other than Anabaena/Nostoc lack the fusion, as do all nearby lineages. The fusion is present in the cluster that includes Rhodopseudomonas palustris, Rhizobium loti, Brucella melitensis, Agrobacterium tumefaciens and Sinorhizobium meliloti. (A. tumefaciens, which is not shown in Figure 9, is virtually identical to S. meliloti). Additional phylogenetically spaced fusions are present in Thermomonospora fusca, Azospirillum brasilense, and Legionella pneumophila. Other fusions that involve trpAa or trpAb homologs also occur in nature, as shown in Table 3, and a degree of care is needed to avoid confusion between them.
Figure 9.
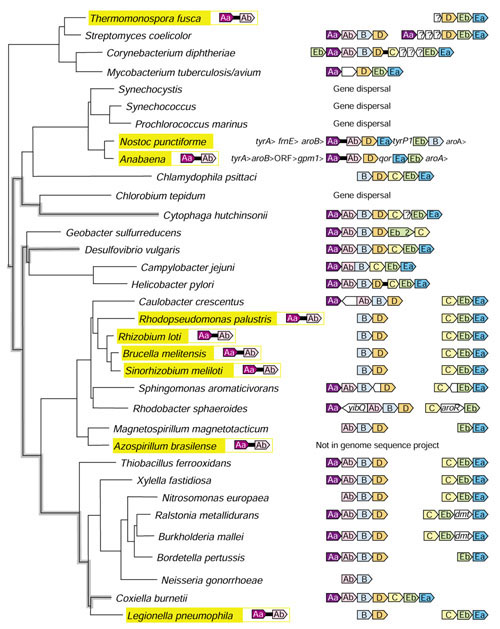
16S rRNA tree showing the phylogenetic distribution (highlighted in yellow) of trpAa•trpAb fusions. The gene fusions unlinked to any other trp genes are shown to the right of the highlighted name. The remaining trp-operon gene organizations are shown at the right. The white arrows indicate gene insertions that encode the following: Thermomonospora, integral membrane protein; Streptomyces, three membrane proteins: Corynebacterium, membrane protein, pantoate 3-alanine ligase (panC), and 3-methyl-2-oxobutanoate hydroxymethyl transferase (panB); Mycobacterium, conserved hypothetical protein; Cytophaga, conserved hypothetical protein; Sphingomonas, conserved hypothetical protein and outer-membrane protein; Rhodobacter, and acetyltransferase yibQ; Ralstonia, DNA methyltransferase (dmt); Burkholdaria, DNA methyltransferase (dmt). In addition aroR in R. sphaeroides is a putative regulatory gene [58]. The lineage relationships of three organisms that have maintained the putative ancestral trp operon are shown with heavy, gray lines.
A phylogenetic tree consisting of all free-standing TrpAa and TrpAb proteins was constructed, together with the corresponding two domains of the TrpAa•TrpAb fusions (available upon request). Surprisingly, each of the 10 fusion domains clustered tightly on the TrpAa and TrpAb trees, to the exclusion of the free-standing TrpAa and TrpAb domains. This is consistent with a single ancestral fusion event, but requires the assumption of multiple LGT events. However, it is surprising that no free-standing domains (that is, close homologs of the original fusion partners) cluster with either of the two sets of 10 fusion domains. This might suggest an alternative to LGT, namely that there has been extreme sequence convergence because of strong selection for appropriate residues mediating domain-domain interactions. If so, it is possible that trpAa•trpAb fusions occurred as a number of independent events, followed by strong convergence.
