

Abstract
The human β-globin locus has been extensively studied as a model of tissue and developmental stage-specific gene expression. Structural mapping of naturally occurring mutations, including transfection and transgenic studies, and the recent finding of intergenic transcripts have suggested that there are cis-acting sequence elements in the Aγ–δ intergenic region involved in regulating γ- and β-globin gene expression. To determine whether previously identified sequences in the Aγ–δ intergenic region are required for appropriate developmental expression of the human β-globin gene cluster, transgenic mice were generated by transfer of yeast artificial chromosomes containing the entire human β-globin locus. Three different deletions of the Aγ–δ intergenic region were introduced, including (i) deletion of the 750-bp Aγ 3′ regulatory element (Aγe), (ii) deletion of 3.2 kb upstream of the δ-globin gene encompassing pyrimidine-rich sequences and the recently described intergenic transcript initiation site, and (iii) deletion of a 12.5-kb fragment encompassing most of the Aγ–δ globin intergenic region. Analysis of multiple transgenic lines carrying these deletion constructs demonstrated that the normal stage-specific sequential expression of the ɛ-, γ-, and β-globin genes was preserved, despite deletion of these putative regulatory sequences. These studies suggest that regulatory sequences required for activation and silencing of the human β-globin gene family during ontogeny reside proximally to the genes and immediately 5′ to the human γ- and β-globin genes.
Hereditary anemias caused by mutations in the α- and β-globin loci are among the most common genetic disorders worldwide (reviewed in ref. 1). The human β-globin locus spans a chromosomal region of >80 kb and contains the locus control region (LCR) and the following five functional genes that are sequentially expressed during development: the embryonic ɛ-, the fetal Aγ- and Gγ-, and the adult δ- and β-globin genes. The LCR directs high-level erythroid-specific expression of the β-globin gene family and encompasses five developmentally stable, erythroid-specific DNase I-hypersensitive sites (5′ HSs 1–5) located 6–18 kb 5′ to the ɛ-globin gene (reviewed in refs. 1 and 2).
In murine models, individual human γ- and β-globin transgenes are expressed at low levels in the absence of the LCR, but are regulated appropriately at fetal and adult stages of development (3–8). The δβ and Aγδβ-thalassemias and deletion forms of hereditary persistence of fetal hemoglobin (HPFH) are naturally occurring mutations associated with the persistent expression of fetal hemoglobin in adult life, albeit at variable levels. The presence of multiple positive and negative regulatory elements in the Aγ–δ globin intergenic sequences could explain the differing phenotypes of seemingly similar structural deletions. One of these elements, a 750-bp region located 3′ to the Aγ-globin gene, was originally defined as an enhancer (Aγe) in transient transfection assays (9–12). More recently, studies in transgenic mice have suggested that sequences in this region may have a role in developmental silencing of the Aγ globin gene (13). A-T rich sequences within this element have been shown to bind the A-T binding protein SATB1, a nuclear matrix- and scaffold-associated DNA-binding protein (9–13).
Other sequences upstream of the δ-globin gene, present in most δβ-thalassemia mutations and deleted in many of the deletion HPFH variants, may also participate in silencing of the γ-globin genes. In transfection assays, the R region, located 2.3–4 kb upstream of the δ-globin gene, had a negative regulatory effect on both the γ- and β-globin promoters (14). Pyrimidine-rich sequences 960 bp 5′ to the δ-globin gene bound a protein complex, PYR, with homology to the yeast SWI/SNF family (15). This complex was primarily present in cells expressing adult globins and is made up of at least four proteins with homology to known mammalian SWI/SNF subunits (15, 16). In transgenic lines produced by transfer of a cosmid construct containing the Aγ–β globin gene region with a 511-bp deletion of these pyrimidine-rich sequences, delays in γ-globin gene silencing and β-globin gene activation were observed (16).
More recently, another potential role for sequences in the intergenic regions has been suggested (17, 18). Stage-restricted intergenic transcripts associated with the human ɛ- and γ-globin genes were detected in primitive erythroid cells. Transcripts associated with the δ- and β-globin genes were detected at embryonic day 12.5 (E12.5) and thereafter (17). In contrast, transcripts from the LCR were detected at all developmental stages. Other studies have described stage-independent intergenic transcripts (18). A transition from a relatively DNase-sensitive to a DNase-insensitive region was detected upstream of the δ-globin genes, suggesting that some sort of chromatin boundary exists 5′ to the δ-globin gene (17). The start site of the 5′ δ-intergenic transcripts maps to HUMHBB 51405 (GenBank accession no. U01317) and overlaps with a minimal region of structural difference between some HPFH and δβ-thalassemia deletions located 2.8–3.7 kb 5′ to the δ-globin gene (18). While all these elements may play important roles in the developmental regulation of globin genes, their regulatory role in the context of the entire β-globin locus has not been examined.
