

Abstract
14-3-3σ proteins regulate numerous cellular processes that are important to cancer development. One of its biological roles involves G2 cell-cycle arrest following DNA damage. It has also been reported that the loss of 14-3-3σ expression via CpG methylation may contribute to malignant transformation by impairing the G2 cell-cycle checkpoint function, thereby allowing an accumulation of genetic defects. However, how the CpG methylation-dependent silencing mechanism works in relation to promoter methylation associated with methyl-CpG-binding proteins (MeCPs) is still unclear. To better understand the mechanism, we first examined the methylation status of the 14-3-3σ promoter-associated CpG islands and 14-3-3σ gene expression in a subset of prostate cancer cell lines using methylation-specific PCR (MSP), a HhaI-based DNA methylation assay, and reverse transcription-PCR (RT-PCR). We found that the 14-3-3σ expression is lost in LNCaP and Tramp-C1 prostate cancer cell lines and that this expression is restored after treatment with epigenetic silencing modifiers 5-aza and TSA. These results imply transcriptional silencing via promoter-associated CpG methylation. Chromatin immunoprecipitation analysis revealed that methyl-CpG-binding protein 2 (MBD2) is associated preferentially to the methylated CpG island in the 14-3-3σ promoter in LNCaP and Tramp-C1 cells but not in 14-3-3σ-expressing PC3 and DU145 cells, which contain an unmethylated CpG island in the 14-3-3σ promoter region. The 14-3-3σ gene silencing due to CpG methylation correlates with binding of MBD2. In addition, the activation of 14-3-3σ gene expression by a combination of 5-aza and TSA also involves the release of the MBD2 from the 14-3-3σ promoter methylated CpG island in LNCaP and Tramp-C1 cells. Furthermore, MBD2 knockdown by siRNA stimulated 14-3-3σ expression in LNCaP cells. We also investigated whether the loss of 14-3-3σ expression in LNCaP and Tramp-C1 cells affects cell proliferation by MTT assays. Interestingly, we observed that 14-3-3σ-inactivated LNCaP and Tramp-C1 cells had markedly decreased cell proliferation and protein expression of proliferation cell nuclear antigen (PCNA) after restoration of 14-3-3σ expression with 5-aza and TSA treatment. On the other hand, the same treatment did not significantly affect 14-3-3σ-active PC3 and DU145 cells, which normally express 14-3-3σ. Finally, 14-3-3σ knockdown by siRNA resulted in increased proliferation in PC3 and DU145 cells. These findings suggest that the transcriptional silencing of the 14-3-3σ gene is caused by promoter CpG island methylation associated with MBD2, and that this may play an important role in prostate cancer progression during the invasive and metastatic stages of the disease.
Keywords: CpG methylation, 14-3-3σ, MBD2, prostate cancer
INTRODUCTION
14-3-3 proteins comprise a family of abundant, highly conserved molecules that form homodimers and heterodimers and bind to over 200 target cellular proteins by phosphoserine-dependent and phosphoserine-independent manners (Dougherty and Morrison 2004; Fu et al., 2000; Mhawech 2005; Tzivion et al., 2001). Binding with 14-3-3 proteins regulates the function of protein ligands by inter- and intra-compartmental sequestration, activation/inactivation of enzymatic activity, and promotion/inhibition of protein interactions (Fu et al., 2000; Hermeking 2003; Tzivion et al., 2001; Tzivion and Avruch 2002; Woodcock et al., 2003). As a result, 14-3-3 proteins modulate numerous cellular processes in all eukaryotic cells (Hermeking 2003; Mhawech 2005; Muslin and Xing 2000). Among the seven human 14-3-3 isoforms (designated β, γ, ε, η, σ, τ and ξ), 14-3-3σ has been linked to cancer most directly due to its critical role in regulating signal transduction pathways, apoptosis, adhesion, cellular proliferation, differentiation and survival (Hermeking 2003; Mhawech 2005; Woodcock et al., 2003; Zhang et al., 2004).
There is growing evidence that loss of 14-3-3σ expression via CpG methylation contributes to invasive and malignant phenotypes. For example, 14-3-3σ has been shown to be highly expressed in normal epithelial cells, but this expression is lost in many high-grade carcinomas of the breast (Ferguson et al., 2000), ovary (Kaneuchi et al., 2004; Mhawech et al., 2005), endometrium (Kaneuchi et al., 2004; Mhawech et al., 2005), skin (Lodygin et al., 2003), lung (Osada et al., 2002; Yatabe et al., 2002), and liver (Iwata et al., 2000). Recently, 14-3-3σ expression was assessed by immunohistochemistry in one hundred and eleven specimens of invasive prostate adenocarcinoma with paired, adjacent high-grade prostatic intraepithelial neoplasia (HGPIN) and normal prostate epithelium. Abundant 14-3-3σ expression was evident in all normal prostatic epithelium, whereas ninety percent of samples of HGPIN and ninety-seven percent of invasive prostatic adenocarcinomas had no or low 14-3-3σ expression (Cheng et al., 2004). This was further substantiated by the results of another recent study showing hypermethylation of promoter-associated CpG islands and silencing of the 14-3-3σ gene in 41 of 41 (100%) primary prostate cancers and prostatic intraepithelial neoplasia (PIN), but not in normal tissues or prostate tissues exhibiting benign hyperplasia (BPH) as determined by methylation-specific PCR (MSP) and immunohistochemical analysis (Lodygin et al., 2004). These findings suggest that loss of 14-3-3σ expression, often due to methylation of promoter-associated CpG islands, is a crucial event associated with disease progression in prostate cancer.
Aberrant cytosine methylation patterns in CpG islands are hallmarks of human cancers, and the methylation of CpG islands in gene promoters acts as a powerful suppressor of transcriptional activity (Baylin and Herman 2000; Jones and Laird 1999; Singal and Ginder 1999). Earlier studies have shown that the methylation of normally unmethylated CpG dinucleotides located in the promoter regions is involved in gene silencing at the transcriptional level (Darwanto et al., 2003; Goodman et al., 2003). The mechanism of gene silencing by methylation of promoter-associated CpG islands varies among gene promoters (Darwanto et al., 2003; Kitazawa et al., 1999) and the pattern of distribution of methylated and unmethylated CpGs is specific to cell-type (De Smet et al., 2004). Increasing our understanding of the molecular mechanisms involved in gene silencing by methylation of promoter-associated CpG islands is an area of active research (Jenuwein and Allis 2001; Jones 1999).
Transcriptional silencing via methylation of promoter-associated CpG islands has been shown to be caused by two mechanisms, one of which is direct interference with the binding of transcriptional factors (Baylin and Herman 2000; Tate and Bird 1993), and the other, recruiting of methyl-CpG-binding proteins (MeCPs), which inhibit the binding of transcriptional factors to the promoter regions (Fujita et al., 1999; Nakagawachi et al., 2003; Nguyen et al., 2001; Yu et al., 2000). Since most transcriptional factors do not have CpG dinucleotides within their binding sites, silencing by methylation of promoter-associated CpG islands is believed to be largely mediated by the binding of MeCPs to methylated CpG dinucleotides. Previous studies have demonstrated that binding of MeCP2 and MBD2 during the process of DNA methylation of several genes that are implicated in human cancers (Bird 2002; Esteller et al., 2001; Jones and Baylin 2002; Lin and Nelson 2003; Sharma et al., 2005; Singal et al., 2001). Indeed, this possibility has been suggested for methylation-dependent transcriptional silencing of 14-3-3σ expression in breast cancer cells (Ferguson et al., 2000).
Since the loss of 14-3-3σ expression via CpG methylation is the leading finding in most cancers and plays a crucial role in the transition of normal epithelium to high-grade carcinoma, it is imperative that we understand what factors are associated with the CpG methylation-dependent transcriptional silencing of 14-3-3σ expression in human cancers. Although recent studies have indicated that the frequent loss of 14-3-3σ via CpG methylation is a key epigenetic mechanism in prostate carcinogenesis and is correlated with disease progression, the precise mechanisms of deranged methylation patterns leading to 14-3-3σ-specific transcriptional silencing in association with promoter methylation and its link to advancement of prostate cancer has not been established. Therefore, a better understanding of the methylation-dependent regulation of 14-3-3σ expression and its biological consequences is of paramount importance. One mechanistic clue is that the overexpression of 14-3-3σ results in G2 cell-cycle arrest in a p53-dependent manner in response to DNA damage and cells that are lacking 14-3-3σ are defective in a stable G2 cell-cycle arrest (Chan et al., 1999). The demethylation of 14-3-3σ-inactivated cells with a combination of 5-aza and TSA leads to activation of 14-3-3σ gene expression, thereby underscoring the importance of MeCPs in maintaining the repressive environment at the 14-3-3σ gene. However, direct evidence is currently lacking. We hypothesized that the CpG methylation-dependent transcriptional silencing of 14-3-3σ may be associated with methyl-CpG-binding proteins in prostate cancer cells and that the loss of 14-3-3σ function causes unregulated cell cycle progression, which in turn, might favor hyperproliferation and tumorigenesis.