Figure 9 shows the individual genomic organization of trp-pathway genes in the 16S rRNA tree sector that is relevant to the trpAa•trpAb fusion. The Anabaena/Nostoc lineage is unique in having trpAa•trpAb linked to other trp-pathway genes and is further unique in having an additional set of freestanding genes encoding TrpAa and TrpAb. Although generally uncommon, complete dispersal of Trp-pathway genes is characteristic of the non-filamentous cyanobacteria, Aquifex aeolicus and Chlorobium tepidum. The ancestral state of trp gene organization has been asserted (G.X., C.B., N.K. and R.J., unpublished work) to be trpAa/Ab/B/D/C/Eb/Ea, an operon organization seen in contemporary Cytophaga hutchinsonii, Desulfovibrio vulgaris and Coxiella burnetii (Figure 9). Dynamic gene reorganization events that involve gene insertions, gene scrambling, gene duplications and gene dispersal are apparent from inspection of Figure 9.
It is expected that LGT would most easily be recognized if it occurred relatively recently before passage of sufficient time for amelioration of alien characteristics to those of the host genome, for example GC content. In the case of each of the known trpAa•trpAb gene fusions, the absence of the gene fusion in a closely related genome implies that the gene-fusion event (or the LGT event) occurred recently, that is, in the one lineage following the time of its separation from the other by speciation. Thus, the acquisition of trpAa•trpAb by Thermomonospora fusca must have occurred by fusion or by LGT relatively recently, that is, after the speciation event that generated the Streptomyces lineage (see Figure 9). In each of the remaining cases of trpAa•trpAb fusion, a relatively near time of fusion shown in Figure 9 origin can be identified. These are defined by points of speciation divergence between Anabaena/Nostoc and other cyanobacteria, between the Rhodopseudomonas/Sinorhizobium cluster (fusion) and Caulobacter (no fusion), between Azospirillum brasilense (fusion) and Magnetospirillum magnetotacticum (no fusion), and between Legionella pneumophila (fusion) and Coxiella burnetii (no fusion).
If any of the trpAa•trpAb fusions, other than the Nostoc/Anabaena pair, have a common origin, similar flanking regions of gene organization might be expected since all of the fusions are of relatively recent origin. On this criterion, only R. loti, B. melitensis, A. tumefaciens and S. meliloti exhibited similarities of flanking-gene organization, and this is phylogenetically congruent. These observations imply that within the span of phylogeny shown in Figure 9, the trpAa•trpAb fusion may have occurred independently as many as seven times.
Interdomain linker regions
In fusion proteins an interdomain linker region of critical length and mobility is important to facilitate specific domain-domain interactions. Fusions of independent origin might be expected to exhibit a variety of linker regions. Particular constraints undoubtedly limit this variety, and such constraints might be more stringent for some domain combinations than others. (In the case of particularly stringent constraints, similar linker regions would not necessarily demonstrate a common origin). Figure 10 shows an alignment of the carboxy-terminal region of the TrpAa domain, the linker region, and the amino-terminal region of the TrpAb domain for all of the fusion proteins depicted in Figure 9 (as well as that from A. tumefaciens).
Figure 10.

Comparison of TrpAa•TrpAb linker regions. The seven independent fusions that are suggested were aligned with free-standing TrpAa and TrpAb proteins in order to visualize the inter-domain linker regions. Amino-acid residue numbering is indicated at the left and right margins.
Only the two operonic fusion proteins from Anabaena and Nostoc and the four rhizobial fusion proteins (Mlo, Bme, Rme and Atu) exhibit linker regions of identical length and obvious similarity. The paralog TrpAa•TrpAb protein of Anabaena sp (Asp_2) seems to have a distinctly different linker, and it maybe that the two fusions in Anabaena arose as two independent events. The partial sequences shown in Figure 10 are spaced to indicate the seven independent events of gene fusion that are suggested.
Function of the Anabaena/Nostoc gene blocks?