Transgenic models have been developed in which the integrity of the human β-globin locus is preserved by transfer of the locus in yeast artificial chromosomes (YACs) (19–24). In the present studies, the role of regulatory sequences in the region between the Aγ- and δ-globin genes was tested (i) by reproducing the 5′ breakpoints of African HPFH-1 and deleting sequences encompassing the PYR-binding region R, (ii) by deleting the entire region between the Aγ- and δ-globin genes, and (iii) by deleting the Aγe. Mutations were made by two-step homologous recombination in a 150-kb YAC carrying the entire human β-globin locus (25). The effects of the deletions on human ɛ-, γ-, and β-globin gene regulation during development were assessed in transgenic lines generated by transfer of the recombinant β-globin YACs.
Materials and Methods
Generation of Deletions in the Aγ–δ Intergenic Region.
The A201F4 yeast strain carrying a 150-kb YAC containing the intact human β-globin locus (25) was modified by one-step gene disruption (26). A LYS2 gene and a neophosphotransferase cassette were inserted into the URA3 gene in the right arm of the YAC. This process generated strain A20NL (for A201F4 neo/LYS2).
Aγe Deletion.
The Aγe was deleted in a 753-bp HindIII fragment between sites at 41364 and 42117, where the numbers indicate the sequence position in the human β-globin locus (GenBank accession no. U01317) HUMHBB. The 5′ flanking fragment was an SspI (38683) to HindIII (41364) fragment. The 3′ flanking fragment was a HindIII (42117) to SspI (44077) fragment. These segments were inserted into the URA3-containing yeast integrating plasmid (YiP) vector pRS-406 (Stratagene). The Aγe-deleted (Aγe−) YiP was linearized by restriction with NheI, and introduced into A20NL by the lithium acetate transformation procedure (27), and recombinants were identified by selection for URA3, LYS2, and TRP1 prototrophy.
Yeast cells with a single copy of the modified β-globin YAC were grown overnight in yeast extract/peptone/dextrose (YPD) medium, and were counterselected for the URA3 gene with 5-fluoroorotic acid (5-FOA) (28). Culture in 5-FOA results in eviction of the YiP containing the URA3 gene, generating either the WT β-globin YAC or a YAC with the targeted deletion of the Aγe. The integrity of the Aγe− YAC was confirmed by restriction mapping and Southern analysis, and by fingerprint analysis of repeat sequences as described (25). (YAC mapping data are shown in Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org.)
D2 and D5 Deletions.
Two overlapping deletions were made in the Aγ–δ intergenic region. The D2 deletion removes 3,169 bp 5′ to the δ-globin gene. The D5 deletion extends 9.3 kb further 5′ and also removes the Aγe (Fig. 1). A URA3-containing D5-YiP was created by inserting DNA sequences that flank the 12.5-kb region to be deleted, into pRS-406. The 5′ segment was a 1,422-bp fragment from XhoI (39942) to HindIII (41364) fragment. The 3′ homologous segment was a 1,343-bp EcoRI (53871) to BamHI (55214) fragment. The D5-YiP was linearized at the unique SalI site in the 3′ fragment and introduced into yeast strain A20NL as described above. Allelic replacement in yeast produced a 12.5-kb deletion from the HindIII site at position 41364 to the EcoRI site at position 53871.
Figure 1.

Structure of the human β-globin locus YAC and deletions of the Aγ–δ intergenic region. (A) The Aγ–δ intergenic region and the position of putative regulatory elements. Open oval, the 3′ Aγ-regulatory element; open rectangle, the pseudo-β-globin gene; striped oval, the R silencer; small black oval, pyrimidine-rich region. Vertical lines “R” designate EcoRI restriction sites. The Aγe−, D2, and D5 deletions are depicted by black lines below the map. Black box 1 depicts the deletion made by Calzolari et al. (38). The “In” indicates the location of the intergenic transcription initiation site (17). The black box 2 depicts the deletion made by O'Neill et al. (16). (B) The map of the human β-globin locus in the A20 neo/lys YAC. The functional genes are depicted as black boxes. Vertical arrows indicate the locations of DNase I-hypersensitive sites. The vertical lines labeled “S” indicate SfiI restriction sites, and “Sa” designates a unique SalI restriction site. The black boxes below the linear map designate the positions of probes used in Southern analysis. (C) A diagram of the β-globin YAC sequences mapped by pulsed-field gel electrophoresis (PFGE) in each of the Aγe−, D2, and D5 lines. The size of the SfiI restriction fragment is indicated on the right.
