

Abstract
In linkage disequilibrium mapping of genetic variants causally associated with phenotypes, spurious associations can potentially be generated by any of a variety of types of population structure. However, mathematical theory of the production of spurious associations has largely been restricted to population structure models that involve the sampling of individuals from a collection of discrete subpopulations. Here, we introduce a general model of spurious association in structured populations, appropriate whether the population structure involves discrete groups, admixture among such groups, or continuous variation across space. Under the assumptions of the model, we find that a single common principle—applicable to both the discrete and admixed settings as well as to spatial populations—gives a necessary and sufficient condition for the occurrence of spurious associations. Using a mathematical connection between the discrete and admixed cases, we show that in admixed populations, spurious associations are less severe than in corresponding mixtures of discrete subpopulations, especially when the variance of admixture across individuals is small. This observation, together with the results of simulations that examine the relative influences of various model parameters, has important implications for the design and analysis of genetic association studies in structured populations.
GENETIC association studies aim to map phenotype-influencing genes by identifying alleles whose presence or absence in individuals correlates with phenotype (Risch and Merikangas 1996; Clark 2003). These studies rely on the fact that remnants of the ancestral genome in which a phenotypically important allele originated can be shared among descendants who carry the phenotype. The presence of the allele in these descendants will therefore be associated not only with the occurrence of the phenotype, but also with the occurrence of alleles inherited from the ancestor at other loci. Thus, a statistical association with a phenotype suggests that an allele directly influences the phenotype or, more probably, that the allele is indirectly associated with the phenotype as a result of shared inheritance and consequent association with true phenotype-influencing alleles. By virtue of the high probability of shared inheritance for alleles physically proximate on a chromosome—due to the relatively low amount of recombination decoupling them over time—in comparison with a low probability for distant alleles or those on different chromosomes, allelic associations are likely to involve alleles that lie close together. Thus, discovery of indirect associations can enable localization of the position of the true directly associated alleles (Pritchard and Przeworski 2001; Zondervan and Cardon 2004).
It is hoped that all alleles observed to be associated with a phenotype have either direct or indirect associations. However, in an instance of the general problem of statistical confounding (Greenland et al. 1999), the existence of an unmeasured variable associated with both genotype and phenotype can be an additional cause of genotype–phenotype associations. The status of an individual for such a variable affects both the genotype and the phenotype of the individual, inducing a genotype–phenotype relationship.
In the genetic mapping context, the primary variable that produces this type of misleading relationship can be thought of as population structure or genetic background. An individual's genetic population of origin, geographical position along a genetic cline, or laboratory strain can affect its probability of having any allele that varies in frequency across groups, as well as the probability of having any varying phenotype. The allele of an individual provides information about the individual's ancestry, which in turn provides information about the phenotype of the individual. This gain in knowledge about phenotype from knowledge of genotype is a statistical association. However, because alleles across the genome have the potential to have this type of association with the phenotype, identifying these associations does not assist in localizing phenotype-influencing alleles. Consequently, an important problem in association mapping is the separation of the spurious associations due to population structure from the indirect and direct associations that permit positional refinement of alleles that affect phenotype (Lander and Schork 1994; Ewens and Spielman 1995; Risch 2000; Pritchard and Donnelly 2001; Thomas and Witte 2002; Ziv and Burchard 2003).
Population-genetic models of the production of spurious associations can provide insight into the circumstances that are likely to increase their frequency and magnitude (Pritchard and Rosenberg 1999; Gorroochurn et al. 2004; Heiman et al. 2004). Such models, together with sampling theory or simulations of finite samples, can provide a framework for measuring the properties of spurious associations in actual populations and in evaluating methods for detecting and avoiding them (Ewens and Spielman 1995; Devlin and Roeder 1999; Wacholder et al. 2000; Pritchard and Donnelly 2001; Freedman et al. 2004; Hinds et al. 2004; Khlat et al. 2004; Marchini et al. 2004; Helgason et al. 2005; Köhler and Bickeböller 2006; Setakis et al. 2006). Here, our focus is on the former topic rather than the latter, that is, on determining at the population-genetic level the characteristics of populations, genotypes, and phenotypes that lead to spurious associations. Thus, we are concerned with what would happen in samples that are large enough that all associations—direct, indirect, and spurious—are detected. We note that while the type of modeling in this article can assist in assessing the relative potential for production of spurious associations across population-genetic scenarios, evaluation of the actual risk of spurious associations for typical association studies further requires that the role of finite sampling and the choice of statistical analysis tool be considered.
Pritchard and Rosenberg (1999) developed a model in which a case-control study is constructed by sampling individuals from a population that consists of a set of discrete underlying subpopulations. We observed that spurious associations could be produced at a locus if the frequency of the phenotype and the allele frequencies at the locus varied across subpopulations. With the same model, Gorroochurn et al. (2004) more precisely characterized the necessary and sufficient conditions for production of spurious associations, finding that under the assumptions of the model, they occur if and only if genotype and phenotype are variable and have nonzero correlation over the space of populations (weighted by sampling scheme). Gorroochurn et al. (2004) also defined a function that measures the extent of spurious association in the model and showed that the magnitude of this function declines with an increase in the number of subpopulations. Using a different modeling approach, Wacholder et al. (2000) previously had made a similar observation.
Given the growing interest in association mapping using samples from admixed and geographically structured populations (Thornsberry et al. 2001; Borevitz and Nordborg 2003; Caicedo et al. 2004; Olsen et al. 2004; Aranzana et al. 2005; Campbell et al. 2005; Flint-Garcia et al. 2005; Camus-Kulandaivelu et al. 2006; Yu et al. 2006), it is important to understand the situations that lead to an elevated false positive rate in admixed and spatially distributed groups that cannot easily be viewed as collections of discrete subpopulations. Here we extend the model of Pritchard and Rosenberg (1999) from a population consisting of discrete subpopulations to admixed and spatial populations. Our general spatial model contains the discrete model as a special case, and it also enables the magnitude of the spurious association problem to be characterized in a flexible class of admixture models. We find that the necessary and sufficient condition of Gorroochurn et al. (2004) extends directly to the general model, and we identify a simple relationship between the extent of spurious association, as measured by the function of Gorroochurn et al. (2004), in an admixed population and that in a corresponding mixture of discrete subpopulations. To further explore the relationship of spurious association in the discrete and admixed settings, as well as to study the roles of model parameters more generally, we perform simulations of spurious association in discrete and admixed populations. Results based on our extended model are then discussed in terms of their implications for genetic association studies in structured populations.
MODELS OF SPURIOUS ASSOCIATION
Consider an association mapping study in a population in which individuals are sampled from points z in a space Z. At present we view Z as geographical space in some number of dimensions; we see later that Z can also represent “admixture space.”
Individuals are tested for a phenotype and for a genotype at a locus of interest. We assume for now that both genotype and phenotype are binary, denoting the alleles by A and non-A (A*) and the phenotypes by D and non-D (D*). A multiallelic locus can be accommodated by focusing on the allele A and grouping the remaining alleles into the A* class. A similar grouping can be made for a phenotype with multiple discrete states; as we show later, a continuous phenotype can also be incorporated.