The gene blocks shown in Figure 2 encode the entire tryptophan pathway (except for trpC), as well as the first two enzymes of the common aromatic pathway, and the key enzyme of tyrosine biosynthesis. Multiple enzymes catalyzing the same reaction have been described in developmental systems where differential regulation of isoenzymes are deployed in different temporal and spatial contexts. Filamentous cyanobacteria (such as Anabaena and Nostoc) subscribe to a developmental program of heterocyst formation that is widely considered the primitive state and that correlates with their exceedingly large genomes. Unicellular cyanobacteria such as Synechocystis, Synechococcus and Prochlorococcus have far smaller genomes and lack the ability to fix nitrogen (heterocyst formation). It, therefore, seems to be a distinct possibility that the gene blocks diagrammed in Figure 2 (as well as additional gene duplicates) are specifically involved in specialized capabilities of Nostoc/Anabaena that do not exist in other cyanobacteria. In terms of the evolutionary scenario, the Anabaena/Nostoc lineage may reflect the ancestral state, and modern unicellular cyanobacteria may be derived genomes that are smaller and more streamlined (reductive evolution).
Conclusions
Operon displacement
Alien genes that may be subject to possible LGT can generally expect a hostile reception in that they lack a history of functional integration with the resident genome. Genes that offer immediate selective advantages (for example, antibiotic resistance) are likely to persist. The acquisition of a completely new functional capability will often require an entire suite of novel genes, and such recruitment is certainly easier to envision if all of the genes arrive en bloc (that is, as an operon). Once a primary biosynthetic pathway, such as that responsible for tryptophan formation, has been established and integrated with the individualistic metabolic circuitry of a given organism, one does not expect facile displacement of resident genes. This should apply even if the incoming genes all coexist as an operon. We have found only two examples of LGT of whole-Trp operons, that of trpAa/Ab/B/D•C/Eb/Ea from the enteric lineage to coryneform bacteria and to Helicobacter, as discussed earlier.
Has there been separate lateral gene transfer of individual genes?
According to the foregoing rationale, isolated genes that participate in multi-step processes would not generally be expected to have much success in LGT. In some cases analog genes encode enzymes that catalyze the same reaction in a multi-step pathway, and one analog gene might conceivably displace another. Lack of enough information about genomic representation of such analog genes can lead to incorrect inferences of LGT. For example, the initial discovery of "plant-type" AroAII in bacteria led to the assumption of LGT from plant to bacterium. Elucidation of the fuller genomic representation of aroAII ([27] and refs therein) demonstrated the origin of aroAII in Bacteria, and plants probably have received aroAII from the Bacteria via endosymbiosis. A similar outcome seems quite possible with respect to the "eukaryotic" fructose-1,6-bisphosphate aldolase in Xylella species. Phylogenetic incongruities that involve such analogs can pose great difficulties in distinguishing LGT from vertical progressions of differential analog losses in different lineages.
Specialized Trp genes not required for primary biosynthesis
In this article we focus on a number of cases where at least several trp genes are linked, thus providing analytical advantages offered by the analysis of more than one gene. These genes are also redundant and phylogenetically incongruent, in contrast to coexisting homolog genes that are part of a full phylogenetically congruent set. Both of the latter are consistent with origin by LGT, but unrecognized ancient paralogy is also possible. In the first case, the homologs coexisting in one organism are xenologs, whereas in the latter case, they are paralogs. A relatively simple example is the trpAa/trpAb pair originally denoted phnA/phnB in Pseudomonas aeruginosa [39]. This comprises an anthranilate synthase that is not strictly required for primary tryptophan biosynthesis and that is uniquely expressed during stationary-phase physiology [40]. Why the generation of anthranilate under these conditions would be of value is unknown, but phylogenetic trees clearly show phnA/phnB to be xenologs originating from the enteric lineage via LGT (G.X. and R.A.J., unpublished data). In this case, genes that function for primary biosynthesis in the donor genome did not displace the corresponding genes in the recipient genome, but have instead been recruited to a specialized function. In Streptomyces coelicolor, trpAa/trpAb/trpB/trpD/aroAII are contained within a large cluster dedicated to antibiotic synthesis [41]. Calcium-dependent antibiotic (CDC) contains tryptophan, and presumably the feedback-resistant variety of enzyme encoded by aroAII ensures enhanced precursor flow to tryptophan during antibiotic production. Detailed studies have not yet been done to see whether the CDC gene cluster originated via LGT or reflects ancient paralogy.