To examine this hypothesis, we assessed the relationship between CpG methylation, mRNA transcriptional level and MeCPs association with the 14-3-3σ gene promoter CpG islands in prostate cancer cells.
MATERIALS AND METHODS
Cell lines and culture conditions
The prostate cancer cell lines LNCaP, DU145, PC3 and Tramp-C1 were obtained from the American Type Culture Collection (Manassas, VA). LNCaP cells were cultured in RPMI medium supplemented with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, and 1.0 mM sodium pyruvate (Invitrogen, Carlsbad, CA). Tramp-C1, PC3 and DU145 cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12K (1:1). Both media contained 10% fetal bovine serum (GIBCO BRL, Lewisville, TX) and 5% penicillin/streptomycin and were maintained in a 37°C incubator in a 5% CO2 humidified atmosphere.
5-aza and trichostatin A (TSA) treatment
Cells were seeded at a density of 1×106 cells/100 mm dish and allowed to attach over 24 h. 5-aza (Sigma, St. Louis, MO) was added to the medium for final concentrations ranging from 0–10 μM for 72 h. At every 24 h interval, fresh medium containing the drug was added. For the synergistic study, cells were first incubated with 10 μM 5-aza for 72 h at 37°C and then 50 nM TSA (Sigma, St. Louis, MO) was added for an additional 16 h. The treated cells were washed once with phosphate buffered saline (PBS). Cells were allowed to recover for 24 h in drug-free medium in a 37 °C incubator in a humidified atmosphere with 5% CO2.
Transfection of siRNAs
To silence 14-3-3σ or MBD2 expression by RNA interference, 150,000 cells per well were seeded in a 6-well plate at least 20 h before transfection. siRNAs against 14-3-3σ, MBD2, or non-specific control (siControl) were transfected using siLentFect transfection reagent (Bio-Rad, Hercules, CA) as per the manufacturer's instructions. Two days post-transfection, the nearly confluent cells were trypsinized and the cells were used for cell proliferation assays, immunoblot analysis, RT-PCR analysis, and ChIP assay. Target sequences of the siRNAs for MBD2 knockdown (derived from the MBD2 gene at base pair 671 of sequence NM_003927) is 5′- aagaggatggattgcccggcc- 3′. The sequences of the siRNAs for 14-3-3σ gene knockdown are as follows: si14-3-3σ1, 5′ – catggcagccttcatgaaa – 3′; si14-3-3σ2, 5′ – cactcttcttgcagctgtt – 3′ and si14-3-3σ3, 5′ – ctctgatcgtaggaattga - 3′. “Smart pool” siRNAs that combined the above 14-3-3σ siRNAs 1-3 targeted against different regions of the 14-3-3σ mRNA sequence (NM_006142) were used for transfection to increase the knockdown effect.
Immunoblot analysis
Cells were washed and lysed in 1X cell lysis buffer (Cell Signaling Technology Inc., Beverly, MA). Protein concentrations were determined on diluted samples using a bicinchoninic acid procedure (Pierce Biochemical Company, Rockford, IL). Equal amounts of protein were separated on SDS-PAGE and transferred to polyvinylidene difluoride membranes (BioRad, Hercules, CA). Membranes were blocked in a PBS solution with 5% non-fat dry milk and incubated with primary antibodies in blocking solution overnight at 4 °C. Membranes were then washed three times in PBS with 0.1% tween-20 (T-PBS) at 10 min intervals. Horseradish peroxidase conjugated secondary antibodies (Biomeda, Foster City, CA) were used for detection of immunoreactive proteins by chemiluminescence (Amersham Biosciences, Piscataway, NJ). The following antibodies were used: anti-MeCP2 (Upstate Biotechnology, Lake Placid, NY), anti-MBD2 (Upstate Biotechnology, Lake Placid, NY), anti-PCNA (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-GAPDH (Abcam, Cambridge, MA). Antibodies against 14-3-3 isoforms, such as 14-3-3β, 14-3-3ξ, 14-3-3ε and 14-3-3σ, were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Protein detection by Immunofluorescence
Cells were cultured on Lab-Tek II chamber slides (Nalge Nunc International, Naperville, IL) and fixed in 4% paraformaldehyde solution for 20 min, permeabilized in 0.2% Triton-X (Sigma) in PBS for 15 min, and blocked in PBS with 1% BSA for 1 h. Primary antibodies specific for 14-3-3σ (Santa Cruz Biotechnology, Santa Cruz, CA) diluted in PBS with 1% BSA and 0.05% Tween 20 were added for 1 h and detected with Texas redconjugated donkey anti-goat antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The cells were covered with mounting medium containing the nuclear counterstain DAPI. Fluorescent images were acquired using a charge-coupled device RT Slider Spot Camera (Diagnostic Instruments Inc, Burroughs Sterling Heights, MI) connected to a microscope (Olympus, Melville, NY) and managed by a computer equipped with the spot RT software v3.5 (Diagnostic instruments, Burroughs Sterling Heights, MI).
Bisulfite modification of genomic DNA
Genomic DNA was isolated from LNCaP, DU145, PC3 and Tramp-C1 prostate cancer cells using the DNeasy tissue kit (Qiagen, Germany). The bisulfite reaction was carried out on 5 μg of genomic DNA with the CpGenome DNA modification kit as recommended by the manufacturer (Chemicon, Temecula, CA). Briefly, genomic DNA was treated with 3.0 mM sodium bisulfite (pH 5.0) and 0.5 mM hydroquinone for 16 h at 50°C. With this treatment, all cytosine residues are converted to uracil in unmethylated DNA, but those that are already methylated (5-methylcytosine) are resistant to this treatment and remain as cytosine. The reaction mixture was then purified with the Promega Wizard Clean-UP Kit (Madison, WI) and desulphanated with 0.3 M sodium hydroxide for 20 min at 40°C. The converted DNA was then precipitated in cold ethanol, dissolved in H20, and stored at −20°C.
Methylation-specific PCR (MSP)
Methylation-specific PCR was performed with 25 μL using 3 units of platinum Taqpolymerase (Invitrogen, Carlsbad, CA) per reaction. We used the following MSP primers: (1) for unmethylated 14-3-3σ sequence: 5′-TTA TTA GAG GGT GGG GTG GAT TGT-3′ (sense) and 5′-CAA CCC CAA ACC ACA ACC ATA A-3′ (antisense); (2) for methylated 14-3-3σ sequence: 5′-GGT TTT TTC GGT TAG TTG CGC GGC G-3′ (sense) and 5′-CCA ACG AAA ACC TCG CGA CCT CCG-3′ (antisense). After denaturation at 95°C for 5 min, 40 PCR-cycles were completed with the bisulfite-treated genomic DNA as a template. 20 μL of the PCR-amplified fragments were loaded onto 2% agarose gels for analysis. Positive controls used for methylation-specific PCR included DNA from normal prostate tissues as unmethylated DNA control and CpGenome Universal methylated DNA as methylated DNA control (Chemicon International). Negative control MS-PCR reactions were performed using water only as template.
Cloning and bisulfite sequencing
To permit DNA sequencing of inclusive CpG sites within the CpG island of the 14-3-3σ gene, PCR products were purified by electrophoresis on 1% agarose gels using the Qiaquick™ gel extraction kit (Qiagen), ligated into pCR2.1pTOPO-TA vectors (Invitrogen), and then introduced into TOP10 One Shot competent bacteria (Invitrogen). Transformed cells were plated on LB agar containing 100 μg/mL ampicillin (Invitrogen) and incubated overnight at 37 °C. Ten individual colonies were selected and each inoculated into 3 mL LB broth containing 100 μg/mL ampicillin (Invitrogen) and grown overnight at 37 °C. The insert containing plasmid DNA was then extracted from the cells using the Eppendorf FastPlasmid mini-prep kit (Eppendorf, Westbury, NY) and resuspended in 30 μL of the included EB buffer. A minimum of six clones selected at random from each DNA was sequenced in automated DNA sequencer.
Promoter methylation analysis
The methylation status of the 14-3-3σ gene promoter was determined using HhaI-based promoter methylation analysis. Genomic DNA (1-2 μg) obtained from LNCaP, DU145, PC3 and Tramp-C1 prostate cancer cells were digested for 3 h with a methylation-sensitive enzyme, HhaI (New England Biolabs Inc., Beverly, MA), in a 20 μL reaction. 2 μL were used as template for 14-3-3σ promoter-specific PCR with the following primers: 5′-CAT GAA AGG CGC CGT GGA GAA-3′ (sense) and 5′-GCT GAT GTC CAT GGC CTC CTG G-3′ (antisense). PCR amplification indicates that the HhaI site within the 14-3-3σ promoter targeted sequence was methylated and protected from digestion.