Suppose that the genotype and the phenotype of an individual are conditionally independent given the position of the individual, z (that is, suppose that there is no direct or indirect association anywhere in Z between the phenotype and alleles at the locus). In other words, genotype and phenotype are unassociated at every point in Z,
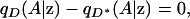 |
(1) |
where qD(A | z) and 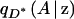 are, respectively, the frequencies of allele A among individuals at point z with phenotype D and among those with phenotype D*.
are, respectively, the frequencies of allele A among individuals at point z with phenotype D and among those with phenotype D*.
The null hypothesis that genotype and phenotype are unassociated in the full space Z is equivalent to
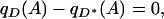 |
(2) |
where qD(A) and 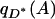 are, respectively, the frequencies of A among individuals sampled in space Z with phenotypes D and D*. The null hypothesis is false if and only if Equation 2 fails to hold. In other words, spurious associations occur when local independence of genotype and phenotype (Equation 1) does not produce global independence (Equation 2). Note that if Z consists of a single point, local independence trivially guarantees global independence, and no spurious associations are produced.
are, respectively, the frequencies of A among individuals sampled in space Z with phenotypes D and D*. The null hypothesis is false if and only if Equation 2 fails to hold. In other words, spurious associations occur when local independence of genotype and phenotype (Equation 1) does not produce global independence (Equation 2). Note that if Z consists of a single point, local independence trivially guarantees global independence, and no spurious associations are produced.
When Equation 2 is not satisfied, depending on the sign of 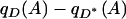 , allele A and phenotype D will be either positively or negatively associated. The absolute deviation
, allele A and phenotype D will be either positively or negatively associated. The absolute deviation 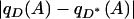 , which we label by Δ, measures the degree to which the null hypothesis of no association is violated (Gorroochurn et al. 2004), with larger values of Δ indicating more severe deviations.
, which we label by Δ, measures the degree to which the null hypothesis of no association is violated (Gorroochurn et al. 2004), with larger values of Δ indicating more severe deviations.
Spatial populations:
Let γ(z) be the prior probability density of sampling an individual from point z in Z, with 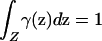 . Denote the frequency of phenotype D at z by p(z) and that of allele A at z by q(z). If the mean frequency of phenotype D with respect to the sampling scheme,
. Denote the frequency of phenotype D at z by p(z) and that of allele A at z by q(z). If the mean frequency of phenotype D with respect to the sampling scheme, 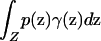 , or the mean frequency of allele A,
, or the mean frequency of allele A, 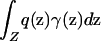 , is 0 or 1, then there can be no association between genotype and phenotype. Henceforth, these trivial scenarios are excluded from consideration.
, is 0 or 1, then there can be no association between genotype and phenotype. Henceforth, these trivial scenarios are excluded from consideration.
Using Bayes' theorem, the probability density that a sampled individual with phenotype D is from location z is
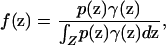 |
(3) |
and the probability density that a sampled individual with phenotype D* is from location z is
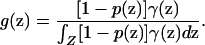 |
(4) |
Applying the local independence of genotype and phenotype, 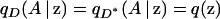 , and
, and
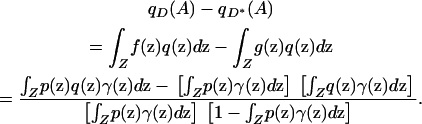 |
(5) |
This equation has two main consequences. First, under the model, Δ is given by the absolute value of the right-hand side of Equation 5. Second, for individuals sampled from the space Z according to sampling scheme γ, spurious associations between genotype and phenotype are produced if and only if the phenotype function p and the allele frequency function q do not satisfy
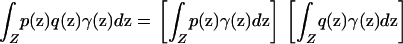 |
(6) |
Note that in the cases in which p or q is constant (except possibly on a set of points Z0 for which the probability of sampling an individual, or 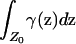 , is zero), Equation 6 is satisfied and no spurious associations occur.
, is zero), Equation 6 is satisfied and no spurious associations occur.
Alternatively, if the variances of p and q with respect to the sampling scheme γ,
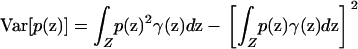 |
(7) |
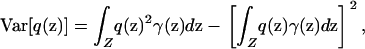 |
(8) |
are nonzero, then 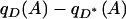 is proportional to the correlation coefficient between p and q with respect to γ,
is proportional to the correlation coefficient between p and q with respect to γ,
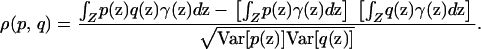 |
(9) |
In other words, for individuals sampled from the space Z according to sampling scheme γ, spurious associations between genotype and phenotype are produced if and only if the phenotype function p and the allele frequency function q are correlated with respect to γ.
Discrete populations:
The spatial model above contains the discrete subpopulation model of Pritchard and Rosenberg (1999) as a special case. Suppose that the space Z can be subdivided into disjoint components Z1, Z2, … , Zn, so that p(z) and q(z), respectively, have the constant values pi and qi in component i, with 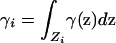 and
and 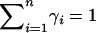 . Thus, each of the n nonoverlapping subunits of Z can be viewed as a discrete subpopulation. In this model, using Equation 5, the absolute deviation Δ simplifies to
. Thus, each of the n nonoverlapping subunits of Z can be viewed as a discrete subpopulation. In this model, using Equation 5, the absolute deviation Δ simplifies to
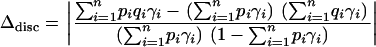 |
(10) |
(Gorroochurn et al., 2004). Consequently, there are no spurious associations if and only if
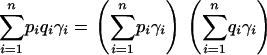 |
(11) |
In the special case of n = 2, in agreement with Pritchard and Rosenberg (1999), Equation 11 reduces to
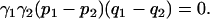 |
(12) |
In other words, in the case of an association study in a population with two underlying discrete subpopulations, spurious associations occur if and only if (i) all subpopulations are sampled, (ii) phenotype frequencies vary across subpopulations, and (iii) genotype frequencies vary across subpopulations.
If n ≥ 3, however, conditions i–iii, although they are necessary to produce spurious associations, are not sufficient (Gorroochurn et al. 2004). For example, consider 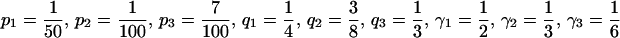 . In this example, three populations are represented. Both allele frequencies and phenotype frequencies vary across populations. However, Equation 11 is satisfied, with both sides equaling
. In this example, three populations are represented. Both allele frequencies and phenotype frequencies vary across populations. However, Equation 11 is satisfied, with both sides equaling 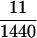 . The effects of the individual subpopulations “cancel” so that no spurious associations occur.
. The effects of the individual subpopulations “cancel” so that no spurious associations occur.
Another scenario of interest is that of uniform sampling: γi = 1/n for each i. Equation 11 then reduces to
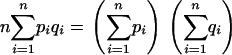 |
(13) |
Admixed populations:
By applying the spatial model to the n − 1 dimensional space 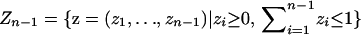 , the model can also be viewed as applicable to a situation in which individuals are allowed to be admixed among n discrete populations. In the space Zn−1, for each i, 1 ≤ i ≤ n − 1, individuals at point (z1, z2, … , zn−1) have a fraction zi of their ancestors from population i, with the fraction zn from subpopulation n equaling
, the model can also be viewed as applicable to a situation in which individuals are allowed to be admixed among n discrete populations. In the space Zn−1, for each i, 1 ≤ i ≤ n − 1, individuals at point (z1, z2, … , zn−1) have a fraction zi of their ancestors from population i, with the fraction zn from subpopulation n equaling 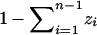 . Each of the n vertices of the simplex Zn−1 corresponds to an individual who is not admixed. For 1 ≤ i ≤ n − 1, an individual strictly in the ith population is located at the vertex ei with the ith coordinate 1 and all other coordinates 0; an individual in the nth population is located at the origin.