In this article, we have discussed at length the Xylella and cyanobacterial gene blocks that seem likely to have specialized functional roles other than primary biosynthesis. The Xylella genes are associated with other genes that presumably dictate a fate for anthranilate other than as a primary precursor of tryptophan. We suspect that selective advantages conferred by this specialized operon accommodated successful LGT to Xylella. The Anabaena/Nostoc supraoperon is reminiscent of the S. coelicolor system in the inclusion of AroAIβ, which might enhance precursor flow to chorismate. Although the Anabaena/Nostoc operon only lacks trpC, its features of gene fusion and gene organization are novel. It might perhaps have an unknown physiological function related to the complex developmental programs unique to heterocystous cyanobacteria. We conclude that in this case the operonic trp genes are ancient paralogs of a dispersed set of trp genes engaged in primary biosynthesis.
Against a backdrop where organisms generally possess highly efficient and integrated pathways of tryptophan biosynthesis, displacement of resident genes by LGT of the corresponding genes is relatively infrequent. Aside from the broadly distributed primary pathway, highly specialized pathways are known that utilize some or all tryptophan-pathway enzymes, and these pathways can originate by recruitment of paralog genes derived from the primary-pathway genes [42]. The genes of such specialized operons may diverge considerably to meet the demands of a novel functional role. In a contemporary organism this might have the status of unrecognized (or recognized) paralogy, as we suggest for the Anabaena/Nostoc gene block. However, such an operon module also has strong potential for xenologous transfer because of its specialized functional potential.
The tryptophan pathway exemplifies the situation where paralogs can be engaged in primary amino-acid biosynthesis (widespread) or in a variety of specialized pathways (narrowly distributed). Aside from the extent to which the specialized pathways may be individually intriguing and important, this study illustrates that case-by-case analysis can distinguish paralogs (or xenologs) from their homologs engaged in primary biosynthesis. This conclusion is encouraging as it shows that both vertical and horizontal events of gene transfer can be deduced to track evolutionary history.
Materials and methods
Dinucleotide frequencies
The CODONW program [43] was used to calculate 3:1 dinucleotide frequencies (third base of a given codon followed by the first base of the next codon). For whole-genome calculations, genome nucleotide sequences (.ffn file) were obtained from GenBank [44]. Perl scripts were used to eliminate the defline and assemble all genomic ORFs together for CODONW calculation. The length (from UNIX wc command) divided by 3 was used to validate the absence of frameshift errors. Pairwise covariation of 3:1 dinucleotide frequencies was assessed by the Spearman rank correlation coefficient [45], a nonparametric rank statistic for testing monotonic relationships. T2 values were kindly provided by Hooper [31].
Codon usage
Codon usage for individual genes was computed with the CDONTREE program [46]. Codon-usage values for whole genomes were obtained from the Codon Usage Database [47,48].
Phylogenetic trees
16S rRNA subtrees were derived from the Ribosomal Database site [49,50]. Unrooted phylogenetic protein trees were derived by input of the indicated homolog amino-acid sequences into the ClustalW program (Version 1.4) [51]. Manual alignment adjustments were made as needed with the assistance of the BioEdit multiple alignment tool of Hall [52]. The refined multiple alignment was used as input for generation of a phylogenetic tree using the program package PHYLIP [53]. The neighbor-joining and Fitch programs [51] were used to obtain distance-based trees. The distance matrix was obtained using Protdist with a Dayhoff Pam matrix. The Seqboot and Consense programs were then used to assess the statistical strength of the tree using bootstrap resampling. Neighbor-joining and Fitch trees yielded similar clusters and arrangement of taxa within them. Bootstrap values indicate the number of times a node was supported in 1,000 resampling replications.