Reverse transcription-PCR analysis
Cellular RNA was isolated from LNCaP, DU145, PC3 and Tramp-C1 prostate cancer cell lines using the Qiagen RNeasy kit. 1 μg of RNA was treated with DNase (10 units/μg of RNA for 1 h) and used as a template for the reverse transcription reaction (RT, 20 μL). The RT reaction mix (Invitrogen, Carlsbad, CA) contained 1 μL (10 pm) of primers. The resultant cDNA was then used in PCR reactions and analyzed by gel electrophoresis. The following primers were used: 5′-ACG ACA AGA AGC GCA TCA TTG-3′ (sense) and 5′-GGC ATC TCC TTC TTG CTG ATG T-3′ (antisense). PCR conditions were as follows: 95°C for 5 min, followed by 40 cycles at 95 °C for 1 min, 55°C for 1 min, and 72°C for 1 min. The final extension was at 72°C for 5 min.
Chromatin Immunoprecipitation (ChIP) and siRNA-coupled ChIP Assays
ChIP analysis was used to determine the binding activity of MeCP2 and MBD2 in the 14-3-3σ promoter in PC3, DU145, LNCaP and Tramp-C1 cells before and after 5-aza treatment. ChIP assays were performed per manufacturer's instructions (17-295, Upstate Biotechnology, Lake Placid, NY). Briefly, prostate cancer cells (∼1×106 cells/100 mm dish) were fixed by adding formaldehyde at a final concentration of 1% and incubating for 10 min at 37°C. The cells were washed twice with ice-cold PBS containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A), harvested, and treated with SDS lysis buffer for 10 min on ice. The resulting lysates were sonicated to shear the DNA to fragment lengths below 1000 bp (amplitude 60%, 4×10s, Fisher Sonic Dismembrator 60, Pittsburgh, PA). From each sonicated sample, 5% was used as the input control for immunoprecipitated fragments. The complexes were immunoprecipitated with antibodies specific for MeCP2 (no. 07-013) and MBD2 (no. 07-198) from Upstate Biotechnology. 10 μL of antibody were used for each immunoprecipitation according to the manufacturer's instructions. Antibody controls were also included for each ChIP assay; no precipitation was observed. The antibody/protein complexes were collected using salmon sperm DNA/protein A agarose slurry and washed several times per manufacturer's instructions. The immune complexes were eluted with 1% SDS and 0.1 M NaHCO3, and the cross-links were reversed by incubation at 65°C for 4 h in the presence of 200 nM NaCl. The samples were treated with proteinase K for 1 h, and the DNA was purified by phenol/chloroform extraction, ethanol precipitation, and resuspended in 30 μL of H2O. Initially, PCR was performed with different number of cycles or dilutions of input DNA to determine the linear range of the amplification; all results shown fall within this range. Following 30 cycles of amplification, PCR products were run on 2% agarose gels and analyzed by ethidium bromide staining. For the siRNA-coupled ChIP assay, LNCaP cells transfected with siRNA against MBD2 and shown knockdown effect were selected. The cells were harvested and the immunoprecipitated chromatin was analyzed by PCR with primers specific to the 14-3-3σ promoter.
MTT Cell proliferation assay
Proliferation of LNCaP, DU145, PC3 and Tramp-C1 cells was assessed with the MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] method using a cell proliferation kit (Chemicon, Temecula, CA). Cells were plated at density of 1×103 cells/well in 96-well plates and cultured under standard tissue culture conditions for 24 h. Cells were treated with 10 μM 5-aza for 72 h followed by 50 nM TSA for an additional 16 h. Treated cells were washed once with PBS. Cells were allowed to recover for 24 h in drug-free medium in a 37°C incubator in a humidified atmosphere with 5% CO2. Then, 10 μL of MTT reagent were added and the cells were incubated for another 4 h at 37°C. Acid-isopropanol (0.04 N HCl/isopropanol) was immediately added to all wells and mixed vigorously so that the dark blue crystals dissolved effectively. Absorbance was measured at 570 nm (Benchmark, BIORAD, Hercules, CA).
RESULTS
Endogenous expression of 14-3-3 proteins in prostate cancer cell lines
We initially examined the expression pattern of 14-3-3 proteins in the prostate cancer cell lines PC3, DU145, LNCaP and Tramp-C1. These cell lines show differential expression of 14-3-3σ proteins. Detectable levels of 14-3-3σ protein were observed only in PC3 and DU145 cells, but not in LNCaP and Tramp-C1 cells. This is consistent with a recent report (Urano et al., 2004) in which the human prostate cancer cell line LNCaP had undetectable protein levels. In contrast, the expression levels of other isoforms of 14-3-3 proteins including 14-3-3β, 14-3-3ξ and 14-3-3ε are easily detectable and do not vary among these cell lines (Fig. 1A).
Figure 1.
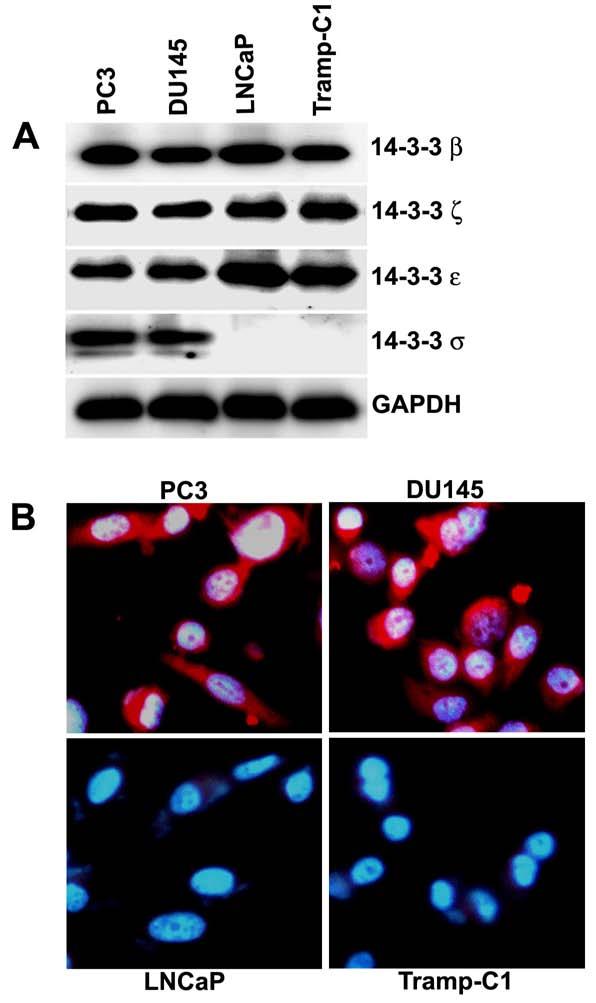
- Immunoblot analysis of 14-3-3 proteins in prostate cancer cell lines PC3, DU145, LNCaP and Tramp-C1. Equal amounts of total cellular protein lysates extracted from each of the cell lines were electrophoresed on SDS-PAGE, subjected to immunoblot with anti-14-3-3β, anti-14-3-3ξ, anti-14-3-3ε, anti-14-3-3σ, and anti-GAPDH antibodies, and followed by chemiluminescence detection. GAPDH was used as a loading control.
- Immunostaining for 14-3-3σ protein expression in PC3, DU145, LNCaP and Tramp-C1 cells with anti-14-3-3σ antibodies (red signal). Nuclei were stained with DAPI (blue signal).
In order to further confirm 14-3-3σ protein expression, immunostaining of 14-3-3σ was performed on PC3, DU145, LNCaP and Tramp-C1 cells. In LNCaP and Tramp-C1 cells, which lack 14-3-3σ protein expression, there was no staining specific for 14-3-3σ. In contrast, PC3 and DU145 cells, which express 14-3-3σ protein, 14-3-3σ-specific staining was observed in the cytoplasm, and to a lesser degree, in the nucleus (Fig. 1B). These indirect immunofluorescence studies confirmed the immunoblot analysis.
