

Abstract
Ultrafast electron diffraction is a unique method for the studies of structural changes of complex molecular systems. In this contribution, we report direct ultrafast electron diffraction study of the evolution of short-lived intermediates in the course of a chemical change. Specifically, we observe the transient intermediate in the elimination reaction of 1,2-diiodotetrafluoroethane (C2F4I2) to produce the corresponding ethylene derivative by the breakage of two carbon–iodine, C—I, bonds. The evolution of the ground-state intermediate (C2F4I radical) is directly revealed in the population change of a single chemical bond, namely the second C—I bond. The elimination of two iodine atoms was shown to be nonconcerted, with reaction time of the second C—I bond breakage being 17 ± 2 ps. The structure of the short-lived C2F4I radical is more favorable to the classical radical structure than to the bridged radical structure. This leap in our ability to record structural changes on the ps and shorter time scales bodes well for many future applications in complex molecular systems.
Significant progress has already been made in the probing of chemical reactions on the fs time scale. In these femtochemistry (1–4) experiments, the nuclear motions on the time scale of bond breaking and bond making are monitored by using an initiation pulse to establish the zero-of-time (clocking) and probing pulses to view the motion; typical probes are optical and IR spectroscopy, photoelectron spectroscopy, mass spectrometry, and nonlinear optical techniques (5). In this laboratory, efforts have been made to include diffraction techniques to map out ultrafast structural changes, especially in complex molecular systems (6, 7). Electron diffraction of molecules in their ground state has been a powerful tool over the past 50 years (8), and both electron and x-ray methods are now being advanced in several laboratories (6, 7, 9–16) for the studies of structural changes. Our focus here and before has so far been on gas-phase ultrafast electron diffraction (UED) of isolated chemical reactions.
Elsewhere, we have reported the latest advance in UED (7), by which major challenges were surmounted: the very low number densities of gas samples, the absence of the long-range order that is present in crystals, which enhances coherent interference, and the challenging task of determining in situ the zero-of-time when diffraction changes are on the ps and sub-ps time scale. With UED, molecular structures (17) and branching ratios (18) of final products have been determined on the ps time scale. The diffraction change before and after the chemical reaction was observed (7). However, no direct observation of transient structural changes in the course of the reaction has so far been reported. In this article, we report such observation of the temporal evolution of short-lived intermediates probed with ultrafast electron diffraction.
EXPERIMENTAL
The UED experiments were performed in the second-generation apparatus developed in this laboratory (7). It is composed of a fs excitation laser, a ps pulsed-electron source, a free-expansion molecular beam, and a two-dimensional single-electron detection (camera) system. All of these are part of a complex apparatus that has been described in more detail elesewhere (7). The electron pulse, the laser pulse, and the molecular beam are arranged in a crossed-beam geometry, and the overlap of the three beams is controlled within 10 μm accuracy. With a flux of ≈7,000 electrons per pulse in our experiment, the corresponding pulse width is ≈8 ps (7). The total temporal resolution, including the contributions from the pump laser pulse width (≈0.7 ps) and the ≈3 ps group-velocity mismatch effect (19) is less than 10 ps (17, 18).
The chemical reaction was initiated with the fs laser pulse (450 μJ at 307 nm) and probed with the ps electron pulses at 18.8 keV (de Broglie wavelength 0.088 Å). The two-dimensional diffraction images at each delay time were recorded in the charge-coupled device camera. Time delays between the fs laser and the ps electron pulses were precisely controlled by a computer-driven translational stage. After the establishment of the time zero by the lensing approach (7), the time-resolved diffraction images at −150, 0, +10, +20, +30 and +330 ps delay times were recorded with ±2 ps accuracy (see below); at each delay time, a series of over 360 diffraction images was collected. For the experiments reported here, the C2F4I2 sample (Lancaster Synthesis, 98%) was further purified by several cycles of freeze-and-thaw. The gas nozzle temperature was 72°C, and the estimated gas density in the scattering volume was ≈10 torr (1 torr = 133 Pa).
RESULTS AND DISCUSSION
The Approach: UED.
In time-resolved experiments with UED, the actual signal being monitored is the net change of a diffraction pattern resulting from a chemical reaction. To follow the course of the fs-initiated structural change in the presence of ground-state species (typically 90%), we followed the diffraction-difference approach developed in our laboratory (17, 18). This approach, compared with simply analyzing the ITot (see below), has critical advantages: elimination of background scattering intensity, enhancement of product contribution to diffraction intensity, and cancellation of systematic errors in the diffraction-difference curve.