. Each of the n vertices of the simplex Zn−1 corresponds to an individual who is not admixed. For 1 ≤ i ≤ n − 1, an individual strictly in the ith population is located at the vertex ei with the ith coordinate 1 and all other coordinates 0; an individual in the nth population is located at the origin.
As in the discrete model, let the allele frequency for nonadmixed individuals from population i be qi, and let their phenotype frequency be pi. For admixed individuals at point z, the genotype frequency is
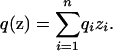 |
(14) |
Using Equation 6, the condition that must be satisfied to avoid spurious associations is
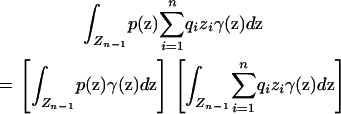 |
(15) |
We can consider a natural phenotypic model for admixed individuals, in which the phenotype frequency at a given point is a linear combination of the phenotype frequencies of the underlying discrete subpopulations:
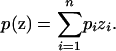 |
(16) |
Following previous treatments with admixture models (Pritchard et al. 2000; Hoggart et al. 2003; Erosheva et al. 2004), suppose further that sampling is Dirichlet-(α1, α2, … ,αn) distributed over Zn−1, where αi > 0 for each i. This multivariate prior accommodates a wide range of possible sampling distributions. Then
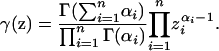 |
(17) |
Denote 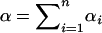 . Using the formulas for moments of a Dirichlet distribution (Lange 1997, p. 44),
. Using the formulas for moments of a Dirichlet distribution (Lange 1997, p. 44),
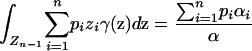 |
(18) |
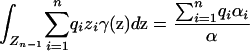 |
(19) |
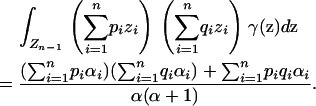 |
(20) |
Applying Equations 18–20 in Equation 15, the necessary and sufficient condition for no spurious associations is
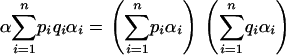 |
(21) |
The correspondence of this formula with Equation 11 means that heuristically, the conditions for spurious associations in an admixed population consisting of individuals admixed among a collection of subpopulations are identical to those that permit spurious associations in a mixed population consisting of individuals sampled only from the subpopulations themselves. In the case of two subpopulations, Equation 21 reduces to
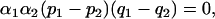 |
(22) |
so that conditions i–iii apply, except that i is replaced with the condition that individuals admixed among both subpopulations must be sampled. In the case of uniform sampling over possible collections of admixture fractions, αi = 1 for each i and Equation 21 reduces to Equation 13, the same condition for no spurious association as in the situation of uniformly sampled discrete subpopulations. More generally, this equality of Equations 21 and 13 holds if αi, which describes the degree to which the distribution of admixture from subpopulation i is concentrated around its mean value αi/α (with larger values of αi indicating less variability of admixture from subpopulation i), is the same for all i.
In the admixture model, using Equation 5, the absolute deviation Δ simplifies to
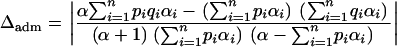 |
(23) |
Using Δdisc and Δadm, it is possible to compare the degree of spurious association in an admixed setting and in the corresponding discrete setting. Consider a mixture of n discrete subpopulations with contributions (γ1, γ2, … , γn) and an admixed population with parameters (α1, α2, … , αn) = (αγ1, αγ2, … , αγn), for positive γi and α. The ith component of a Dirichlet-(α1, α2, … , αn) random vector with 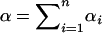 has beta-(αi, α − αi) distribution. As the mean of this beta distribution, αi/α, equals γi, the admixed population has the same relative contributions from the various subpopulations as the discrete mixture, regardless of the value of α. The difference between the two scenarios is that in the admixed population, contributions from multiple sources occur within individuals, and in the discrete mixture these contributions occur across individuals. Simplifying Equations 10 and 23,
has beta-(αi, α − αi) distribution. As the mean of this beta distribution, αi/α, equals γi, the admixed population has the same relative contributions from the various subpopulations as the discrete mixture, regardless of the value of α. The difference between the two scenarios is that in the admixed population, contributions from multiple sources occur within individuals, and in the discrete mixture these contributions occur across individuals. Simplifying Equations 10 and 23,
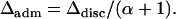 |
(24) |
Because α > 0, the deviation from the null, and thus the severity of spurious associations, is smaller in the admixture model than in the corresponding discrete model. Note that large values of α indicate relatively localized sampling in admixture space, whereas small values suggest that most individuals are close to vertices of the space and are only slightly admixed. Thus, admixed populations in which individuals have similar admixture will have large α and consequently will produce relatively few spurious associations in comparison with corresponding mixtures of discrete subpopulations.
The assumption of Dirichlet-distributed sampling can be interpreted to mean that individuals in a population are uniformly distributed over the set of possible admixture combinations, but that sampling from this population is weighted by a Dirichlet distribution. Perhaps more appropriate to actual populations is a view in which the underlying population of individuals actually satisfies a Dirichlet distribution of admixture combinations and in which random sampling of individuals from the population leads to a Dirichlet distribution of admixture combinations in a sample.
Continuous traits:
We have so far assumed that the trait that is to be mapped is discrete. However, a small modification can be made to make the theory above applicable to quantitative traits. Instead of viewing p(z) as the frequency of phenotype D at location z, we can view p(z) as the mean trait value at z. We can then repeat the development by investigating the conditional probability of sampling location given genotype rather than conditional on phenotype, as is done above and in Pritchard and Rosenberg (1999) and Gorroochurn et al. (2004). In this alternate perspective, rather than using Equation 1, local independence of genotype and phenotype occurs if for every z,
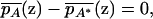 |
(25) |
where 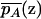 and
and 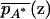 are the mean trait values among individuals at point z with alleles A and A*, respectively. Global independence occurs if
are the mean trait values among individuals at point z with alleles A and A*, respectively. Global independence occurs if
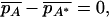 |
(26) |
where 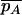 and
and 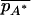 are the mean trait values among individuals with alleles A and A* in the full space Z. The theory above then proceeds similarly, and in particular, the same condition for the occurrence of spurious association is obtained, except with p(z) corresponding to the mean trait value instead of the phenotype frequency.
are the mean trait values among individuals with alleles A and A* in the full space Z. The theory above then proceeds similarly, and in particular, the same condition for the occurrence of spurious association is obtained, except with p(z) corresponding to the mean trait value instead of the phenotype frequency.
EXAMPLES
We have found that the same general condition for spurious associations—first identified by Gorroochurn et al. (2004) for the discrete subpopulation case—applies whether the population of interest is spatially distributed, composed of subpopulations, or composed of individuals admixed among subpopulations, and regardless of whether the trait of interest is continuous or discrete. Loosely speaking, under the model, spurious associations occur if and only if genotype and phenotype are variable and correlated over the space of populations. To illustrate this principle, we now consider several heuristic examples, investigating the potential for production of spurious associations in each scenario.