Methylation status of 14-3-3σ gene promoter in prostate cancer cell lines
To determine whether methylation of the 14-3-3σ promoter region was responsible for the loss of expression of this gene, we examined the methylation status of the 14-3-3σ promoter region in PC3, DU145, LNCaP and Tramp-C1 cells using MSP. Genomic DNA was treated with bisulfite under conditions that convert unmethylated cytosine to uracil, while 5-methylcytosine remains unchanged (Frommer et al., 1992). Two MSP primer sets were designed to specifically amplify either methylated (M) or unmethylated (U) bisulfite-modified sequence in the 14-3-3σ promoter-associated CpG islands (Fig. 2A). Amplification with either the methylated or unmethylated set of primers results in either the presence or absence of a PCR product depending on the methylation status of the CpG dinucleotides interrogated by that primer pair. PCR products obtained with the two MSP primer sets, M and U, on bisulfite-modified DNA from PC3, DU145, LNCaP and Tramp-C1 cells are shown (Fig. 2B). In PC3 and DU145 cells, which express 14-3-3σ, the PCR products were only visible in the unmethylated lane representing no methylation at the 14-3-3σ CpG islands. However, in the LNCaP and Tramp-C1 cells, prominent PCR products were obtained specific for methylated CpG islands with the methylated primer set but no products were detected with the unmethylated primer set. These MSP analysis results suggest the presence of dense methylation of the 14-3-3σ promoter in LNCaP and Tramp-C1 cells.
Figure 2.
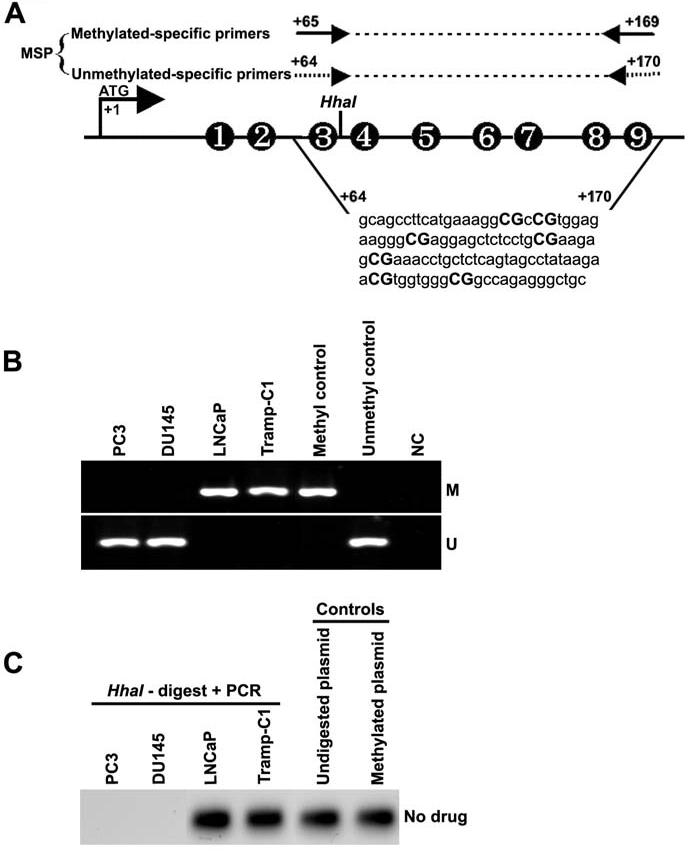
- Schematic map of the 14-3-3σ promoter-associated CpG island region indicating the location of PCR primers used for MSP analysis. Each of the CpG dinucleotide residues with respect to the transcriptional start site ATG (+1) is shown as numbers in circles from 1 to 9, which can serve as potential DNA methylation sites. The nucleotide sequence located in the promoter region of the human 14-3-3σ gene from +64 to +170 and CpG dinucleotides are indicated in bold.
- Bisulfite-modified DNA derived from prostate cancer cell lines PC3, DU145, LNCaP and Tramp-C1 were amplified with 14-3-3σ primers specific for methylated (top) and unmethylated DNA (bottom). Methylation-specific primers generated the PCR products labeled with “M”. Those labeled with “U” were generated by primers specific for unmethylated DNA. Positive and negative controls were described in Materials and Methods.
- HhaI-based DNA methylation assay of the 14-3-3σ promoter. Genomic DNA was prepared from prostate cancer cell lines PC3, DU145, LNCaP and Tramp-C1. HhaI cut within the 14-3-3σ PCR primer target site, thereby preventing PCR amplification in unmethylated samples. SssI-methylated, 14-3-3σ plasmid DNA and undigested 14-3-3σ plasmid DNA were used as positive controls.
To further confirm the methylation status of 14-3-3σ in PC3, DU145, LNCaP and Tramp-C1 cells, we determined the precise methylation status of the 14-3-3σ CpG island by HhaI-based methylation assay. A set of primers was designed to amplify 14-3-3σ promoter region spanned with CpG dinucleotides and used to confirm promoter methylation. This primer target within the 14-3-3σ promoter contains a restriction site for the methylation-sensitive enzyme, HhaI. When methylated, this site is protected from digestion, and a PCR product spanning it can be amplified. If unmethylated, the HhaI site cut within the 14-3-3σ promoter primer target site, prevents PCR amplification. HhaI site was unmethylated in genomic DNA from PC3 and DU145 cells, which show undetectable PCR products. However, prominent PCR products were observed in genomic DNA from LNCaP and Tramp-C1 cells, suggesting that HhaI site was methylated (Fig. 2C). Undigested and Sss-I methylated plasmid DNAs were used as positive controls. LNCaP and Tramp-C1 cells, but not PC3 and DU145 cells, showed CpG methylation of the 14-3-3σ promoter with both techniques, which correlated with 14-3-3σ protein expression.
Treatment of LNCaP and Tramp-C1 cells with the epigenetic gene silencing modifiers 5-aza and TSA results in activation of 14-3-3σ expression
To determine the contribution of 14-3-3σ promoter methylation to the expression of this gene, we treated the promoter-methylated prostate cancer cell lines with varying concentrations of 5-aza and TSA. At the end of the treatment, RNA was isolated for RT-PCR analysis of 14-3-3σ expression. Amplification products were analyzed by gel electrophoresis. Formaldehyde gels were used to determine the integrity and quantity of RNA used in the PCR reaction. The 14-3-3σ RT-PCR primers used in these experiments generate a 105 bp PCR product. Conditions for optimal transcriptional induction after combined treatment with 5-aza and trichostatin A were determined by RT-PCR using 14-3-3σ gene-specific primers. According to RT-PCR analysis, treatment with 10 μM 5-aza for 72 h and 50 nM TSA for an additional 16 h restored an optimal level of 14-3-3σ mRNA expression in LNCaP and Tramp-C1 cells (Fig. 3A). These changes in 14-3-3σ expression following treatment with 5-aza plus TSA are consistent with the notion that promoter-associated CpG methylation repressed 14-3-3σ expression in LNCaP and Tramp-C1 cells. In order to test for an inherent effect of this drug on 14-3-3σ expression in cells that normally express 14-3-3σ, we also examined the effects of 5-aza plus TSA in PC3 and DU145 cells. No significant change in 14-3-3σ expression levels was observed in PC3 and DU145 cells after drug treatment (Fig. 3B). These results demonstrate that 14-3-3σ expression in the prostate cancer cell lines LNCaP and Tramp-C1 is directly regulated by 14-3-3σ promoter methylation. The treatment of prostate cancer cell lines LNCaP and Tramp-C1 with 5-aza plus TSA results in reactivation of 14-3-3σ expression, indicating a causal role for hypermethylation in the loss of 14-3-3σ expression.
Figure 3.

- mRNA expression of 14-3-3σ was analyzed using reverse transcription-PCR in LNCaP, DU145, PC3 and Tramp-C1 prostate cancer cells treated with either DMSO (control) or 10 μM 5-aza followed by 50 nM TSA, as described in Materials and Methods. GAPDH mRNA was co-amplified as a loading control and expression standard.
- Bar diagram showing densitometry quantified data of 14-3-3σ/GAPDH mRNA ratios from three independent experiments. Each bar represents triplicate analyses of mean ± SD where significant difference from cells treated with vehicle alone (control) is represented by an asterisk * (P <0.05).
- MSP assay on DNA isolated from 5-aza/TSA-treated or untreated cell lines PC3, DU145, LNCaP and Tramp-C1. Methylation-specific primers generated the PCR products labeled with “M”. Those labeled with “U” were generated by primers specific for unmethylated DNA. Positive and negative controls were described in Materials and Methods.
- HhaI-based DNA methylation assay on DNA isolated from 5-aza and TSA-treated PC3, DU145, LNCaP and Tramp-C1 cells. SssI-methylated, 14-3-3σ plasmid DNA and undigested 14-3-3σ plasmid DNA were used as positive controls.
- Bisulfite sequencing analysis of 14-3-3σ promoter CpG island methylation in 5-aza/TSA treated or untreated prostate cancer cells. Seven potential CpG sites (3/+81 to 9/+156, genomic positions 8721, 8724, 8736, 8751, 8758, 8787, and 8796; Accession No. AF029081) were analyzed by methylation-specific PCR followed by cloning and DNA sequencing. Methylated cytosines remain cytosines (cm), whereas unmethylated cytosines are converted thymidine (T). Absence of 14-3-3σ expression (−) and presence of 14-3-3σ expression (+). Results are representative of at least 4 different DNA sequence analyses.