The diffraction data were analyzed according to our previous procedure (17, 18, 20). The two-dimensional diffraction images from the charge-coupled device were first converted to one-dimensional intensity curves by calculating the average intensity as a function of pixel radius from the primary beam center for each diffraction image. Then, the total raw scattering intensity, ITot(s), was obtained by further averaging of all the images at each time delay and converting the pixel radius to the momentum transfer parameter (s): s = (4π/λ)sin(θ/2), where λ is the de Broglie wavelength of the electrons and θ is the scattering angle.
In Fig. 1, we present a charge-coupled device image, together with a modified molecular scattering intensity, sM(s), and the corresponding radial distribution function, f(r), for the parent molecule C2F4I2 (data at −150 ps). The sM(s) curve was calculated with the structural parameters (21) obtained by conventional gas-phase electron diffraction. The sM(s) is given by:
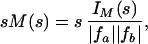 |
1 |
where fa and fb are the atomic scattering amplitudes (I and F atoms in our case), and IM is the molecular scattering intensity. IM is composed of interference terms from all atom–atom pairs and contains the molecular structural information: IM = ITot (s) − IB(s), where IB(s) is the background intensity profile. In the calculation, the theoretical sM(s) was normalized to the experimental sM(s) with a scaling factor.
Figure 1.

(Left) The two-dimensional diffraction image of C2F4I2 molecule recorded with the ultrashort electron pulses. An intensity profile across the image is also displayed at the bottom. The inset shows the structures of the two isomers. (Right) The experimental sM(s) and f(r) curves at t = −150 ps (red lines). The blue lines represent the theoretical calculations (see text). The different internuclear separations relevant to the chemical reaction under study are also shown in f(r).
The corresponding radial distribution curve, which provides the relative density of internuclear distances in a molecule, was generated by Fourier transforming sM(s) using the standard equation (22, 23):
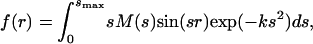 |
2 |
where the constant k (k = 0.02 Å2) is the known damping coefficient included for a limited s range. For consistency, we compared our results at −150 ps for the ground-state structure of the two isomers (anti and gauche) of C2F4I2 with that of static diffraction experiments (21). The fraction of anti and gauche isomers was obtained by a least-square fitting and found to be 75% and 25%, respectively. This result is in excellent agreement with the value of 76% and 24% obtained (at our gas temperature of 72°C) from the temperature-dependence curve of Hedberg and coworkers (21).
To monitor the transient structural change, we introduced the temporally resolved diffraction difference as ΔI(t; tref; s), where tref refers to the reference time: ΔI(t; tref; s) is simply ITot(t; s) minus ITot(tref; s). Typically, we scan diffraction from, say, −150 ps to +330 ps, with an accuracy of 2 ps. Thus we can easily obtain ΔI(t; −150 ps; s), which displays the evolution as a function of time and in reference to the −150-ps diffraction (the ground state structure). It follows from the definition of ITot (= IM + IB) that ΔI will give IM(t) −IM(tref). In so doing, the background signal is eliminated, thus focusing on the molecular structure (IM) under consideration. Furthermore, because IM for a chemical reaction has contributions from both reactant and product structures, ΔI gives, as demonstrated below, the product(s) contribution as it evolves with time; even if the fraction of product(s) is small, this approach avoids the dominance of the reactant diffraction in the signal.
For the sake of comparison with theory, molecular structural calculations were performed using the Jaguar 3.0 program (24). The LAV3P basis set (25) was used for the I atom and 6–31G* for the C and F atoms. For the parent molecule, both restricted Hartree-Fock and density functional theory (B3PW91) were used to obtain the optimized structures. With restricted Hartree-Fock, the optimized structures of C2F4I2 reproduce the experimental structures (21) very well (the deviation was at most 0.01 Å for bond length and 1° for angle); when density functional theory is used, the deviation was somewhat larger. For the anti and gauche radicals, the structures optimized at the restricted Hartree-Fock level of theory were used for diffraction data analysis. Compared with the parent molecule, the radicals have a longer C—I bond, but a shorter C—C bond. Single-point calculations with the localized second-order Møller–Plesset (MP2) method at the restricted Hartree-Fock optimized structures showed that the anti structure is ≈2 kcal/mol more stable than the gauche structure of the radical. The symmetrically bridged radical was found to be a transition state with ≈62 kcal/mol higher energy than the anti species.