Construction of the D2 deletion was achieved by using a similar strategy by inserting 5′ and 3′ sequences flanking the 3.2-kb deletion into the YiP pRS406 generating the D2-YiP. The 5′ flanking fragment in the D2-YiP extended from XbaI (49459) to EcoRI (50701). The 3′ flanking fragment was the same as in the D5 construct. The D2-YiP was linearized at the unique SalI site in the 3′ homologous sequence and transferred into the A20NL yeast strain as outlined above. The new yeast strains containing the deleted form of the β-globin YAC were designated A20D5, A20D2, and A20Aγe−.
YAC DNA Isolation and Microinjection.
YAC DNA was purified as described (19). The purified A20D5, A20D2, and A20Aγe− YACs were microinjected into fertilized oocytes from B6D2 F1 animals (The Jackson Laboratory) as described (29). Transgenic animals were screened by PCR analysis of DNA, using primers in the human γ- and β-globin genes, and then by conventional and long-range restriction mapping as described (25).
Structural Analysis of YAC Sequences in Transgenic Mice.
The structure and copy number of the β-globin YAC locus in transgenic animals were determined by Southern analysis of EcoRI-restricted genomic DNA and SfiI-restricted chromosomal DNA from F1 animals as described (19, 22). To normalize samples for differences in DNA loading, Southern blots were hybridized with a 32P-labeled 2.1-kb EcoRI fragment specific for the mouse Nrf1 gene as an internal control (31).
Primer Extension Analysis.
Total RNA was isolated from embryonic yolk sac (E8.5–E11.5), fetal liver (E12.5–E16.5), or blood (E18.5 and later) of transgenic embryos or animals, using RNAzol-B solution (Cinna/Biotex Laboratories, Houston). Primer extension analysis was performed with primers for mβh1, mβmaj, mɛy, Huγ, and Huβ, as described (19). The primer for human embryonic globin gene was 5′-GCCTCTTCCACATTCATCTTG-3′. Primer extension products were quantified as described (19).
Results
Structure/function correlations of HPFH and δβ-thalassemia have been difficult to establish, because the deletions are overlapping, and they remove extensive sequences 3′ to the locus as well (reviewed in ref. 1). In this analysis, three deletions were constructed in the Aγ–δ intergenic region that reproduce naturally occurring breakpoints, or delete specific elements, but do not alter the promoters or the genes of the human β-globin locus. Fig. 1 shows a map of the modified YAC, designated A20NL, with the location of the deletions made and probes used to map recombinant YACs and transgenic lines.
Construction of Aγe− YAC and Structural Analysis of YAC Sequences in Transgenic Lines.