To determine whether this reactivation was associated with a change in the methylation pattern of the 14-3-3σ promoter, genomic DNA was isolated from drug-treated prostate cancer cells and subjected to MSP analysis as well as HhaI-based methylation assay. We obtained consistent results with both techniques. As shown in Fig. 3C, treatment of both cell lines LNCaP and Tramp-C1 showed almost complete demethylation as determined by MSP. We also confirmed the precise demethylation status of the 14-3-3σ CpG islands at promoter region using a HhaI-based methylation assay (Fig. 3D). Furthermore, the DNA sequence analysis of the methylation-specific PCR products revealed that the amplified region was completely methylated at C residues followed by G residues in cell lines LNCaP and Tramp-C1, whereas none of these sites were methylated in the 14-3-3σ expressing cell lines PC3, DU145 and 5-aza/TSA treated cell lines LNCaP and Tramp-C1 (Fig. 3E). These results strongly suggest a direct role of CpG island methylation in this region for transcriptional silencing of 14-3-3σ gene expression. Collectively, our findings are in agreement with numerous studies demonstrating that promoter-associated CpG island methylation interferes with transcription, resulting in the absence of mRNA and protein expression (Galm et al., 2005; Konduri et al., 2003; Park et al., 2004).
Involvement of methyl-CpG binding proteins in 14-3-3σ silencing
Silencing mediated by CpG methylation correlates with the binding of MeCPs on methylated promoters (Bird and Wolffe 1999; Ego et al., 2005; Sharma et al., 2005; Singal and Ginder 1999). MeCP2 and MBD2 have been shown to bind to methylated CpG residues in any sequence context and are implicated in methylation-mediated transcriptional silencing of several genes in eukaryotic cells (Ego et al., 2005). The 14-3-3σ promoter region contains dense CpG dinucleotides and this region could potentially bind to MeCPs. It is possible that methylation-mediated transcriptional silencing of the 14-3-3σ gene promoter may involve interaction with a methyl-CpG binding protein, such as MBD2 or MeCP2.
To assess whether MBD2 and MeCP2 are involved in CpG methylation-dependent 14-3-3σ gene silencing, we performed ChIP assays in 14-3-3σ-expressing prostate cancer cell lines with an unmethylated promoter (PC3 and DU145) and a methylated promoter of 14-3-3σ non-expressing prostate cancer cell lines (LNCaP and Tramp-C1). ChIP is a powerful technique used to test for the presence of certain DNA-binding proteins that modulate transcriptional characteristics of the specific region of DNA with which they are associated. Using antibodies against MeCP2 and MBD2 proteins, formaldehyde cross-linked protein-chromatin complexes were immunoprecipitated from 14-3-3σ expressing or non-expressing cells. After immunoprecipitation, the DNA was released, and the amount of 14-3-3σ specific DNA that was released from the immunoprecipitates was analyzed by PCR. As shown in Fig. 4A & B, ChIP assay showed that MBD2 is strongly associated with the methylated CpG island in the 14-3-3σ promoter in LNCaP and Tramp-C1 cells. However, there was no significant difference of MeCP2 association between the unmethylated and methylated 14-3-3σ promoters. For PC3 and DU145 cells, which contain unmethylated CpG islands in the 14-3-3σ promoter and express 14-3-3σ, ChIP analysis revealed that there was no significant level of MBD2 association with the unmethylated CpG island in the 14-3-3σ promoter, consistent with active 14-3-3σ transcription (Fig. 4B). These findings indicate that MBD2 was associated with the methylated 14-3-3σ promoter-associated CpG Island, and thus, interferes with transcriptional activity of the methylated promoter. Consistent with this notion, a previous report suggests that methyl-CpG-binding proteins are involved in suppressing 14-3-3σ transcription in breast cancer cells (Ferguson et al., 2000).
Figure 4.

- Assessment of MBD2 and MeCP2 binding on the 14-3-3σ promoter by ChIP analyses. The bound MeCPs fraction (ChIP) shows binding to the 14-3-3σ promoter. Crosslinked chromatin was carefully prepared from 14-3-3σ-expressing (PC3 and DU145) and non-expressing (LNCaP and Tramp-C1) prostate cancer cells, sonicated to shear DNA fragments below 1000 bp, and immunoprecipitated with the indicated antibodies. Sheared chromatin fractions above this upper limit were not processed for ChIP mapping of the 14-3-3σ promoter. The immunoprecipitates were subjected to PCR analysis using primer pairs spanning the 14-3-3σ promoter-associated CpG island (depicted schematically in Fig. 2A). Aliquots of chromatin taken before immunoprecipitation were used as “Input” controls whereas chromatin eluted from immunoprecipitations lacking antibody were used as “no antibody” (−Ab) controls.
- Quantification of the MBD2 and MeCP2 ChIP fractions on the 14-3-3σ promoter. ChIP quantification was plotted as a ratio of bound MBD2 or MeCP2 ChIP (no antibody control: bound) to input signal and expressed as bound MBD2 or MeCP2 on the y-axis. Each bar represents the mean ± SD of three independent assays where significant differences from 14-3-3σ-expressing (PC3 and DU145) cells, which exhibited low or undetectable bound MBD2 on the 14-3-3σ promoter, are represented by asterisks * (P <0.05).
- Immunoblot of MeCP2 and MBD2 proteins. Equal amounts (100 μg) of protein from cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for MeCP2, MBD2 and GAPDH. GAPDH was utilized as a loading control.
- Bar diagram showing densitometry quantified data of MBD2 or MeCP2/GAPDH protein expression ratios from three independent experiments. Each bar represents triplicate analyses of mean ± SD.
MBD2 and MeCP2 expression in the four prostate cell lines was confirmed by immunoblot analysis (Fig. 4C). 14-3-3σ expressing prostate cancer cell lines PC3 and DU145, which contain an unmethylated promoter, showed lower expression of MBD2 than did the 14-3-3σ-inactive, promoter-methylated Tramp-C1 cells. However, a low level of MBD2 was observed in LNCaP cells, which also do not express 14-3-3σ and have promoter methylation (Fig. 4D). PC3 cells express 14-3-3σ showed higher levels of MeCP2 expression than LNCaP and Tramp-C1 cells. In contrast, DU145 cells showed a lower level of MeCP2 expression (Fig. 4D). However, there was no significant correlation between the four cell lines in terms of endogenous MBD2 and MeCP2 protein expression and 14-3-3σ gene silencing with the methylation of promoter-associated CpG island.
Efficient dissociation of MBD2 proteins from the silenced 14-3-3σ promoter is observed after treatment with 5-aza and TSA
We examined the effects of 5-aza and TSA, epigenetic silencing modifiers, on MBD2 associated with the 14-3-3σ promoter. Treatment of LNCaP and Tramp-C1 cells with 5-aza and TSA caused a significant reduction in MBD2 occupancy of the 14-3-3σ promoter as determined by the ChIP assay (Fig. 5A & 5B). Our results indicate that treatment of cells with 5-aza and TSA demethylates the 14-3-3σ promoter-associated CpG islands and precipitates release of MBD2 from the target promoter, suggesting that induced demethylation interferes with the recruitment of MBD2 and abolishes the silencing effect of 14-3-3σ.
Figure 5.

- ChIP assay of MBD2. The formaldehyde crosslinked chromatin was immunoprecipitated with antibodies specific for MBD2. Purified DNA was amplified for the 14-3-3σ promoter as described in Fig. 4. MBD2 dissociate from the 14-3-3σ promoter-associated CpG island following treatment of LNCaP and Tramp-C1 cells with 5-aza and TSA, as described in Materials and Methods. Controls show input genomic DNA before the addition of antibody and eluants from no antibody immunoprecipitations.
- Quantification of the MBD2 ChIP fractions on the 14-3-3σ promoter as described in Fig. 4. Each bar represents triplicate analyses of mean ± SD where significant difference from cells treated with vehicle alone (control) is represented by an asterisk * (P <0.05).
- MBD2 protein expression in total cell lysates from 5-aza and TSA-treated and untreated PC3, DU145, LNCaP and Tramp-C1 cells. Equal amounts (100 μg) of protein from control or treated cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for MBD2 and GAPDH. GAPDH was utilized as a loading control.
- Bar diagram showing densitometry quantified data of MBD2/GAPDH protein expression ratios from three independent experiments. Each bar represents triplicate analyses of mean ± SD.