Asymmetric cline:
Consider a population spatially distributed on a line segment, along which allele frequencies vary linearly from one end to the other and along which phenotype also varies monotonically (Figure 1). An example of such a situation would be a latitudinal or altitudinal gradient, in which the frequency or value of a trait increases (or decreases) with latitude or altitude. Because allele frequencies and phenotype vary in a correlated manner in this scenario, Equation 6 will not be satisfied, and spurious associations will occur.
Figure 1.—

Asymmetric cline. Allele frequency varies monotonically with latitude, as might be expected in a species in which individuals disperse or migrate along a latitudinal gradient. Phenotype frequency varies to a greater extent, as might be expected, for example, if temperature, represented by the shading, produces selective pressure in favor of one particular phenotype. The genotype and phenotype curves are constants plus functions asymmetric around the center of the range of latitudes. In this example, with uniform sampling by latitude, spurious associations will occur.
A variant of this example is what might be termed a discrete cline, in which the latitudinal or altitudinal gradient is a set of regions in which genotype and phenotype are piecewise constant. Allele frequency and phenotype frequency are step functions, perhaps with spatial gaps between “steps.” Each step corresponds to a discrete population; similarly to the case of a continuous cline, spurious associations occur if and only if the genotypic and phenotypic step functions are correlated.
One special case is a situation in which sampling is uniform over the line segment, phenotype frequency equals a constant plus a function asymmetric around the center of the segment, and allele frequency is a second constant plus a second asymmetric function. Assuming that both asymmetric functions are nonzero over at least some of the length of the segment, Equation 6 can be applied to demonstrate that spurious associations must occur. As a corollary, discrete clines necessarily produce spurious associations if both phenotype and genotype equal constants plus asymmetric functions.
Symmetric cline:
Consider another population distributed along a line segment, but now, suppose that phenotype varies symmetrically around the midpoint of the segment. An example of this situation would be individuals along a symmetric transect perpendicular to a mountain range, with the peak at the center (Figure 2), or a latitudinal gradient bisected by the equator. Allele frequencies at random loci may still vary in a linear fashion from one end of the segment to the other, but now, the latitudinal or altitudinal gradient places one extreme of the phenotype in the middle of the range and the other extreme at both endpoints. In this case, genotype and phenotype vary in an uncorrelated manner, and spurious associations might not occur.
Figure 2.—

Symmetric cline. Allele frequency varies monotonically with altitude, as might be expected in a species in which individuals disperse or migrate along a mountain slope. Phenotype frequency varies to a greater extent, as might be expected, for example, if altitude produces selective pressure in favor of one particular phenotype. The genotype curve is a constant plus an asymmetric function, and the phenotype curve is a constant plus a symmetric function. In this example, with a sampling scheme symmetric around the mountain peak, no spurious associations will occur.
A corresponding special case is a situation in which sampling is uniform over the line segment, phenotype frequency equals a constant plus a function symmetric around the center of the line segment, and allele frequency is a constant plus an asymmetric function. In this case, Equation 6 can be applied to demonstrate that spurious associations are evaded. As a corollary, discrete clines do not produce spurious associations if phenotype is a constant plus a symmetric function and genotype is a constant plus an asymmetric function. The same is true if the roles of phenotype and genotype are reversed.
SIMULATION PROCEDURE
Recall that under the null hypothesis of no association between genotype and phenotype, the absolute deviation Δ equals 0 and that larger values of the absolute deviation Δ indicate that the null hypothesis is more severely violated through an increase both in the number and in the magnitude of spurious associations. As a result, an investigation of how Δ depends on model parameters can uncover the major influences on the severity of spurious associations. We therefore studied the determinants of Δ using simulations of discrete and admixed populations.
Allele frequencies across populations were simulated using the F model of Falush et al. (2003), which assumes that populations descend from a common ancestral population. We employed a special case of the F model for biallelic loci, in which the Dirichlet distribution for allele frequencies used by Falush et al. (2003) collapses to a beta distribution, as was studied by Marchini and Cardon (2002). In this special case, alleles (A, A*) at a biallelic locus have frequencies 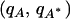 in the ancestral population, and n descendant populations are considered. For descendant population i, allele frequencies are drawn independently from a beta-
in the ancestral population, and n descendant populations are considered. For descendant population i, allele frequencies are drawn independently from a beta-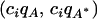 distribution, where ci = (1 − Fi)/Fi and Fi is a parameter (analogous to Fst) that measures the level of genetic drift of population i from the ancestor on a scale from 0 to 1. We used the same level of divergence for each descendant population, or Fi = F for each i.
distribution, where ci = (1 − Fi)/Fi and Fi is a parameter (analogous to Fst) that measures the level of genetic drift of population i from the ancestor on a scale from 0 to 1. We used the same level of divergence for each descendant population, or Fi = F for each i.
We considered several fixed values of F (0.01, 0.02, 0.05, 0.10, and 0.20) and assumed that the ancestral frequency qA was uniformly distributed between 0.05 and 0.95. For each value of F and each of several values of the number of populations n (2, 3, 4, 5, 10, 20, and 40), 10,000 loci were simulated. These simulated allele frequencies were then utilized in a variety of ways.
First, for each F, to investigate the influence of n on Δ in the discrete population case, a set of phenotype frequencies (p1, … , pn) was simulated for each set of simulated allele frequencies. This procedure used the F model with the same value of F as was used for the allele frequencies. The ancestral phenotype frequency (henceforth denoted p) was fixed at a specific value, and in each replicate it was additionally required that the phenotype frequency be in the interval [0.01, 0.99] in at least one of the descendant populations. The number of simulated loci required to obtain 10,000 loci that satisfied this requirement was generally <11,000, except with n ≤ 5 and p ≤ 0.05, for which it was as large as ∼50,000. For this analysis, the contribution γi from population i was assumed to equal 1/n for each i. The simulated values of the frequencies of the first allele—the values of qi—were then inserted into Equation 10 together with the simulated values of pi and the fixed values of γi.
To investigate the influence of γ1 on Δ in the two-subpopulation discrete model, for each value of F the 10,000 values of q1 and q2 from the simulations above with n = 2 were used. In this analysis, phenotype frequencies p1 and p2 were fixed rather than simulated with the F model. The simulated genotype frequencies and the fixed phenotype frequencies were then inserted along with specific values of γ1 and γ2 into Equation 10.
Finally, to examine the roles of the mean and variance of admixture in the two-subpopulation admixture model—as well as to compare the admixed and discrete models—for each F, the 10,000 values of q1 and q2 from the simulations of the discrete model with n = 2 were used as the allele frequencies in the subpopulations among which individuals were admixed. Using the moment formulas for a Dirichlet distribution (Lange 1997, p. 44), the mean admixture from subpopulation 1, denoted E, and the variance of admixture V—which is equal for both admixture fractions, the one for subpopulation 1 and the one for subpopulation 2—are related to the model parameters α1 and α2 by
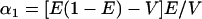 |
(27) |
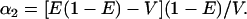 |
(28) |
Because α1, α2 > 0, E is constrained to the interval from 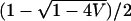 to
to 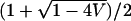 , and V must be in (0, E(1 − E)). A variety of values of E and V—and consequently of α1 and α2—were considered. For each (α1, α2), the simulated genotype frequencies, as well as fixed phenotype frequencies and fixed values of α1 and α2, were inserted into Equation 23.
, and V must be in (0, E(1 − E)). A variety of values of E and V—and consequently of α1 and α2—were considered. For each (α1, α2), the simulated genotype frequencies, as well as fixed phenotype frequencies and fixed values of α1 and α2, were inserted into Equation 23.