Diffraction of Transient Structures.
For the chemical reaction investigated here, we consider its two elementary steps with the different structures involved:
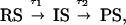 |
3 |
where the reactant structure (RS), in this case C2F4I2, forms the intermediate in τ1, whereas the intermediate structure (IS), C2F4I, transforms in time τ2 to the final product structure (PS), C2F4. Following the approach outlined above, we can now obtain ΔI(t; −150 ps; s). In Fig. 2, we show the experimental results for the corresponding ΔsM(s) and Δf(r) as a function of time; all data were referenced to tref = −150 ps. For each ΔI, the corresponding ΔsM(s) and Δf(r) curves were obtained through Eqs. 1 and 2, respectively. The ground-state structure, which is the data for −150 ps, is shown in Fig. 1. Note that the contribution in the radial distribution function from the F⋅⋅⋅I and C⋅⋅⋅I internuclear separations is evident near ≈3 Å. The I⋅⋅⋅I internuclear separation is at ≈5 Å (anti), and that of C—F is at ≈1.3 Å. In Fig. 2, the increase of the peak intensity at ≈3 Å with time is clear, while the peak intensity at ≈5 Å remains constant after 10 ps.
Figure 2.

The experimental diffraction-difference ΔsM(t; −150 ps; s) (Left) and Δf(t; −150 ps; r) (Right) curves at different reaction times referenced to the parent C2F4I2 data at −150 ps (solid lines). The shaded lines are the theoretical difference curves. The reaction time t is indicated, and the relevant internuclear distances are also displayed.
To follow the time evolution of individual structures, we obtained ΔI(t; +10 ps; s). As before, ΔI(t; +10 ps; s) = IM(t) −IM(+10 ps), and, therefore,
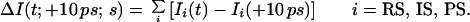 |
4 |
It is evident that the evolution of the structures in the reaction can be observed, as these three terms for reactant, intermediate, and product structure are distinct in some of their internuclear separations, and their contribution to the diffraction difference is therefore unique. For example, the change in IRS will be most pronounced at the I⋅⋅⋅I internuclear separation, while changes in IIS and IPS will be observed at the C—I, C⋅⋅⋅I and F⋅⋅⋅I distances. The dynamic time scale of the process is also evident. If, for example, the reaction proceeds with τ1 ≪ τ2 (τ1 < tref = 10 ps) in a nonconcerted pathway, then ΔI(t; +10 ps; s) will arise only from IIS and IPS (Eq. 4) with the depletion of IIS being evident at C—I, C⋅⋅⋅I, and F⋅⋅⋅I separations. Because the population of other internuclear separations, C—F, C—C, and F⋅⋅⋅F, is essentially unchanged, their contribution to the evolution in time of ΔI(t; +10 ps; s) will be zero. In Fig. 3, we display the theoretical simulations for the reaction studied, for the dynamical time scales, τ1 = 200 fs and τ2 = 17 ps.
Figure 3.

(Upper) The temporal evolution of the reactant, the intermediate, and the final product. The changes of the individual internuclear separations and the associated molecular structures are also shown. (Lower) Diffraction difference curves, labeled by the time of the reaction (t) and the reference time (tref).
Fig. 4 displays the Δf(t; +10 ps; r) diffraction data for the investigated reaction. The temporal evolution of the C—I, C⋅⋅⋅I, and F⋅⋅⋅I internuclear separations is clearly observed. In Fig. 5, the change of the population of the ground-state C2F4I intermediate and C2F4 product with time shows that the second bond breakage occurs in 17 ± 2 ps. The measurements were made both for the buildup of C2F4 structure and the decay of C2F4I structure (Fig. 5). The first bond breakage must occur on a time scale shorter than 10 ps, and this is consistent with the spectroscopic detection of I atoms on the fs time scale (26, 27); it occurs in ≈200 fs. In Fig. 4, it is seen that the intensity of peaks corresponding to depletion of C—I (≈2.2 Å), C⋅⋅⋅I (≈3.1 Å) and F⋅⋅⋅I (≈2.9 and ≈3.1 Å) nuclear separations is gradually increasing with time because of the secondary C—I bond breakage. The absence of an I⋅⋅⋅I component (≈5 Å) in Δf(r) confirms that the population of unreacted parent molecules remains constant after 10 ps (see Fig. 5) and is, therefore, absent in the difference curves. Hence, the reaction dynamics of C2F4I2 is a two-step nonconcerted process involving the intermediate C2F4I radical ground-state structure.