Identification of linker regions
Fusion proteins were aligned (ClustalW) with one another and with the assemblage of free-standing proteins corresponding to the amino-terminal and the carboxy-terminal domains of the fusion proteins. The boundaries of each domain were defined by the last highly conserved residues of the amino-terminal domain and the early highly conserved residues of the carboxy-terminal domain. The Conserved Domain Database was useful as a reference guide [54,55].
Comparative genome analysis
Most of the comparative genome analysis was carried out using the database and tools of ERGO [56].
Acknowledgments
Acknowledgements
G.X. was partially supported in this work through the STDGEN project at Los Alamos National Laboratory (NIH/NIAIDAGY1-A1-8228-05). We thank Sean Hooper (Department of Molecular Evolution, Uppsala University, Sweden) for assistance with dinucleotide frequency calculations. We are indebted to A. Osterman of Integrated Genomics, Inc. (Chicago, IL) for provision of access to ERGO [56]. This is Florida Agricultural Experiment Station Journal series no. R-09159.
References
- Margulis L. Symbiosis in Cell Evolution. San Francisco: WH Freeman; 1981. [Google Scholar]
- Gray MW. Evolution of organellar genomes. Curr Opin Genet Dev. 1999;9:678–687. doi: 10.1016/s0959-437x(99)00030-1. [DOI] [PubMed] [Google Scholar]
- Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 1998;14:442–444. doi: 10.1016/s0168-9525(98)01553-4. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Martin W. Mosaic bacterial chromosomes: a challenge en route to a tree of genomes. BioEssays. 1999;21:99–104. doi: 10.1002/(SICI)1521-1878(199902)21:2<99::AID-BIES3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Syvanen M. On the occurrence of horizontal gene transfer among an arbitrarily chosen group of 26 genes. J Mol Evol. 2002;54:258–266. doi: 10.1007/s0023901-0007-z. [DOI] [PubMed] [Google Scholar]
- Kyrpides NC, Olsen GJ. Archaeal and bacterial hyperthermophiles: horizontal gene exchange or common ancestry? Trends Genet. 1999;15:298–299. doi: 10.1016/s0168-9525(99)01811-9. [DOI] [PubMed] [Google Scholar]
- Stiller JW, Hall BD. Lateral gene transfer, genome surveys, and the phylogeny of prokaryotes. Science. 1999;286:1443. [Google Scholar]
- Kurland CG. Something for everyone: Horizontal gene transfer in evolution. EMBO Rep. 2000;1:92–95. doi: 10.1093/embo-reports/kvd042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, White O, Peterson J, Eisen JA. Microbial genes in the human genome: lateral transfer or gene loss? Science. 2001;292:1903–1906. doi: 10.1126/science.1061036. [DOI] [PubMed] [Google Scholar]
- Stanhope MJ, Lupas A, Italia MJ, Koretke KK, Volker C, Brown JR. Phylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates. Nature. 2001;411:940–944. doi: 10.1038/35082058. [DOI] [PubMed] [Google Scholar]
- Glansdorff N. About the last common ancestor, the universal life-tree and lateral gene transfer: a reappraisal. Mol Microbiol. 2000;38:177–185. doi: 10.1046/j.1365-2958.2000.02126.x. [DOI] [PubMed] [Google Scholar]
- Woese CR. Interpreting the universal phylogenetic tree. Proc Natl Acad Sci USA. 2000;97:8392–8396. doi: 10.1073/pnas.97.15.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. Limitations of compositional approach to identifying horizontally transferred genes. J Mol Evol. 2001;53:244–250. doi: 10.1007/s002390010214. [DOI] [PubMed] [Google Scholar]
- Koski LB, Morton RA, Golding GB. Codon bias and base composition are poor indicators of horizontally transferred genes. Mol Biol Evol. 2001;18:404–412. doi: 10.1093/oxfordjournals.molbev.a003816. [DOI] [PubMed] [Google Scholar]