Reduction of global protein expression may account in part for the dissociation of MBD2 from the 14-3-3σ promoter in 5-aza and TSA-treated LNCaP and Tramp-C1 cells. However, immunoblot analysis showed that the overall levels of MBD2 in LNCaP and Tramp-C1 cells did not drastically change after 5-aza and TSA treatment (Fig. 5C & 5D), suggesting that the chromatin alterations detected by ChIP analysis were region-specific rather than global. Consistent with this notion, the global levels of MBD2 was unaltered after 5-aza and TSA treatments in PC3 and DU145 cells. These findings suggest that MBD2 is involved in 14-3-3σ gene silencing through a methylated promoter.
MBD2 knockdown by small interfering RNA implicates MBD2 in CpG methylation-dependent 14-3-3σ silencing
To further confirm that the MBD2 was responsible for promoter CpG island methylation-mediated silencing of 14-3-3σ, the endogenous levels of the MBD2 were knockdown in LNCaP cells by treatment with specific siRNAs capable of degrading mRNA transcripts in a target specific manner. The siRNAs that specifically targeted to MBD2 exhibited a significant reduction in MBD2 mRNA (Fig. 6A, top) and MBD2 protein expression (Fig. 6A, bottom). In MBD2 knockdown cells, a stimulation of 14-3-3σ expression is observed when compared with the non-specific siRNA-transfected LNCaP cells (Fig. 6A & B). We then analyzed the specific binding levels of MBD2 proteins to the methylated region of 14-3-3σ promoter CpG island. The siRNA coupled-ChIP assay in LNCaP cells revealed that knockdown of MBD2 resulted in failure of recruitment of MBD2 on the 14-3-3σ promoter-associated CpG island (Fig. 6B, top). In addition, in MBD2 knockdown cells, 5-aza and TSA treatments had a lower effect on MBD2 binding to the 14-3-3σ promoter when compared with the siControl transfected LNCaP cells (Fig. 6B-C, top). Furthermore, the knockdown of MBD2 is not compensated by the binding of other methyl binding protein MeCP2 at the methylated region of 14-3-3σ promoter (Fig. 6B-C, bottom). These findings supports the conclusion that MBD2 binding is specific to the methylated region of 14-3-3σ promoter CpG island in LNCaP cells, the lack of repression activity for methylated region of 14-3-3σ promoter CpG island in LNCaP cells with knockdown of MBD2 levels strongly suggest that MBD2 likely mediates CpG methylation-dependent silencing of 14-3-3σ.
Figure 6.

- LNCaP cells were transfected with siRNA against MBD2. Specific knockdown of MBD2 mRNA (top) and MBD2 protein expression (bottom) were monitored by reverse-transcription PCR and immunoblot analysis, respectively.
- Bar diagram showing densitometry quantified data of MBD2/GAPDH (top) and MeCP2/GAPDH protein expression ratios (bottom) from three independent experiments. Each bar represents triplicate analyses of mean ± SD where significant difference from controls is represented by an asterisk * (P <0.05).
Biological consequences of loss of 14-3-3σ in prostate cancer cell lines
The biological meaning of the CpG methylation-based loss of 14-3-3σ has been analyzed in many human cancers (Cheng et al., 2004; Ferguson et al., 2000; Lodygin et al., 2003; Lodygin et al., 2004; Lodygin and Hermeking 2005; Mhawech 2005; Reichelt and Magin 2002; Urano et al., 2004). These studies demonstrated that epigenetic silencing of 14-3-3σ via CpG methylation in many cancer cells presumably contributes to hyperproliferation due to an inability to maintain cell-cycle arrest when 14-3-3σ is inactivated. Therefore, we asked whether the restoration of 14-3-3σ using epigenetic silencing modifiers 5-aza and TSA in 14-3-3σ-inactivated cell lines LNCaP and Tramp-C1 would affect cell proliferation. We used the MTT assay to assess the effect of 5-aza and TSA treatment (or lack thereof) on prostate cancer cell proliferation. As shown in Fig 7A, 5-aza plus TSA treatment in LNCaP and Tramp-C1 cells resulted in a 51.7% and 55.2% inhibition of proliferation as compared to the untreated controls. In contrast, treatment with 5-aza and TSA did not significantly affect the degree of cell proliferation in DU145 and PC3 cells (Fig. 7A). Immunoblot analysis of proliferating cell nuclear antigen (PCNA), a cell proliferation marker, confirmed by the MTT assay results (Fig. 7B-C). These findings suggest that loss of 14-3-3σ expression can induce prostate cancer cell proliferation.
Figure 7.
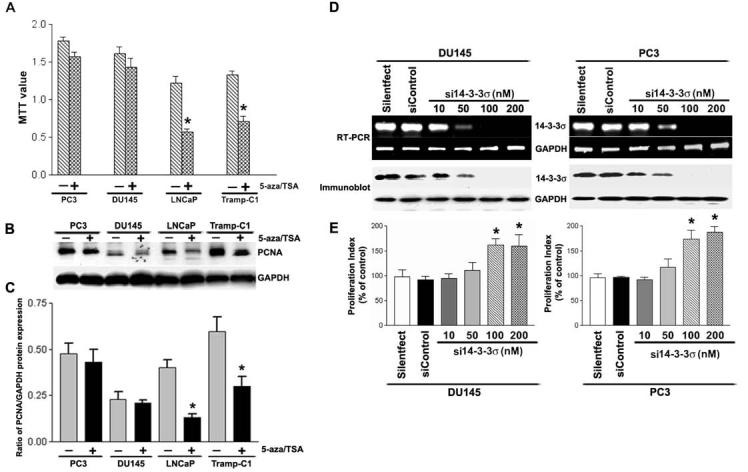
- Results of the MTT assay showed that 5-aza and TSA (epigenetic 14-3-3σ silencing modifiers) caused significant inhibition of proliferation in LNCaP and Tramp-C1 cells. There was no significant difference following treatment with 5-aza and TSA in PC3 and DU145 cells. Each bar represents triplicate analyses of mean ± SD where significant difference from controls is represented by an asterisk * (P <0.05).
- Immunoblot analysis of PCNA (a cellular proliferation marker) in prostate cancer cells before and after treatment with 5-aza and TSA. Equal amounts (10 μg) of protein from control and treated cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for PCNA and GAPDH. GAPDH was used as a loading control.
- Bar diagram showing densitometry quantified data of PCNA/GAPDH protein expression ratios from three independent experiments. Each bar represents triplicate analyses of mean ± SD where significant difference from cells treated with vehicle alone (control) is represented by an asterisk * (P <0.05).
- DU145 and PC3 cells were transfected with siRNA against 14-3-3σ. Specific knockdown of 14-3-3σ mRNA (top) and 14-3-3σ protein expression (bottom) were monitored by reverse-transcription PCR and immunoblot analysis, respectively.
- Proliferation index of DU145 and PC3 cells transfected with either 14-3-3σ specific siRNA or nonspecific siRNA control was revealed by MTT assay. Each bar represents triplicate analyses of mean ± SD where significant difference from controls is represented by an asterisk * (P <0.05).
To further confirm that silencing of 14-3-3σ contributes to hyperproliferation in prostate cancer cells, we silenced the expression of 14-3-3σ in PC3 and DU145 cells by using siRNA molecules. We verified specific knockdown effects of 14-3-3σ mRNA by reverse transcription-PCR (Fig. 7D, top). Immunoblot analysis of cell extracts was carried out to determine whether decreased mRNA expression, as observed, correlated with decreased translation of the gene product. A similar trend was observed by immunoblot assay as well (Fig. 7D, bottom). No effects of RNAi were observed on the expression of GAPDH, which was used as an internal control for specificity and loading at mRNA levels as well as protein levels. In addition, cells transfected with non-specific siRNA control also showed that RNAi-directed 14-3-3σ knockdown is specific (Fig. 7D). We then examined the effect of RNAi-directed knockdown of 14-3-3σ expression on cell proliferation in both PC3 and DU145 cells. As shown in Fig. 7E, with efficient 14-3-3σ knockdown, there was a significant increase in cell proliferation when compared with the silentfect alone or nonspecific siRNA-transfected PC3 cells. A similar trend was observed in DU145 cells as well (Fig. 7E). Consistent with the clinical data shown earlier (Cheng et al., 2004), these evidence suggest that the loss of 14-3-3σ expression promotes cell proliferation in prostate cancer cells.
DISCUSSION
Prostate cancer remains the most common solid tumor malignancy in men and the second leading cause of cancer death in the United States. It is a disease with a complex etiology influenced by several factors, including genetic, biological, lifestyle and environmental factors. The precise mechanisms that predispose, precipitate and maintain the cellular transformation process during tumorigenesis are poorly defined and still being intensely studied. As such, the factors that lead to the development of metastasis and profoundly influence the course and outcome of this disease are of great interest. Although effective treatments such as radical prostatectomy, radiotherapy, and chemotherapy are available for organ-confined prostate cancer, metastatic prostate cancer remains essentially incurable. Genetic changes in prostate cells significantly affect cell proliferation and behavior, thereby contributing to the development and progression of malignant cancers. For instance, loss of 14-3-3σ gene expression is associated with increased proliferation and metastasis in many cancers (Ferguson et al., 2000; Tanaka et al., 2004; Lodygin and Hermeking 2005; Cheng et al., 2004).