Figure 4.

(Top) The experimental diffraction-difference Δf(t; +10 ps; r) curves at different reaction times referenced to tref = +10 ps (solid lines). Shaded lines are the theoretical difference curves. (Middle and Bottom) The comparison between theoretical and experimental Δf(t; +8 ps; r) for the two structures, the bridged (Middle), and the classical, non-bridged (Bottom) structure. The experimental data in the lower two panels are identical; the difference in appearance is because of values of zero points of ΔI determined by the theoretical analysis (see refs. 8, 17, and 18). The reaction time t is indicated on each curve.
Figure 5.

The fraction (%) change of transient C2F4I radicals (diamond), final product C2F4 (closed circle), and parent C2F4I2 (open square), as a function of delay time. The error bars are one standard deviation. The solid curves are from the solution of the kinetic model, Eq. 3, with τ2 = 17 ± 2 ps for C2F4 data. For the decay of C2F4I, τ2 was found to be 18 ps, but the error bars are somewhat bigger. In the fitting, the first C—I bond breakage was taken as a step function, since it occurs in ≈200 fs; convolution with our time resolution was included (see text). The inset shows the clocking (zero-of-time) with better than 2 ps accuracy. Note that the fraction of reacted parent molecule detected at the time zero is half of the final fraction, and this is entirely consistent with the fact that the first bond breakage is much shorter than 10 ps. This observation also confirms the accurate clocking (2 ps) of time zero in these experiments, as shown in the inset.
The maximum internal energy for the radical is ≈42 kcal/mol (see ref. 27); the barrier for the second bond breakage is ≈15 kcal/mol (27). By using state detection on the fs time scale, a typical reaction time was measured to be 25 ps at 277 nm (27) and 69 ps at 307 nm (26). At our two-photon energy, the 17-ps time is entirely consistent with the range of available energy for the barrier crossing. We determined the percentage of the radicals undergoing further dissociation to be 82%, again consistent with the available energy that exceeds both the I and I* thresholds.
Two more interesting observations can be made. First, we can compare the rates observed by molecular structural changes with time (i.e., certain internuclear separations) and those observed by probing spectroscopic states. Of significance for the comparison is intramolecular vibrational-energy redistribution. The distribution of vibrational motions projected on the C—I reaction coordinate, monitored in these diffraction experiments, is different. This difference raises an important point regarding the role of intramolecular vibrational-energy redistribution and the nature of reaction trajectories (28). Such studies are under consideration, together with studies of the wavelength and intensity dependencies.
The second observation concerns the structure of the intermediate. The bridged radical model (29) has been postulated to explain the stereoselectivity observed in many reactions involving the haloethyl radicals. To explore this possibility, ΔI(t; +10 ps; s) difference curves were also fitted with the bridged radical structure rather than the classical radical structures (anti and gauche). The fitting with the classical radical gives more satisfactory results than that with the bridged model. To confirm this conclusion further, another set of data was also collected with only four time points (−90, 0, 8, and 188 ps) to increase the total integration time and signal-to-noise ratio at each delay time. As shown in Fig. 4, our experimental results for this set of data are also consistent with the classical structure. It is interesting to note that the time scale for C—C rotation relative to that of bond breakage may be the dominant effect for stereochemistry, in contrast with real molecular structural rearrangement.
Future experiments will include further refinements, i.e., better R values (see ref. 17) of structures and increased number of atoms, using the newly built third generation apparatus with enhanced sensitivity (by three orders of magnitude) and increased spatial resolution (by an order of magnitude).
Acknowledgments
This research was supported by a grant from the Air Force Office of Scientific Research and the Office of Naval Research. We thank the two colleagues who carefully reviewed the manuscript and made valuable comments.