- Ragan MA. On surrogate methods for detecting lateral gene transfer. FEMS Microbiol Lett. 2001;201:187–191. doi: 10.1111/j.1574-6968.2001.tb10755.x. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Reconciling the many facets of lateral gene transfer. Trends Microbiol. 2002;10:1–4. doi: 10.1016/s0966-842x(01)02282-x. [DOI] [PubMed] [Google Scholar]
- Ragan M. Detection of lateral gene transfer among microbial genomes. Curr Opin Genet Dev. 2001;11:620–626. doi: 10.1016/s0959-437x(00)00244-6. [DOI] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Jensen RA. Dynamic diversity of the tryptophan pathway in the chlamydiae: reductive evolution and a novel operon for tryptophan recapture. Genome Biol. 2002;3:research0005.1–0005.13. doi: 10.1186/gb-2002-3-9-research0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henner D, Yanofsky C. Biosynthesis of aromatic amino acids. In: Sonenshein AL, Hoch J, Losick R, editor. In Bacillus subtilis and other Gram-positive Bacteria: Biochemistry, Physiology, and Molecular Genetics. Washington, DC: ASM Press; 1993. pp. 269–280. [Google Scholar]
- Subramaniam PS, Xie G, Xia T, Jensen RA. Substrate ambiguity of 3-deoxy-D-manno-octulosonate 8-phosphate synthase from Neisseria gonorrhoeae in the context of its membership in a protein family containing a subset of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthases. J Bacteriol. 1998;180:119–127. doi: 10.1128/jb.180.1.119-127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RA, Xie G, Bonner CA. The correct phylogenetic relationship of KdsA (3-deoxy-D-manno-octulosonate 8-phosphate synthase) with one of two independently evolved classes of AroA (3-deoxy-D-arabino-heptulosonate 7-phosphate synthase. J Mol Evol. 2002;54:416–423. doi: 10.1007/s00239-001-0031-z. [DOI] [PubMed] [Google Scholar]
- Gosset G, Bonner CA, Jensen RA. Microbial origin of plant-type 2-keto-3-deoxy-D-arabino-heptulosonate 7-phosphate synthases, exemplified by the chorismate-and tryptophan-regulated enzyme from Xanthomonas campestris. J Bacteriol. 2001;183:4061–4070. doi: 10.1128/JB.183.13.4061-4070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Jensen RA. Cyclohexadienyl dehydrogenase from Pseudomonas stutzeri exemplifies a widespread type of tyrosine-pathway dehydrogenase in the TyrA protein family. Comp Biochem Physiol C Toxicol Pharmacol. 2000;125:65–83. doi: 10.1016/s0742-8413(99)00090-0. [DOI] [PubMed] [Google Scholar]
- Fitch WM. Homology: a personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/s0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- Van Vliet F, Boyen A, Glansdorff N. On interspecies gene transfer: the case of the argF gene of Escherichia coli. Ann Inst Pasteur Microbiol. 1988;139:493–496. doi: 10.1016/0769-2609(88)90111-1. [DOI] [PubMed] [Google Scholar]
- Hooper SD, Berg OG. Detection of genes with atypical nucleotide sequence in microbial genomes. J Mol Evol. 2002;54:365–375. doi: 10.1007/s00239-001-0051-8. [DOI] [PubMed] [Google Scholar]
- Mohamed MES, Zaar A, Ebenau-Jehle C, Fuchs G. Reinvestigation of a new type of aerobic benzoate metabolism in the proteobacterium Azoarcus evansii. J Bacteriol. 2001;183:1899–1908. doi: 10.1128/JB.183.6.1899-1908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schühle K, Jahn M, Ghisla S, Fuchs G. Two similar gene clusters coding for enzymes of a new type of aerobic 2-aminobenzoate (anthranilate) metabolism in the bacterium Azoarcus evansii. J Bacteriol. 2001;183:5268–5278. doi: 10.1128/JB.183.18.5268-5278.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun DH, Bonner CA, Gu W, Xie G, Jensen RA. The emerging periplasm-localized subclass of AroQ chorismate mutases, exemplified by those from Salmonella typhimurium and Pseudomonas aeruginosa. Genome Biol. 2001;2:research0030.1–0030.16. doi: 10.1186/gb-2001-2-8-research0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri LEN, Keating TA, Patel HM, Walsh CT. Assembly of the Pseudomonas aeruginosa nonribosomal peptide siderophore pyochelin: In vitro reconstitution of aryl-4,2-bisthiazoline synthetase activity from PchD, PchE, and PchF. Biochemistry. 1999;38:14941–14954. doi: 10.1021/bi991787c. [DOI] [PubMed] [Google Scholar]