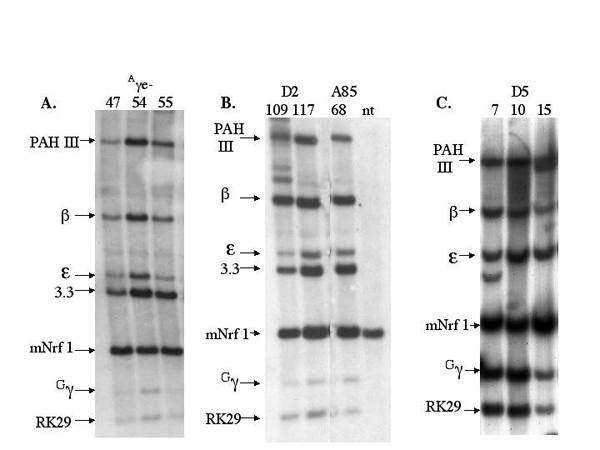
The Aγe is located on a 750-bp HindIII fragment 3′ to the Aγ-globin gene. A URA3-containing YiP was constructed with sequences flanking the Aγe, and two-step allelic replacement was performed in the A20NL strain. The structure of the Aγe-deleted (A20Aγe−) β-globin YAC was confirmed by Southern analysis (see Figs. 5 and 6, which are published as supporting information on the PNAS web site). Three transgenic lines were obtained carrying intact copies of the Aγe-deleted YAC and are as follows: Aγe−.47; Aγe−.54; and Aγe−.55. Probes for the LCR (pAHIII and 3.3), the human globin genes, and sequences 3′ to the β-globin gene all identified the correct EcoRI restricted fragments in Southern analysis. pulsed-field gel electrophoresis (PFGE) of SfiI-restricted murine chromosomal DNA was performed to assess the structural integrity of the YAC transgenic loci, as previously described (Fig. 2; ref. 19). Line Aγe−.47 contains a single intact copy of the locus (Figs. 1C and 2). The larger 150-kb band reflects incomplete digestion of the SfiI site proximal to the ɛ-globin gene (Fig. 2). In line Aγe−.54 there were two intact copies, and a third partial copy extending from the LCR through Gγ (Figs. 1C and 2). The complete copy or copies of the Aγe− YAC in lines 47 and 54 extends from the 1.8-kb EcoRI probe 5′ of the LCR to the RK29 probe 3′ to the β-globin gene (Figs. 1C and 2). Line Aγe−.55 had a single intact copy of the locus but expression was difficult to detect and this line was not further analyzed (Fig. 2).
Figure 2.

Long-range Southern mapping of the Aγe−, D2, and D5 transgenic mouse lines. Chromosomal DNA, embedded in agarose blocks, was digested with SfiI and resolved by PFGE. DNA probes depicted in Fig. 1B were hybridized with Southern blots. With the exception of the 3.3-kb probe, all probes hybridized with the same SfiI fragment(s), demonstrating the integrity of the YAC transgene. The 3.3-kb probe hybridizes with a 10-kb SfiI fragment (see Fig. 1B). Each panel depicts Southern analysis of pulsed-field gels loaded with DNA from each of the lines as indicated.
Analysis of Globin Gene Expression in Aγe− YAC Transgenic Lines.
Primer extension analysis of samples from embryonic yolk sac, fetal liver, or blood of transgenic embryos or animals was performed and products were quantified as described (22). All six of the murine and human globin primers (mβh1, mβmaj, mɛy, Huγ, Huβ, and Huɛ) were included in each reaction (Fig. 3). Primer extension analysis of samples from WT line A85.68 appears at the top of Fig. 3. In lines Aγe−.47 and Aγe−.54, the human γ-globin genes were activated in the blood islands of the yolk sac in concert with the human ɛ-globin gene and mɛy and βh1. The levels of γ-globin gene expression in E16 fetal liver and adult blood samples were not higher in the Aγe− lines than in line A85.68 (Fig. 3). Activation of adult human β- and mouse β-globin gene expression between E10.5 and E12.5 of gestation was observed both in the Aγe− lines and in line A85.68 in fetal liver samples (Fig. 3). Thus, deletion of the Aγe did not alter the timing of either β-globin gene activation or of γ-globin gene silencing when compared with WT β-globin YAC line A85.68. These data are in agreement with similar studies in other Aγe β-globin YAC transgenic mice generated with an Aγ-deleted construct derived from the same A201F4 YAC (24).
Figure 3.

Primer extension analysis of Aγe-deleted lines. Autoradiographs of primer extension analysis of RNA prepared from yolk sac (E8.5–E11.5), fetal liver (E12.5–E16.5), or blood (E18.5 and E20.5). Bands specific for each gene product were quantified by PhosphorImager (Molecular Dynamics) analysis. To the right of each primer extension analysis is a graphic representation showing the mean of at least three experiments. The mRNA level of the human ɛ-, γ-, or β-globin genes is expressed as the fraction ɛ, γ, or β/(ɛ + γ + β) on the y axis. The developmental time points at which tissues were harvested are indicated on the x axis. Arrows indicate the position of each murine or human mRNA-extension product. A85.68 is a transgenic line carrying a single copy of the WT locus.
Construction of Deletions of the Aγ–δ Globin Gene Region in the A20NL YAC.