ABBREVIATION
- UED
ultrafast electron diffraction
References
- 1.Sundström V, editor. Femtochemistry and Femtobiology: Ultrafast Reaction Dynamics at Atomic-Scale Reaction. Singapore: World Scientific; 1997. [Google Scholar]
- 2.Chergui M, editor. Ultrafast Chemical and Physical Processes in Molecular Systems. Singapore: World Scientific; 1996. [Google Scholar]
- 3.Manz J, Wöste L, editors. Femtosecond Chemistry. New York: VCH; 1995. [Google Scholar]
- 4.Zewail A H. Femtochemistry: Ultrafast Dynamics of the Chemical Bond. Singapore: World Scientific; 1994. [Google Scholar]
- 5.Castleman, A. W., Jr., ed. (1998) J. Phys. Chem. A102, 4021–4404.
- 6.Williamson J C, Zewail A H. Proc Natl Acad Sci USA. 1991;88:5021–5025. doi: 10.1073/pnas.88.11.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williamson J C, Cao J, Ihee H, Frey H, Zewail A H. Nature (London) 1997;386:159–162. [Google Scholar]
- 8.Hargittai I, Hargittai M, editors. Stereochemical Applications of Gas-Phase Electron Diffraction. New York: VCH; 1988. [Google Scholar]
- 9.Ischenko A A, Schäfer L, Luo J Y, Ewbank J D. J Phys Chem. 1994;98:8673–8678. [Google Scholar]
- 10.Elsayed-Ali H E, Mourou G A. Appl Phys Lett. 1988;52:103–104. [Google Scholar]
- 11.Ráksi F, Wilson K R, Jiang Z, Ikhlef A, Côté C Y, Kieffer J-C. J Chem Phys. 1996;104:6066–6069. [Google Scholar]
- 12.Schoenlein R W, Leemans W P, Chin A H, Volfbeyn P, Glover T E, Balling P, Zolotorev M, Kim K-J, Chattopadhyay S, Shank C V. Science. 1996;274:236–238. doi: 10.1103/PhysRevLett.77.4182. [DOI] [PubMed] [Google Scholar]
- 13.Perman B, Šrajer V, Ren Z, Teng T, Pradervand C, Ursby T, Bourgeois D, Schotte F, Wulff M, Kort R, et al. Science. 1998;279:1946–1950. doi: 10.1126/science.279.5358.1946. [DOI] [PubMed] [Google Scholar]
- 14.Tomov I V, Chen P, Rentzepis P M. J Appl Crystallogr. 1995;28:358–362. [Google Scholar]
- 15.Larsson J, Heimann P A, Lindenberg A M, Schuck P J, Bucksbaum P H, Lee R W, Padmore H A, Wark J S, Falcone R W. Appl Phys A. 1998;66:587–591. [Google Scholar]
- 16.Rischel C, Rousse A, Uschmann I, Albouy P-A, Geindre J-P, Audebert P, Gauthier J-C, Förster E, Martin J-L, Antonetti A. Nature (London) 1997;390:490–492. [Google Scholar]
- 17.Cao J, Ihee H, Zewail A H. Chem Phys Lett. 1998;290:1–8. [Google Scholar]
- 18.Ihee H, Cao J, Zewail A H. Chem Phys Lett. 1997;281:10–19. [Google Scholar]
- 19.Williamson J C, Zewail A H. Chem Phys Lett. 1993;209:10–16. [Google Scholar]
- 20.Dantus M, Kim S B, Williamson J C, Zewail A H. J Phys Chem. 1994;98:2782–2796. [Google Scholar]
- 21.Thomassen H, Samdal S, Hedberg K. J Am Chem Soc. 1992;114:2810–2815. [Google Scholar]
- 22.Hedberg K, Iwasaki M. Acta Crystallogr. 1964;17:529–533. [Google Scholar]
- 23.Bartell L S. In: Physical Methods of Chemistry. Weissberger A, Rossiter B W, editors. New York: Wiley; 1972. [Google Scholar]
- 24.jaguar 3.0 (1997) (Schrödinger, Inc., Portland, OR).
- 25.Hay P J, Wadt W R. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 26.Khundkar L R, Zewail A H. J Chem Phys. 1990;92:231–242. [Google Scholar]
- 27.Zhong D, Ahmad S, Zewail A H. J Am Chem Soc. 1997;119:5978–5979. [Google Scholar]
- 28.Møller K B, Zewail A H. Chem Phys Lett. 1998;295:1–10. [Google Scholar]
- 29.Skell P S, Traynham J G. Acc Chem Res. 1984;17:160–166. [Google Scholar]