Great effort has been devoted to understanding the relevance of aberrant CpG-methylation patterns in human cancers and their role(s) in controlling gene transcription in human malignancies (Esteller 2005; Lodygin and Hermeking 2005). Promoter-associated CpG island methylation of cancer-related genes is a frequent event in human cancers, including prostate cancer (McKie et al., 2005). Indeed, 14-3-3σ promoter methylation has been associated with loss of expression in many human cancers (Ferguson et al., 2000; Iwata et al., 2000; Kaneuchi et al., 2004; Lodygin et al., 2003; Mhawech et al., 2005; Osada et al., 2002; Yatabe et al., 2002). A recent study has also reported the loss of 14-3-3σ expression by CpG methylation in most prostate adenocarcinomas and HGPIN lesions (Cheng et al., 2004). The pattern of 14-3-3σ expression exhibited an inverse correlation with that of disease progression, which prompted us to study the molecular mechanisms underlying 14-3-3σ gene silencing.
Several findings presented here provide evidence that promoter-associated CpG island methylation is a major cause of 14-3-3σ gene silencing in LNCaP and Tramp-C1 cells (Fig. 1-3). In this paper, we have utilized both MSP, bisulfite genomic sequencing and HhaI assay, which provide information about potential CpG residues in the 14-3-3σ promoter region, were used for precise methylation analysis (Fig. 2). Consistent with previous reports (Lodygin et al., 2004; Mhawech et al., 2005), we demonstrated here that demethylation of the 14-3-3σ promoter-associated CpG island directly contributes to 14-3-3σ gene expression (Fig. 3).
We further investigated the repressive mechanism that mediates silencing of 14-3-3σ expression in LNCaP and Tramp-C1 cells, which have a methylated promoter-associated CpG island. Promoter-associated CpG-methylation, along with MeCPs and HDACs, has been identified as a major epigenetic event associated with the loss of cancer-related gene expression during tumor progression (Fraga et al., 2005; Sharma et al., 2005). In general, CpG methylation is thought to be involved in the stable repression of gene expression while the direct binding of MeCPs to methylated DNA appears to be a major mechanism of transcriptional repression (Konduri et al., 2003; Sharma et al., 2005; Singal et al., 2001). These MeCPs contain a minimal region that mediates selective binding to DNA-containing 5-methylcytosine and also contains a transcriptional repressor domain that permits interaction with transcriptional repressor protein complex-associated HDACs. MeCPs can thus act as a CpG island methylation-dependent transcriptional repressor by binding transcriptional regulatory sequences carrying 5-methylcytosine and recruiting HDACs. For this reason, MeCP-mediated transcriptional repression of the gene promoter has been found to be alleviated by treatment with 5-aza and TSA, inhibitors of HDACs. Several lines of evidence have demonstrated that certain numbers of the methyl-CpG-binding proteins, such as MeCP2 and MBD2, are involved in methylation-based gene repression and also affect chromatin structure (Konduri et al., 2003; Sharma et al., 2005; Singal et al., 2001). Indeed, treatment of 14-3-3σ non-expressing breast cancer cells with trichostatin A (TSA), a histone deacetylase inhibitor, leads to reactivation of 14-3-3σ, thereby suggesting that chromatin condensation is responsible for suppressing 14-3-3σ transcription in breast cancer cells (Ferguson et al., 2000).
In this study, ChIP assays detected MBD2 selectively bound to methylated 14-3-3σ promoter CpG sequences in LNCaP and Tramp-C1 prostate cancer cells (Fig. 4). Consistent with these results, a previous report has shown that the MBD2 associates with glutathione-S transferase P1 (GSTP1), a gene promoter, in LNCaP cells (Singal et al., 2001). Furthermore, several earlier studies have shown that the spread of methylation across CpG islands is linked to MeCP-binding (Konduri et al., 2003; Sharma et al., 2005; Singal et al., 2001). Recent evidence also indicates that the ability of MeCPs to bind to methylated DNA provides the capacity to direct silencing (Sharma et al., 2005; Stirzaker et al., 2004). Moreover, silencing conferred by MBD2 and DNA methylation in LNCaP and Tramp-C1 cells was relieved by treatment with 5-aza and TSA (Fig. 5). These findings indicate that MBD2 has an important role in recruiting transcription-repressive machinery to the methylated promoter, thereby suppressing the transcriptional activation of 14-3-3σ. Confirming these findings, the siRNA-directed knockdown of MBD2 triggers stimulation of 14-3-3σ expression in LNCaP cells (Fig. 6). Taken together, these several lines of evidence suggests that MBD2 is an important factor in the methylation-mediated silencing of the 14-3-3σ. Immunoblots show that the MBD2 expression levels between prostate cancer cells are biased with barely detectable expression levels except in Tramp-C1 cells (Fig. 5). Patra et al. (2003) have demonstrated that MBD2 and MeCP2 expression occurs at the mRNA level, but not at the protein level, in DU145 and PC3 cells. Two explanations for this difference are possible: (1) differences in the standard and the specificity of antibodies used against MeCP2 and MBD2; or (2) differences in protein level. The latter is more likely because we also observed low or barely detectable protein expression for MeCP2 and MBD2 in three of the prostate cancer cell lines.
14-3-3σ functions to modulate multiple intracellular signaling pathways. Its exact mechanism of action is complex, involved in several fundamental pathways, which control both apoptosis (BAD, Bcl-Xs, Bak and Bax) and cell cycle progression (Cdc25, p53, p27 and Wee1). 14-3-3σ is likely to be integrated with survival signals, leading to a context-dependent sensitivity to apoptosis and cell cycle checkpoint control (Hermeking 2003). Failure to activate checkpoint pathways may cause the development of hyperproliferative, more invasive/malignant phenotypes (Chan et al., 1999). The proliferation index and PCNA levels support the notion that methylation of the promoter and silencing of the 14-3-3σ contribute to hyperproliferation in prostate cancer cells (Fig. 7A-C). Further evidence to support this conclusion comes from the examination of proliferation index in 14-3-3σ knockdown PC3 and DU145 cells, which shows increased proliferation (Fig. 7D & E). Consistent with these results, previous studies have reported a correlation between the loss of 14-3-3σ expression and high tumor grade in human prostate cancer tissues (Lodygin et al., 2003; Lodygin et al., 2004; Lodygin and Hermeking 2005; Mhawech 2005; Reichelt and Magin 2002; Tanaka et al., 2004).
So far, most therapeutic strategies aimed at the reactivation of epigenetically-silenced genes in human cancer cells have targeted DNMTs or HDACs. However, the clinical use of nucleoside DNMT inhibitors, 5-aza and 5-aza-C (5-azacytidine), has been complicated by myelotoxicity, global hypomethylation, and other side effects (Santini et al., 2001). In fact, hypomethylation of CpG islands after treatment with 5-aza has been shown to be responsible for overexpression of tumor-promoting genes and oncogenes, including uPA, ras and myc (Pakneshan et al., 2003; Pakneshan et al., 2004). Since MBD2 acts to inhibit transcription of 14-3-3σ and other tumor-suppressor genes inactivated by CpG methylation in cancers, new cancer treatments or preventive strategies should potentially target the MBD2 transcriptional repression pathway. A recent study lends further support to our results indicating that MBD2 is not only a repressive regulator, but also an active transcriptional regulator (Ego et al., 2005). The potential treatment value of using the reactivation of silenced genes in cancer cells using the MBD2 pathway should be investigated further.
In summary, we have delineated the molecular mechanisms by which 14-3-3σ expression is silenced in LNCaP and Tramp-C1 prostate cancer cells. Our data suggest that methyl-CpG-binding protein (MBD2) participates in the inhibition of transcription from hypermethylated 14-3-3σ promoters in prostate cancer cells. We also demonstrated that silencing of 14-3-3σ contributes to hyperproliferation in prostate cancer cells. Collectively, our results suggest that the loss of 14-3-3σ via CpG methylation may play an important role in prostate cancer progression.
ACKNOWLEDGEMENTS
We thank Shellee Abraham for preparing the manuscript and Diana Meister and Sushma Jasti for manuscript review.