Two deletion mutants within the Aγ–δ intergenic region, D2 and D5 (Fig. 1A), were also constructed in the A20NL YAC by two-step allelic replacement as described above. The D2 deletion encompasses 3,169 bp extending to the EcoRI site immediately upstream of the δ-globin gene promoter HUMHBB (50701–53871; Fig. 1A). This deletion removes the PYR sequences that bind the trans-acting PYR complex in in vitro binding assays (15, 16). It also removes sequences close to the 5′ breakpoints of Italian HPFH 5, the δβ-thalassemia identified in African-American individuals (32, 33), and the initiation site for intergenic transcripts 5′ to the δ-globin gene (ref. 17; Fig. 1A). In the 12.5-kb [HUMHBB (41364–53871)] deletion, the entire Aγ–δ intergenic globin region was deleted (Fig. 1A). The structural integrity of the deleted β-globin YACs, designated A20D5 and A20D2, was confirmed by Southern analysis (Fig. 5; ref. 25).
Structural Analysis of β-Globin YAC Sequences in Transgenic Lines Carrying the D5 and D2 Deletions.
Microinjection produced three independent β-globin YAC transgenic lines for each of the deletion constructs. The A20D2 lines were designated D2.109, D2.117, and D2.123. The D2.123 line had internal rearrangements in the YAC and was not studied further (data not shown). The A20D5 lines were designated D5.7, D5.10, and D5.15. Long-range mapping of SfiI-restricted mouse chromosomal DNA identified a single copy of the A20D2 YAC in lines D2.109 and D2.117 extending from the LCR through sequences in probe RK29, located 30 kb 3′ to the β-globin gene (Figs. 1C and 2). In line D5.7, probes pAHIII, ɛ, γ, β, RK29, and DF10 all hybridized to a 210-kb SfiI fragment, whereas probes ɛ, γ, β, RK29, and DF10 also hybridized with a 160-kb SfiI fragment (Figs. 1C and 2). These results are consistent with the integration of two tandem copies of the β-globin locus, where one copy contains the intact locus and a second copy extends from the ɛ-globin gene to the region 30 kb 3′ to the adult β-globin (Fig. 1C). In line D5.10, several SfiI fragments were consistently observed that hybridized with probes spanning from pAHIII to RK29 consistent with a locus copy number of 4 (Figs. 1C and 2). The precise arrangement of each of the four copies of the β-globin locus could not be definitively established. In line D5.15, all probes hybridized to a single 170-kb SfiI fragment, whereas sequences in probes 1.8RI, 0.7B/RI, and 3.3RI 5′ to the LCR were not present (Figs. 1C and 2). Because of partial digestion at the SfiI site 5′ and proximal to the ɛ-globin gene, two bands, differing by 10 kb in length (170 and 180 kb) are visible in the autoradiograph (Fig. 2). Thus, lines D2.109 and D2.117 each contained a single copy of the A20D2 YAC. Probes for the LCR (pAHIII, 3.3RI), the ɛ-, Gγ-, and β-globin genes, and sequences 3′ to β-globin gene (RK29) all identified the correct EcoRI fragments on Southern analysis and confirmed the copy numbers demonstrated by PFGE (Fig. 6).
Primer Extension Analysis of Globin Gene Expression in D2 and D5 Deletion Lines.
The developmental expression of the human ɛ-, γ-, and β-globin genes was compared in the A20D2, A20D5, and A85.68 lines by plotting the mean of pooled results of the primer extension analyses from several animals at each time point (Fig. 4). In all of the D2- and D5-derived lines, expression of the ɛ- and mɛy-globin genes coincided. Activation of the human and murine adult β-globin genes occurred at E11.5–E12.5. Human γ-globin gene expression was detected in E8.5 yolk sac samples (Fig. 4). Between E11.5 and E12.5, the relative levels of both the ɛ- and γ-globin gene decreased, although expression of the human γ-globin gene persisted. In all A20D2- and A20D5-derived lines, the level of γ-globin expression did not exceed 4% (1.5–3.9%) of mouse β-globin expression (when corrected for copy number) at E14.5. At this developmental time point the WT line A85.68 expresses γ-globin at 3.3% of mouse β-globin per copy. Furthermore, the pattern of β-globin gene expression between E11.5 and E16.5 was the same for A85.68, D2.109, D2.117, D5.7, and D5.10 transgenic lines (Fig. 4). Thus, neither the 3.2-kb D2 deletion nor the 12.5-kb D5 deletion of the Aγ–δ intergenic region significantly altered the developmental timing of the human β-globin gene expression during ontogeny, compared with the WT β-globin YAC line A85.68.
Figure 4.