SIMULATION RESULTS
Number of discrete subpopulations:
Figure 3 shows the influence on Δdisc of the number of populations n, illustrating that as n increases while the population divergence F and the ancestral phenotype frequency p are held constant, the fraction of simulations with a large deviation generally decreases. Thus, spurious associations in a population consisting of many distinct subgroups are likely to be rarer and less severe than in a mixed population containing only a few subgroups (Gorroochurn et al. 2004). This result, that as the number of subpopulations increases, the effects of these populations tend to destructively interfere, can be explained by the fact that with only two subpopulations, if genotype and phenotype frequency vary across populations, the genotype and phenotype functions necessarily must be correlated. As the number of subpopulations increases, however, the chance decreases that two sets of numbers—the genotype and phenotype frequencies—have nontrivial correlation. A similar effect was also seen in the simulations of Wacholder et al. (2000). In those simulations, as the number of subpopulations in a discrete subpopulation model increased, the influence of population structure on the estimation of relative risk of disease decreased.
Figure 3.—

Distribution of the absolute deviation from the null hypothesis (Δdisc, Equation 10) in a discrete subpopulation model. The fractions of 10,000 simulations that exceed given levels of absolute deviation are plotted for seven choices of the number of populations n and four combinations of the ancestral phenotype frequency p and population divergence F. (A) p = 0.05, F = 0.02; (B) p = 0.05, F = 0.10; (C) p = 0.20, F = 0.02; (D) p = 0.20, F = 0.10.
Genetic divergence across discrete subpopulations:
A comparison of Figure 3, A and B, or of Figure 3, C and D, illustrates that increasing the divergence F across populations while holding n and p constant increases the fraction of simulations for which Δdisc exceeds a given value. Thus, greater population divergence produces a greater magnitude of spurious association. As this effect was seen in all simulations and the value of F did not influence the qualitative relationships between other parameters and Δ, simulations focusing on these other parameters are displayed only for single values of F. Among the values chosen are 0.10, which corresponds roughly to the magnitude of intercontinental divergences between pairs of human populations (Akey et al. 2002; Ramachandran et al. 2005; Weir et al. 2005), and 0.01 and 0.02, which correspond to many human population divergences within continents (Ramachandran et al. 2005; Rosenberg et al. 2005).
Phenotype frequency divergence across discrete subpopulations:
An increase in the ancestral phenotype frequency p while holding the level of population divergence and the number of subpopulations constant was observed to decrease the amount of spurious association. This result is evident from a comparison of the corresponding graphs in Figure 3, A and C, or in Figure 3, B and D.
Fixing the number of subpopulations at two, the role of phenotype frequency divergence can be examined in greater detail by using specific values for the phenotype frequencies in the two subpopulations, p1 and p2, rather than by simulating values from the F model (as in Figure 3). Figure 4 shows the distribution of Δdisc with n = 2 and F = 0.01, for fixed values of p1 and p2. A comparison of Figure 4, A and B, illustrates that if the phenotype frequency p1 is held constant, spurious association is more severe if p2 has a greater difference from p1. A similar result is observed by comparing Figure 4, B and C, which have the same value of p2 but different values of p1.
Figure 4.—

Distribution of the absolute deviation from the null hypothesis (Δdisc, Equation 10) in a discrete subpopulation model with two subpopulations. The fractions of 10,000 simulations that exceed given levels of absolute deviation are plotted for seven choices of the fractional contribution of subpopulation 1, or γ1, and three combinations of the phenotype frequencies p1 and p2 in the two subpopulations. All three plots use population divergence F = 0.01. (A) p1 = 0.01, p2 = 0.02; (B) p1 = 0.01, p2 = 0.20; (C) p1 = 0.10, p2 = 0.20.
Note, however, that the similarity of Figure 4, A and C, which is based on the same value for p1/p2, suggests that this ratio is a major determinant of Δdisc. Examination of Equation 10 demonstrates that if the allele frequencies q1 and q2 and the population contributions γ1 and γ2 are held constant, Δdisc can be written
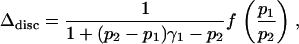 |
(29) |
where f(p1/p2) is a function of the ratio p1/p2. Thus, holding p1/p2 constant (and not equal to 1), as p1 and p2 vary, Δdisc changes only by a multiplicative factor that is fairly close to 1 under most reasonable choices of p1, p2, and γ1. For given values of p1/p2 and γ1, this factor is largest when p1 and p2 are largest, so that for a given ratio of prevalences between subpopulations, more frequent phenotypes will produce more spurious association. Consequently, a graph with a given value of γ1 in Figure 4C—which has larger p1 and p2 than does Figure 4A, with the same ratio p1/p2—produces slightly greater values of Δdisc than does the corresponding graph in Figure 4A.
The dependence of Δdisc largely on the ratio p1/p2 can help explain why in Figure 3, A and C, and in Figure 3, B and D, rarer phenotypes led to a greater degree of spurious association. In the simulations used to generate Figure 3, the phenotype frequency varied across subpopulations according to the F model. Thus, the greater degree of spurious association for rarer phenotypes in Figure 3 is a consequence of the fact that pairs of small frequencies under the F model will tend to have ratios farther from 1 than will pairs of large frequencies. Only if the ratio of phenotype frequencies is held constant do more frequent phenotypes produce more spurious association.
Relative contributions of two discrete subpopulations:
Similarly to the observations of Khlat et al. (2004), Figure 4 shows that Δdisc is usually greater if both populations have nontrivial contributions and smaller if one population dominates the sample and one is nearly absent. This observation is sensible, as the limiting case in which one of the populations has contribution 0 produces no spurious associations.
However, as can be shown using Equation 10, the mixture that produces the maximal degree of spurious association does not occur when the contributions of the two populations are exactly equal, and the position of the maximum depends on the phenotype frequencies. Consider fixed values of p1, p2, q1, and q2, and write γ2 = 1 − γ1 so that Δdisc is thought of as a function of γ1:
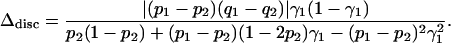 |
(30) |
Setting the derivative of this function with respect to γ1 equal to zero, it can be shown that the maximum of Δdisc occurs at
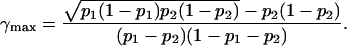 |
(31) |
The allele frequencies q1 and q2 do not appear in this expression, so that the location of the maximal Δdisc depends only on the phenotype frequencies. Figure 5 shows the median of Δdisc for each of the choices of p1 and p2 plotted in Figure 4, locating the values of γ1 that produce the highest median. These points, marked by circles, match the values obtained from Equation 31.
Figure 5.—

Median absolute deviation from the null hypothesis (Δdisc, Equation 10) in a discrete subpopulation model with two subpopulations. The median deviation over 10,000 simulations is plotted against γ1, the fraction contributed by subpopulation 1. Three combinations of the phenotype frequencies p1 and p2 in the two populations are considered, and the population divergence parameter F is set to 0.01. The maximal deviation predicted by Equation 31 occurs at the values marked with circles. For (p1, p2) = (0.01, 0.02), 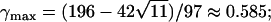 for (0.01, 0.20),
for (0.01, 0.20), 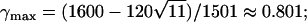 for (0.10, 0.20), γmax =
for (0.10, 0.20), γmax = 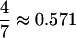 .
.