- Patel HM, Walsh CT. In vitro reconstitution of the Pseudomonas aeruginosa nonribosomal peptide synthesis of pyochelin: characterization of backbone tailoring thiazoline reductase and N-methyltransferase activities. Biochemistry. 2001;40:9023–9031. doi: 10.1021/bi010519n. [DOI] [PubMed] [Google Scholar]
- Guindon S, Perrière S. Intragenomic base content variation is a potential source of biases when searching for horizontally transferred genes. Mol Biol Evol. 2001;18:1838–1840. doi: 10.1093/oxfordjournals.molbev.a003972. [DOI] [PubMed] [Google Scholar]
- Koski L, Golding GB. The closest BLAST hit is often not the nearest neighbor. J Mol Evol. 2001;52:540–542. doi: 10.1007/s002390010184. [DOI] [PubMed] [Google Scholar]
- Essar DW, Eberly L, Hadero A, Crawford IP. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa : interchangeability of the two anthranilate synthases and evolutionary implications. J Bacteriol. 1990;172:884–900. doi: 10.1128/jb.172.2.884-900.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrodi DV, Bonsall RF, Delaney SM, Soule MJ, Phillips G, Thomashow LS. Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PA01. J Bacteriol. 2001;183:6454–6465. doi: 10.1128/JB.183.21.6454-6465.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryding JJ, Anderson TB, Champness WC. Regulation of the Streptomyces coelicolor calcium-dependent antibiotic by absA, encoding a cluster-linked two-component system. J Bacteriol. 2002;184:794–805. doi: 10.1128/JB.184.3.794-805.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RA. Enzyme recruitment in the evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Peden J. CodonW as a freeware release for codon usage analysis. http://www.molbiol.ox.ac.uk/cu/culong.html#Codonw
- GenBank Database http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html
- Lehmann EL, D'Abrera HJM. Nonparametrics: Statistical Methods Based on Ranks, Rev Edn. Englewood Cliffs, NJ: Prentice-Hall; 1998. pp. 292–323. [Google Scholar]
- Pesole G, Attimonelli M, Liuni S. A backtranslation method based on codon usage strategy. Nucleic Acids Res. 1988;16:1715–1728. doi: 10.1093/nar/16.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28:292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codon Usage Database http://www.kazusa.or.jp/codon
- Maidak BL, Cole JR, Lilburn TG, Parker CT, Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribosomal Database Project II http://rdp.cme.msu.edu/html
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. Biological Sequence Alignment Editor for Windows 95/98/NT, 509 Edn. Raleigh: North Carolina State University; http://www.mbio.ncsu.edu/BioEdit/bioedit.html [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny inference package (version 3.2). Cladistics. 1989;5:164–166. [Google Scholar]
- Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acid Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI Conserved Domain Database http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml
- ERGO http://wit.integratedgenomics.com/ERGO
- Xie G, Forst C, Bonner C, Jensen RA. Significance of two distinct types of tryptophan synthase beta chain in Bacteria, Archaea and higher plants. Genome Biol. 2002;3:research0004.1–0004.13. doi: 10.1186/gb-2001-3-1-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie C, Simmons AE, Kaplan S. Multiple chromosomes in bacteria: The yin and yang of trp gene localization in Rhodobacter sphaeroides 2.4.1. Genetics. 1999;153:525–538. doi: 10.1093/genetics/153.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]