Primer extension analyses of the D2- and D5-deleted transgenic lines. (A Upper) Autoradiographs of primer extension products resolved on acrylamide gels. The transgenic line from which tissues were harvested is indicated above each gel. The developmental time point of tissue harvest is indicated above each lane. Specific extension products are indicated by arrows and labeled to the right and left of each autoradiograph. Graphic representations of the PhosphorImager analysis are shown below the autoradiograph. (B) Graphic representation of developmental expression of D2.109 and D.117 are shown individually and the mean expression of multiple experiments from both transgenic lines is also shown.
Discussion
In these studies, the effects of several deletions in the human Aγ–δ intergenic region on the expression of the β-globin gene family in YAC transgenic mice were analyzed. A number of previous studies suggested that DNA sequences that participate in silencing the fetal γ-globin genes or activation of β-globin genes were located in the Aγ–δ intergenic region. Thus, we hypothesized that making a deletion of either the Aγe, the 3.2-kb fragment upstream of the δ-globin gene, or the 12.5-kb Aγ–δ intergenic fragment would impair fetal globin gene silencing or affect β-globin gene activation. The normal regulation of the human β-globin gene family we observed in mice with the Aγe deletion reproduces the results of a previous study in Aγe-deleted YAC transgenic mice (24). Despite the deletion of putative regulatory elements in the Aγ–δ intergenic region, a total of five D2 and D5 transgenic lines reported here exhibited a pattern of human globin gene expression during ontogeny indistinguishable from that observed in four β-globin YAC transgenic lines carrying the WT locus (19, 22).
YAC transgenic mice carrying the entire WT β-globin locus are attractive models of developmental gene regulation, as they exhibit more reproducible globin gene switching (19, 22) than do transgenic mice carrying smaller β-globin constructs (9, 30, 34, 35), and they maintain the normal spatial organization of the locus. However, because mice express only embryonic and adult β-globin genes, the extent to which murine transcription factors recapitulate the normal control of the human β-globin locus remains an important question. A number of studies of transgenic lines demonstrate that murine trans-acting factors, at least partially, reproduce the regulation of the human β-like globin genes. Thus, β-globin YAC transgenic mice carrying a T → A transition at position −117 of Aγ gene promoter, associated with HPFH in human populations, also exhibit persistent human fetal globin gene expression (21). Studies of the phenotypes of mutations introduced into the LCR or the 5′ ɛ silencer of β-globin YAC transgenic mice have also provided new insights into the mechanism of globin gene regulation (23, 36, 37). These studies suggest that mutations in the human β-globin locus in YAC transgenic mice recapitulate many of the effects of naturally occurring mutations in the endogenous human locus.
Our results contrast with other studies of deletions 5′ to the δ-globin gene (16, 17, 38). In a study by Calzolari et al. (38), a 2.5-kb deletion was made [HUMHBB (49834–52338)] that extends further 5′ in the Aγ–δ intergenic region than does the D2 deletion, and it overlaps the D2 deletion by 1.6 kb (Fig. 1A, solid bar 1). Position-effect variegation was observed primarily in the single-copy line that was integrated near centromeric DNA, but position effects were also evident in the multicopy line (38). Nuclear runoff assays demonstrated decreased and delayed transcription of the β-globin gene in the single-copy line (38). However, in our studies the D5 deletion removes all of the sequences in the Calzolari deletion, and no effect on the timing of γ-globin silencing or β-activation was observed (Fig. 4). We cannot rule out the possibility that only the specific deletion made in the Calzolari study delays β-globin activation, whereas larger deletions obscure the phenotype. Although the initiation site of 5′ δ intergenic transcripts identified by Gribnau et al. (17) is deleted in the D2- and D5-derived lines, delayed β-globin gene activation was not observed in the five transgenic lines. This finding suggests that the initiation of intergenic transcripts from this site 5′ to the δ-gene is not functionally required for appropriate stage-restricted activation of gene expression.