To more completely understand how γmax depends on p1 and p2, Figure 6 plots the function in Equation 31, showing that for most choices of p1 and p2, γmax is close to 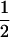 . Figure 6 also illustrates that for phenotypes with frequencies <50% in both subpopulations, the maximal Δdisc occurs at a value of γ1 for which the low-prevalence subpopulation is overrepresented, with contributions of this subpopulation being greatest when its phenotype frequency is extremely small.
. Figure 6 also illustrates that for phenotypes with frequencies <50% in both subpopulations, the maximal Δdisc occurs at a value of γ1 for which the low-prevalence subpopulation is overrepresented, with contributions of this subpopulation being greatest when its phenotype frequency is extremely small.
Figure 6.—

The value of γ1 that maximizes the absolute deviation Δdisc as a function of the phenotype frequencies p1 and p2 in a two-subpopulation discrete model (γmax, Equation 31). From lightest to darkest, the shadings represent values in [0, 0.15), [0.15, 0.3), [0.3, 0.45], (0.45, 0.55), [0.55, 0.7], (0.7, 0.85], and (0.85, 1], with the shading for (0.45, 0.55) occupying most of the plot. The function is not defined for p2 = p1 or p2 = 1 − p1 (although its limit is 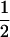 when approaching these diagonals).
when approaching these diagonals).
Discrete and admixed models:
Figure 7 shows that an admixed population produces fewer spurious associations than does the corresponding discrete population, comparing a discrete model with two subpopulations, which respectively contribute 20 and 80% of the individuals, to admixture models in which the mean admixture is 20%. The fraction of simulations with large absolute deviations is considerably greater in the discrete model than in any of the admixture scenarios. This observation follows directly from Equation 24.
Figure 7.—

Distribution of the absolute deviation from the null hypothesis in an admixture model (Δadm, Equation 23). The fractions of 10,000 simulations that exceed given levels of absolute deviation are plotted for various choices of the variance across individuals of the admixture fraction for population 1 in a two-subpopulation model with admixture contributions 0.2 and 0.8 for the two subpopulations; the curves with mean admixture 0.2 and variance 0.01 use similar values to estimates for European ancestry in African–Americans (Patterson et al. 2004, mean of 0.21, standard deviation of 0.11). For comparison, Δdisc for a mixture of discrete subpopulations is also shown. Both the top and the bottom plots use a level of population divergence of F = 0.10, and they differ only in the phenotype frequencies of the two underlying subpopulations. (A) p1 = 0.05, p2 = 0.10; (B) p1 = 0.10, p2 = 0.05.
Note that the connection between the admixed and discrete models via Equation 24 has the additional consequence that the level of spurious association in the admixture model depends on a function of the ratio p1/p2 in the same way as in the corresponding discrete model. Although the function differs because it must subsume the factor of 1/(α + 1), the multiplier, 1/[1 + (p2 − p1)(α1/α) − p2] in the admixture model, is the same.
Variance of admixture:
Holding the phenotype frequencies and the level of population divergence fixed, the severity of spurious associations is observed to be larger in admixture models with larger variance of individual admixture. This can be seen by comparing the various graphs within Figure 7A or 7B or by comparing corresponding graphs in Figures 8, A and B. The result is sensible, as an increase in this variance increases the heterogeneity of the population, contributing to a greater potential for spurious association.
Figure 8.—

Distribution of the absolute deviation from the null hypothesis in an admixture model (Δadm, Equation 23). The fractions of 10,000 simulations that exceed given levels of absolute deviation are plotted for various choices of the mean and variance across individuals of the admixture fraction for population 1 in a two-subpopulation model. For comparison, the mixture of discrete subpopulations that maximizes Δdisc is also shown, as is a uniform distribution of admixture. Both the top and the bottom plots use a level of population divergence of F = 0.10 and phenotype frequencies p1 = 0.05 and p2 = 0.10, and they differ only in the variances of the admixture fraction. (A) V = 0.04. (B) V = 0.01.
Equation 24 can be used to precisely determine the nature of the dependence of Δadm on the variance of admixture. For the two-population case, using α = α1 + α2 together with Equations 27 and 28, Equation 24 simplifies to
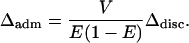 |
(32) |
Thus, in the two-population admixture model, for fixed mean admixture E, the level of spurious association increases linearly with the variance of admixture. This result explains why in Figure 7 the fraction of simulations with variance V that exceeds a cutoff C in absolute deviation is equal to the fraction of simulations in the graph with variance 2V that exceeds 2C.
Mean admixture:
Finally, with all other parameters held constant, the mean individual admixture also affects the severity of spurious associations, although its effect on Δadm is smaller than that of the variance, as is shown in Figure 8.
Equation 32 can be used to understand the influence of mean admixture on Δadm. The Δdisc term in Equation 32 applies to the corresponding discrete model, that is, the discrete model in which γ1 = E and γ2 = 1 − E. Thus, as a function of E, Δadm reduces to
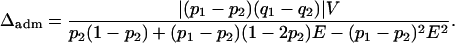 |
(33) |
This equation is simpler than Equation 30 for the discrete model. With p1, p2, q1, q2, and V held constant, it can be shown that the function has no maximum between the minimal and maximal mean admixture. Thus, the largest values of Δadm occur as E approaches one of these boundaries, 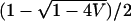 or
or 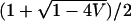 .
.
Recall that in the discrete two-subpopulation model, for phenotypes rarer than 50% in both subpopulations, Δdisc was maximal at a value where the contribution from the low-prevalence subpopulation exceeded 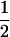 . Without loss of generality, suppose that subpopulation 1 is the low-prevalence subpopulation. For
. Without loss of generality, suppose that subpopulation 1 is the low-prevalence subpopulation. For 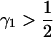 , if p1,
, if p1, 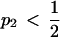 , it can be shown that transposing the contributions of the two populations (that is, switching the values of γ1 and γ2) reduces Δdisc. Because Δadm = Δdisc up to a factor symmetric about the line
, it can be shown that transposing the contributions of the two populations (that is, switching the values of γ1 and γ2) reduces Δdisc. Because Δadm = Δdisc up to a factor symmetric about the line 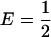 , Δadm has the same property that if
, Δadm has the same property that if 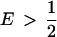 and p1,
and p1, 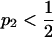 , transposing the contributions of the two populations reduces Δadm. Thus, the factor of V/[E(1 − E)] in Δadm does not affect the side of the line
, transposing the contributions of the two populations reduces Δadm. Thus, the factor of V/[E(1 − E)] in Δadm does not affect the side of the line 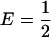 on which the values of E with higher Δadm occur. In other words, although the effect of E in the admixture model differs from that of γ1 in the discrete model, the E-values with highest Δ—similarly to the discrete model—involve a larger contribution from the low-prevalence subpopulation. This result is reflected in Figure 7, in which the extent of spurious association is greater when the larger admixture contribution is from the low-prevalence subpopulation (Figure 7B) rather than from the high-prevalence subpopulation (Figure 7A).
on which the values of E with higher Δadm occur. In other words, although the effect of E in the admixture model differs from that of γ1 in the discrete model, the E-values with highest Δ—similarly to the discrete model—involve a larger contribution from the low-prevalence subpopulation. This result is reflected in Figure 7, in which the extent of spurious association is greater when the larger admixture contribution is from the low-prevalence subpopulation (Figure 7B) rather than from the high-prevalence subpopulation (Figure 7A).