Our results also differ from the data of O'Neill et al. (16), in which a 511-bp fragment encompassing a PYR element 5′ of the δ-globin gene (Fig. 1A, solid bar 2) was deleted from a 29-kb genomic fragment extending from the Aγ-globin gene through the β-globin gene and linked to a 2.5-kb micro (μ)-LCR construct. Three mouse lines, two with one copy of the locus, and one with three copies of the locus, demonstrated a delay in the silencing of the Aγ-globin gene, and/or a delay in the initiation of β-globin gene expression (16). However, the extent to which delayed γ-silencing and impaired β-activation occurred varied among the three lines, again suggesting position-of-integration effects. There are several possible explanations for the different results of these analyses. In our studies, a YAC containing the entire β-globin locus with its native structure was used, as opposed to the μLCR construct tested by O'Neill et al. (16). When the μLCR is linked to the Aγ-globin gene, the overall structure of the transgenic construct is significantly different from the normal arrangement of the locus, particularly with respect to distance from the LCR. The PYR region deleted in these studies was suggested to have a locus-opening function that allowed transcription factors to bind the DNA (16). The PYR complex that binds this region also binds to DNA sequences within both γ-globin genes. It is possible, particularly in our transgenic mice with the D5 deletion, that the δ- and β-globin genes are brought into close proximity of these PYR-binding sites, thus obscuring the effect of deleting the 5′ δ PYR region. Our results are consistent with the evidence that in the endogenous murine or human β-globin loci, regulatory sequences essential for stage-restricted expression are located proximally to each of the genes. Sequences in other regions of the locus may play a modulating role in gene expression but are not required for sequential activation and silencing of the genes.
Supplementary Material
Acknowledgments
We thank Jennifer Gregonis and Julie Vargas for technical assistance, Niki Nibbe for assistance with manuscript preparation, and Y. W. Kan for his ongoing support. This work was supported by the Northern California Comprehensive Sickle Center, which is funded by National Institutes of Health Grant PO1-HL20985 (to K.M.L.G.). The transgenic facility at the University of California, San Francisco, is funded in part by the Lucille Markey Foundation.
Abbreviations
- YAC
yeast artificial chromosome
- LCR
locus control region
- HPFH
hereditary persistence of fetal hemoglobin
- YiP
yeast integrating plasmid
- En
embryonic day n
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Stamatoyannopoulos G, Grosveld F. In: The Molecular Basis of Blood Diseases. Stamatoyannopoulos G, Marjerus P W, Perlmutter R M, Varmus H, editors. Philadelphia: Saunders; 2001. pp. 135–182. [Google Scholar]
- 2.Cunningham J M, Jane S M. Semin Hematol. 1996;33:9–23. [PubMed] [Google Scholar]
- 3.Costantini F, Radice G, Magram J, Stamatoyannopoulos G, Papayannopoulou T, Chada K. Cold Spring Harbor Symp Quant Biol. 1985;50:361–370. doi: 10.1101/sqb.1985.050.01.046. [DOI] [PubMed] [Google Scholar]
- 4.Townes T M, Lingrel J B, Chen H Y, Brinster R L, Palmiter R D. EMBO J. 1985;4:1715–1723. doi: 10.1002/j.1460-2075.1985.tb03841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kollias G, Wrighton N, Hurst J, Grosveld F. Cell. 1986;46:89–94. doi: 10.1016/0092-8674(86)90862-7. [DOI] [PubMed] [Google Scholar]
- 6.Chada K, Magram J, Costantini F. Nature. 1986;319:685–689. doi: 10.1038/319685a0. [DOI] [PubMed] [Google Scholar]
- 7.Ryan T M, Behringer R R, Martin N C, Townes T M, Palmiter R D, Brinster R L. Genes Dev. 1989;3:314–323. doi: 10.1101/gad.3.3.314. [DOI] [PubMed] [Google Scholar]
- 8.Collis P, Antoniou M, Grosveld F. EMBO J. 1990;9:233–240. doi: 10.1002/j.1460-2075.1990.tb08100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dillon N, Grosveld F. Nature. 1991;350:252–254. doi: 10.1038/350252a0. [DOI] [PubMed] [Google Scholar]
- 10.Stamatoyannopoulos G, Josephson B, Zhang J W, Li Q. Mol Cell Biol. 1993;13:7636–7644. doi: 10.1128/mcb.13.12.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bodine D M, Ley T J. EMBO J. 1987;6:2997–3004. doi: 10.1002/j.1460-2075.1987.tb02605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham J M, Purucker M E, Jane S M, Safer B, Vanin E F, Ney P A, Lowrey C H, Nienhuis A W. Blood. 1994;84:1298–1308. [PubMed] [Google Scholar]
- 13.Li Q, Stamatoyannopoulos J A. Mol Cell Biol. 1994;14:6087–6096. doi: 10.1128/mcb.14.9.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitale M, Di Marzo R, Calzolari R, Acuto S, O'Neill D, Bank A, Maggio A. Biochem Biophys Res Commun. 1994;204:413–418. doi: 10.1006/bbrc.1994.2474. [DOI] [PubMed] [Google Scholar]
- 15.O'Neill D, Bornschlegel K, Flamm M, Castle M, Bank A. Proc Natl Acad Sci USA. 1991;88:8953–8957. doi: 10.1073/pnas.88.20.8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Neill D, Yang J, Erdjument-Bromage H, Bornschlegel K, Temmpst P, Bank A. Proc Natl Acad Sci USA. 1999;96:349–354. doi: 10.1073/pnas.96.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P. Mol Cell. 2000;5:377–386. doi: 10.1016/s1097-2765(00)80432-3. [DOI] [PubMed] [Google Scholar]
- 18.Plant K E, Routledge S J, Proudfoot N J. Mol Cell Biol. 2001;21:6507–6514. doi: 10.1128/MCB.21.19.6507-6514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaensler K M, Kitamura M, Kan Y W. Proc Natl Acad Sci USA. 1993;90:11381–11385. doi: 10.1073/pnas.90.23.11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson K R, Clegg C H, Huxley C, Josephson B M, Haugen H S, Furukawa T, Stamatoyannopoulos G. Proc Natl Acad Sci USA. 1993;90:7593–7597. doi: 10.1073/pnas.90.16.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson K R, Li Q L, Clegg C H, Furukawa T, Navas P A, Norton E J, Kimbrough T G, Stamatoyannopoulos G. Proc Natl Acad Sci USA. 1995;92:5655–5659. doi: 10.1073/pnas.92.12.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porcu S, Kitamura M, Witkowska E, Zhang Z, Mutero A, Lin C, Chang J, Gaensler K M L. Blood. 1997;90:4602–4609. [PubMed] [Google Scholar]
- 23.Liu Q, Bungert J, Engel J D. Proc Natl Acad Sci USA. 1997;94:169–174. doi: 10.1073/pnas.94.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Q, Tanimoto K, Bungert J, Engel J D. Proc Natl Acad Sci USA. 1998;95:9944–9949. doi: 10.1073/pnas.95.17.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaensler K M L, Burmeister M, Brownstein B H, Taillon-Miller P, Myers R M. Genomics. 1991;10:976–984. doi: 10.1016/0888-7543(91)90188-k. [DOI] [PubMed] [Google Scholar]
- 26.Rothstein R J. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 27.Schiestl R H, Gietz R D. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 28.Sikorski R S, Boeke J D. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- 29.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 226–248. [Google Scholar]
- 30.Lloyd J A, Krakowsky J M, Crable S C, Lingrel J B. Mol Cell Biol. 1992;12:1561–1567. doi: 10.1128/mcb.12.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan J Y, Han X L, Kan Y W. Proc Natl Acad Sci USA. 1993;90:11371–11375. doi: 10.1073/pnas.90.23.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camaschella C, Serra A, Gottardi E, Alfarano A, Revello D, Mazza U, Saglio G. Blood. 1990;75:1000–1005. [PubMed] [Google Scholar]
- 33.Anagnou N P, Papayannopoulou T, Stamatoyannopoulos G, Nienhuis A W. Blood. 1985;65:1245–1251. [PubMed] [Google Scholar]
- 34.Enver T, Raich N, Ebens A J, Papayannopoulou T, Costantini F, Stamatoyannopoulos G. Nature. 1990;344:309–313. doi: 10.1038/344309a0. [DOI] [PubMed] [Google Scholar]
- 35.Strouboulis J, Dillon N, Grosveld F. Genes Dev. 1992;6:1857–1864. doi: 10.1101/gad.6.10.1857. [DOI] [PubMed] [Google Scholar]
- 36.Peterson K R, Clegg C H, Navas P A, Norton E J, Kimbrough T G, Stamatoyannopoulos G. Proc Natl Acad Sci USA. 1996;93:6605–6609. doi: 10.1073/pnas.93.13.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bungert J, Dave U, Lim K C, Lieuw K H, Shavit J A, Liu Q, Engel J D. Genes Dev. 1995;9:3083–3096. doi: 10.1101/gad.9.24.3083. [DOI] [PubMed] [Google Scholar]
- 38.Calzolari R, McMorrow T, Yannoutsos N, Langeveld A, Grosveld F. EMBO J. 1999;18:949–958. doi: 10.1093/emboj/18.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}