DISCUSSION
In this article, we have constructed a model that extends the discrete subpopulation model of Pritchard and Rosenberg (1999) to describe the occurrence of spurious associations in discrete, admixed, or spatially distributed populations. Generalizing a result of Gorroochurn et al. (2004), we have found in that in the general model, false positives occur when the genotype and phenotype functions are correlated with respect to the sampling scheme, but not otherwise.
In a special case of the model—the discrete subpopulation case—our analysis also demonstrates that for two subpopulations, the maximal amount of spurious association occurs not for an equal subpopulation mixture, but for a particular combination that involves a greater contribution from the low-prevalence subpopulation. Our analysis also shows that in the two-subpopulation discrete model, a key determinant of the extent of spurious association is the ratio of phenotype frequencies between the two underlying subpopulations.
In agreement with Wacholder et al. (2000) and Gorroochurn et al. (2004), the severity of spurious associations generally decreases with the number of underlying subpopulations, suggesting that in the most highly mixed human populations, spurious associations are likely to be less common than in more moderately mixed groups. Thus, an article by Helgason et al. (2005), which examined the potential for spurious associations in Iceland using simulations based on a division of the Icelandic population into two rather than many subgroups, may have overstated the risk of false positives in that population.
In the admixed case the model suggests that the spurious association problem is not as great as in the discrete case. If individual ancestry is highly variable in an admixed population, however, the problem increases in severity. Consequently, concern about spurious associations may be most justified in populations such as Hispanic or Latino groups, for which individuals with a very wide range of ancestry combinations may be included in the same population (Choudhry et al. 2006).
Similarly to the discrete case, in the admixed case, severity of spurious association is greater if the low-prevalence population is the major contributor to the admixed population. Thus, in African–Americans, who have ∼20% European–American admixture (Patterson et al. 2004), spurious associations are more likely for phenotypes more common in European–Americans than in African–Americans, in comparison with phenotypes that are less prevalent in European–Americans. This result is convenient, as the phenotypes that are more likely to produce spurious associations in African–Americans—those with greater frequency in European–Americans—are less likely to be studied in an African–American sample.
It is noteworthy that the numerator of the quantity that indicates the magnitude of spurious association in our general model, Δ, takes the form of a covariance between frequencies of a phenotype and of an allele A at a particular locus. Suppose that the “phenotype” of interest was the presence in individuals of a certain allele B at a second locus distant from the first in the genome, so that genotypes at the two loci were locally independent throughout a structured population. Then Δ, with its numerator equaling the absolute value of the difference between the probability of having both alleles A and B and the product of the probability of having A and the probability of having B, would take the form of a coefficient of linkage disequilibrium (LD) between the two loci. Consequently, because any phenotype that had the presence of allele B as a necessary and sufficient predictor would have the same frequencies as allele B, LD in the full population between the two locally independent loci—that is, Δ ≠ 0 for the two loci—would indicate a spurious association between allele A and any phenotypes causally produced by allele B (the same would occur with the roles of A and B reversed). In other words, LD between distant loci in a genome indicates that population structure will produce spurious associations with any phenotype that has a perfect causal relation with any locus that experiences a pattern of genomewide LD with other loci. If in a structured population this kind of genomewide LD occurs for some loci, it is likely to occur for most loci, and thus it can be considered extremely likely that when some allele in the genome is actually responsible for a phenotype, population structure will produce spurious associations between the phenotype and alleles spread throughout the genome. Thus, if the phenotype of interest has a causal allele, the occurrence of LD between many distant pairs of markers genomewide is nearly a sufficient condition for the production of spurious associations with the phenotype. If on the other hand the phenotype has no causal alleles, it cannot be assumed that there is an allele in the genome whose frequency profile is equivalent to that of the phenotype; thus, LD throughout the genome has no bearing on whether Δ ≠ 0 for the phenotype and some allele in the genome and, consequently, on whether or not spurious associations are produced.
It is important to clarify the limitations of our analysis. First, we have discussed the necessary and sufficient conditions for the production of spurious association under a specific model. As conditions such as Equation 6 are not likely to be satisfied in practice in actual populations, their primary use is for clarifying the conceptual basis for the production of spurious association, so that methods for avoiding spurious associations can be founded on appropriate assumptions. In particular, although spurious associations have frequently been understood to arise in discrete populations from variation in allele and phenotype frequencies across populations, our analysis and that of Gorroochurn et al. (2004) demonstrate that this does not provide a fully accurate picture of the basis for production of spurious association and that a view in which spurious associations arise from a (weighted) correlation between genotype and phenotype frequencies in “population space” is more appropriate.
Second, our model implicitly assumes that all individuals at any point in population space are independent draws from a frequency distribution. Consequently, pairs of individuals from the same place in population space are assumed to be equally unrelated. This assumption is often reasonable, as the probability of sampling pairs of closely related individuals is generally small. Especially for a spatial setting, however, in which geographically proximate individuals may very well be close relatives, explicit consideration of the variation in levels of relationship among individuals (Kennedy et al. 1992; Voight and Pritchard 2005; Yu et al. 2006) has the potential to provide an improved model for the production of spurious associations.
Third, the analysis of our model has focused only on the population-genetic scenarios that produce spurious associations, assuming that sample sizes are large enough that all associations, real and spurious, are detectable. As our study concerns the level of spurious association in population-genetic models and not the actual risk of spurious association that results from the collection of finite samples from real populations, the conclusions that can be drawn relate to the relative severity of spurious association in different settings and not to the actual type I error rates that would be obtained in populations from the application of specific statistical procedures. Thus, it is possible to conclude from the model (for example) that mixtures of many groups will generally produce fewer spurious associations than will mixtures of fewer groups, that admixed populations will produce fewer spurious associations than will corresponding discrete populations, and that a key parameter in predicting the level of spurious association is the variability of admixture. This does not mean that it can be concluded that multisource mixtures and admixed populations will not produce spurious associations. It does, however, suggest that modeling studies that have utilized a discrete mixture of two populations may have overstated the risk of spurious association for the more realistic settings of admixed populations and mixtures of many groups.
Note that in nonhuman organisms, unlike in the usual scenario for humans, association studies may intentionally utilize geographically distributed samples, as phenotypic variation is required for mapping and may be small within local populations. In these cases, particularly if sampling includes small numbers of individuals from each of many sites, rather than many individuals from each of a few sites, a spatial perspective may have greater potential than discrete or admixed models to describe the genetic variation that leads to spurious associations. Thus, the incorporation of models of spatially distributed groups into procedures that evade spurious associations is an important step toward the development of linkage disequilibrium mapping strategies in natural populations.
Acknowledgments
We thank David Balding and an anonymous reviewer for comments on an earlier draft of this article and Jonathan Pritchard for sharing computer code for simulating the F model. This research was supported by a Burroughs Wellcome Fund career award in biomedical sciences to N.A.R and by National Institutes of Health Center of Excellence in Genomic Science grant P50HG002790 from the National Human Genome Research Institute.
References
- Akey, J. M., G. Zhang, K. Zhang, L. Jin and M. D. Shriver, 2002. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12: 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranzana, M. J., S. Kim, K. Zhao, E. Bakker, M. Horton et al., 2005. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1: 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz, J. O., and M. Nordborg, 2003. The impact of genomics on the study of natural variation in Arabidopsis. Plant Physiol. 132: 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo, A. L., J. R. Stinchcombe, K. M. Olsen, J. Schmitt and M. D. Purugganan, 2004. Epistatic interaction between Arabidopsis FRI and FLC flowering time genes generates a latitudinal cline in a life history trait. Proc. Natl. Acad. Sci. USA 101: 15670–15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, C. D., E. L. Ogburn, K. L. Lunetta, H. N. Lyon, M. L. Freedman et al., 2005. Demonstrating stratification in a European American population. Nat. Genet. 37: 868–872. [DOI] [PubMed] [Google Scholar]
- Camus-Kulandaivelu, L., J.-B. Veyrieras, D. Madur, V. Combes, M. Fourmann et al., 2006. Maize adaptation to temperate climate: relationship with population structure and polymorphism in the Dwarf8 gene. Genetics 172: 2449–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry, S., N. E. Coyle, H. Tang, K. Salari, D. Lind et al., 2006. Population stratification confounds genetic association studies among Latinos. Hum. Genet. 118: 652–664. [DOI] [PubMed] [Google Scholar]
- Clark, A. G., 2003. Finding genes underlying risk of complex disease by linkage disequilibrium mapping. Curr. Opin. Genet. Dev. 13: 296–302. [DOI] [PubMed] [Google Scholar]
- Devlin, B., and K. Roeder, 1999. Genomic control for association studies. Biometrics 55: 997–1004. [DOI] [PubMed] [Google Scholar]
- Erosheva, E., S. Fienberg and J. Lafferty, 2004. Mixed-membership models of scientific publications. Proc. Natl. Acad. Sci. USA 101: 5220–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., and R. S. Spielman, 1995. The transmission/disequilibrium test: history, subdivision, and admixture. Am. J. Hum. Genet. 57: 455–464. [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia, S. A., A.-C. Thuillet, J. Yu, G. Pressoir, S. M. Romero et al., 2005. Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J. 44: 1054–1064. [DOI] [PubMed] [Google Scholar]
- Freedman, M. L., D. Reich, K. L. Penney, G. J. McDonald, A. A. Mignault et al., 2004. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 36: 388–393. [DOI] [PubMed] [Google Scholar]
- Gorroochurn, P., S. E. Hodge, G. Heiman and D. A. Greenberg, 2004. Effect of population stratification on case-control association studies II. False-positive rates and their limiting behavior as number of subpopulations increases. Hum. Hered. 58: 40–48. [DOI] [PubMed] [Google Scholar]
- Greenland, S., J. M. Robins and J. Pearl, 1999. Confounding and collapsibility in causal inference. Stat. Sci. 14: 29–46. [Google Scholar]
- Heiman, G. A., S. E. Hodge, P. Gorroochurn, J. Zhang and D. A. Greenberg, 2004. Effect of population stratification on case-control association studies: I. Elevation in false positive rates and comparison to confounding risk ratios (a simulation study). Hum. Hered. 58: 30–39. [DOI] [PubMed] [Google Scholar]
- Helgason, A., B. Yngadóttir, B. Hrafnkelsson, J. Gulcher and K. Stefánsson, 2005. An Icelandic example of the impact of population structure on association studies. Nat. Genet. 37: 90–95. [DOI] [PubMed] [Google Scholar]
- Hinds, D. A., R. P. Stokowski, N. Patil, K. Konvicka, D. Kershenobich et al., 2004. Matching strategies for genetic association studies in structured populations. Am. J. Hum. Genet. 74: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart, C. J., E. J. Parra, M. D. Shriver, C. Bonilla, R. A. Kittles et al., 2003. Control of confounding of genetic associations in stratified populations. Am. J. Hum. Genet. 72: 1492–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, B. W., M. Quinton and J. A. M. van Arendonk, 1992. Estimation of effects of single genes on quantitative traits. J. Anim. Sci. 70: 2000–2012. [DOI] [PubMed] [Google Scholar]
- Khlat, M., M.-H. Cazes, E. Génin and M. Guiguet, 2004. Robustness of case-control studies of genetic factors to population stratification: magnitude of bias and type I error. Cancer Epidemiol. Biomarkers Prev. 13: 1660–1664. [PubMed] [Google Scholar]
- Köhler, K., and H. Bickeböller, 2006. Case-control association tests correcting for population stratification. Ann. Hum. Genet. 70: 98–115. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and N. J. Schork, 1994. Genetic dissection of complex traits. Science 265: 2037–2048. [DOI] [PubMed] [Google Scholar]
- Lange, K., 1997. Mathematical and Statistical Methods for Genetic Analysis. Springer, New York.
- Marchini, J. L., and L. R. Cardon, 2002. Discussion on the meeting on ‘statistical modelling and analysis of genetic data’. J. R. Stat. Soc. B 64: 740–741. [Google Scholar]
- Marchini, J., L. R. Cardon, M. S. Phillips and P. Donnelly, 2004. The effects of human population structure on large genetic association studies. Nat. Genet. 36: 512–517. [DOI] [PubMed] [Google Scholar]
- Olsen, K. M., S. S. Halldorsdottir, J. R. Stinchcombe, C. Weinig, J. Schmitt et al., 2004. Linkage disequilibrium mapping of Arabidopsis CRY2 flowering time alleles. Genetics 167: 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, N., N. Hattangadi, B. Lane, K. E. Lohmueller, D. A. Hafler et al., 2004. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 74: 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and P. Donnelly, 2001. Case-control studies of association in structured or admixed populations. Theor. Popul. Biol. 60: 227–237. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and N. A. Rosenberg, 1999. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 65: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, S., O. Deshpande, C. C. Roseman, N. A. Rosenberg, M. W. Feldman et al., 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. USA 102: 15942–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch, N., and K. Merikangas, 1996. The future of genetic studies of complex human diseases. Science 273: 1516–1517. [DOI] [PubMed] [Google Scholar]
- Risch, N. J., 2000. Searching for genetic determinants in the new millenium. Nature 405: 847–856. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., S. Mahajan, S. Ramachandran, C. Zhao, J. K. Pritchard et al., 2005. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 1: 660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setakis, E., H. Stirnadel and D. J. Balding, 2006. Logistic regression protects against population structure in genetic association studies. Genome Res. 16: 290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D. C., and J. S. Witte, 2002. Population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol. Biomarkers Prev. 11: 513–520. [PubMed] [Google Scholar]
- Thornsberry, J. M., M. M. Goodman, J. Doebley, S. Kresovich, D. Nielsen et al., 2001. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28: 286–289. [DOI] [PubMed] [Google Scholar]
- Voight, B. F., and J. K. Pritchard, 2005. Confounding from cryptic relatedness in case-control association studies. PLoS Genet. 1: 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacholder, S., N. Rothman and N. Caporaso, 2000. Population stratification in epidemiologic studies of common genetic variants and cancer: quantification of bias. J. Natl. Cancer Inst. 92: 1151–1158. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., L. R. Cardon, A. D. Anderson, D. M. Nielsen and W. G. Hill, 2005. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 15: 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J., G. Pressoir, W. H. Briggs, I. V. Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38: 203–208. [DOI] [PubMed] [Google Scholar]
- Ziv, E., and E. G. Burchard, 2003. Human population structure and genetic association studies. Pharmacogenomics 4: 431–441. [DOI] [PubMed] [Google Scholar]
- Zondervan, K. T., and L. R. Cardon, 2004. The complex interplay among factors that influence allelic association. Nat. Rev. Genet. 5: 89–100. [DOI] [PubMed] [Google Scholar]