Abbreviations
- ChIP
(chromoatin immunoprecipitation)
- HDAC
(histone deacetylase)
- MBD2
(methyl-CpG-binding protein 2)
- MSP
(methylation-specific PCR)
- RNAi
(RNA interference)
- siRNA
(small interfering RNA)
- 5-aza
(5-aza-2′-deoxycytidine)
- TSA
(trichostatin A)
- RT
(reverse transcription)
- TPBS
(tween-20 (0.1%) phospate buffered saline)
- PBS
(phospate buffered saline)
- PIN
(prostatic intraepithelial neoplasia)
- BSA
(bovine serum albumin)
Footnotes
This research was supported by National Cancer Institute Grant CA 75557, CA 92393, CA 95058, CA 116708 and N.I.N.D.S. NS47699 and Caterpillar, Inc., OSF Saint Francis Medical Center, Peoria, IL (to J.S.R.).
REFERENCES
- Baylin SB, Herman JG. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- Bird A. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- Cheng L, Pan CX, Zhang JT, Zhang S, Kinch MS, Li L, Baldridge LA, Wade C, Hu Z, Koch MO, Ulbright TM, Eble JN. Clin Cancer Res. 2004;10:3064–3068. doi: 10.1158/1078-0432.ccr-03-0652. [DOI] [PubMed] [Google Scholar]
- Darwanto A, Kitazawa R, Maeda S, Kitazawa S. Cancer Sci. 2003;94:442–447. doi: 10.1111/j.1349-7006.2003.tb01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet C, Loriot A, Boon T. Mol Cell Biol. 2004;24:4781–4790. doi: 10.1128/MCB.24.11.4781-4790.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Morrison DK. J Cell Sci. 2004;117:1875–1884. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- Ego T, Tanaka Y, Shimotohno K. Oncogene. 2005;24:1914–1923. doi: 10.1038/sj.onc.1208394. [DOI] [PubMed] [Google Scholar]
- Esteller M. Annu Rev Pharmacol Toxicol. 2005;45:629–656. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- Esteller M, Risques RA, Toyota M, Capella G, Moreno V, Peinado MA, Baylin SB, Herman JG. Cancer Res. 2001;61:4689–4692. [PubMed] [Google Scholar]
- Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal PA, Stampfer MR, Sukumar S. Proc Natl Acad Sci U S A. 2000;97:6049–6054. doi: 10.1073/pnas.100566997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. Proc Natl Acad Sci U S A. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Subramanian RR, Masters SC. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- Fujita N, Takebayashi S, Okumura K, Kudo S, Chiba T, Saya H, Nakao M. Mol Cell Biol. 1999;19:6415–6426. doi: 10.1128/mcb.19.9.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galm O, Suzuki H, Akiyama Y, Esteller M, Brock MV, Osieka R, Baylin SB, Herman JG. Oncogene. 2005 doi: 10.1038/sj.onc.1208599. e-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- Goodman PA, Burkhardt N, Juran B, Tibbles HE, Uckun FM. Oncogene. 2003;22:2504–2514. doi: 10.1038/sj.onc.1206313. [DOI] [PubMed] [Google Scholar]
- Hermeking H. Nat Rev Cancer. 2003;3:931–943. doi: 10.1038/nrc1230. [DOI] [PubMed] [Google Scholar]
- Iwata N, Yamamoto H, Sasaki S, Itoh F, Suzuki H, Kikuchi T, Kaneto H, Iku S, Ozeki I, Karino Y, Satoh T, Toyota J, Satoh M, Endo T, Imai K. Oncogene. 2000;19:5298–5302. doi: 10.1038/sj.onc.1203898. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jones PA. Trends Genet. 1999;15:34–37. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jones PA, Laird PW. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Kaneuchi M, Sasaki M, Tanaka Y, Shiina H, Verma M, Ebina Y, Nomura E, Yamamoto R, Sakuragi N, Dahiya R. Biochem Biophys Res Commun. 2004;316:1156–1162. doi: 10.1016/j.bbrc.2004.02.171. [DOI] [PubMed] [Google Scholar]
- Kitazawa S, Kitazawa R, Maeda S. J Biol Chem. 1999;274:28787–28793. doi: 10.1074/jbc.274.40.28787. [DOI] [PubMed] [Google Scholar]
- Konduri SD, Srivenugopal KS, Yanamandra N, Dinh DH, Olivero WC, Gujrati M, Foster DC, Kisiel W, Ali-Osman F, Kondraganti S, Lakka SS, Rao JS. Oncogene. 2003;22:4509–4516. doi: 10.1038/sj.onc.1206695. [DOI] [PubMed] [Google Scholar]
- Lin X, Nelson WG. Cancer Res. 2003;63:498–504. [PubMed] [Google Scholar]
- Lodygin D, Diebold J, Hermeking H. Oncogene. 2004;23:9034–9041. doi: 10.1038/sj.onc.1208004. [DOI] [PubMed] [Google Scholar]
- Lodygin D, Hermeking H. Cell Res. 2005;15:237–246. doi: 10.1038/sj.cr.7290292. [DOI] [PubMed] [Google Scholar]
- Lodygin D, Yazdi AS, Sander CA, Herzinger T, Hermeking H. Oncogene. 2003;22:5519–5524. doi: 10.1038/sj.onc.1206854. [DOI] [PubMed] [Google Scholar]
- McKie AB, Douglas DA, Olijslagers S, Graham J, Omar MM, Heer R, Gnanapragasam VJ, Robson CN, Leung HY. Oncogene. 2005;24:2166–2174. doi: 10.1038/sj.onc.1208371. [DOI] [PubMed] [Google Scholar]
- Mhawech P. Cell Res. 2005;15:228–236. doi: 10.1038/sj.cr.7290291. [DOI] [PubMed] [Google Scholar]
- Mhawech P, Benz A, Cerato C, Greloz V, Assaly M, Desmond JC, Koeffler HP, Lodygin D, Hermeking H, Herrmann F, Schwaller J. Mod Pathol. 2005;18:340–348. doi: 10.1038/modpathol.3800240. [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Xing H. Cell Signal. 2000;12:703–709. doi: 10.1016/s0898-6568(00)00131-5. [DOI] [PubMed] [Google Scholar]
- Nakagawachi T, Soejima H, Urano T, Zhao W, Higashimoto K, Satoh Y, Matsukura S, Kudo S, Kitajima Y, Harada H, Furukawa K, Matsuzaki H, Emi M, Nakabeppu Y, Miyazaki K, Sekiguchi M, Mukai T. Oncogene. 2003;22:8835–8844. doi: 10.1038/sj.onc.1207183. [DOI] [PubMed] [Google Scholar]
- Nguyen CT, Gonzales FA, Jones PA. Nucleic Acids Res. 2001;29:4598–4606. doi: 10.1093/nar/29.22.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada H, Tatematsu Y, Yatabe Y, Nakagawa T, Konishi H, Harano T, Tezel E, Takada M, Takahashi T. Oncogene. 2002;21:2418–2424. doi: 10.1038/sj.onc.1205303. [DOI] [PubMed] [Google Scholar]
- Pakneshan P, Szyf M, Farias-Eisner R, Rabbani SA. J Biol Chem. 2004;279:31735–31744. doi: 10.1074/jbc.M401669200. [DOI] [PubMed] [Google Scholar]
- Pakneshan P, Xing RH, Rabbani SA. FASEB J. 2003;17:1081–1088. doi: 10.1096/fj.02-0973com. [DOI] [PubMed] [Google Scholar]
- Park J, Song SH, Kim TY, Choi MC, Jong HS, Kim TY, Lee JW, Kim NK, Kim WH, Bang YJ. Oncogene. 2004;23:3474–3480. doi: 10.1038/sj.onc.1207470. [DOI] [PubMed] [Google Scholar]
- Patra SK, Patra A, Zhao H, Carroll P, Dahiya R. Biochem Biophys Res Commun. 2003;302:759–766. doi: 10.1016/s0006-291x(03)00253-5. [DOI] [PubMed] [Google Scholar]
- Reichelt J, Magin TM. J Cell Sci. 2002;115:2639–2650. doi: 10.1242/jcs.115.13.2639. [DOI] [PubMed] [Google Scholar]
- Santini V, Kantarjian HM, Issa JP. Ann Intern Med. 2001;134:573–586. doi: 10.7326/0003-4819-134-7-200104030-00011. [DOI] [PubMed] [Google Scholar]
- Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Mol Endocrinol. 2005;19:1740–1751. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- Singal R, Ginder GD. Blood. 1999;93:4059–4070. [PubMed] [Google Scholar]
- Singal R, van Wert J, Bashambu M. Cancer Res. 2001;61:4820–4826. [PubMed] [Google Scholar]
- Stirzaker C, Song JZ, Davidson B, Clark SJ. Cancer Res. 2004;64:3871–3877. doi: 10.1158/0008-5472.CAN-03-3690. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Hatada T, Kobayashi M, Mohri Y, Tonouchi H, Miki C, Nobori T, Kusunoki M. Int J Oncol. 2004;25:1591–1597. [PubMed] [Google Scholar]
- Tate PH, Bird AP. Curr Opin Genet Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- Tzivion G, Avruch J. J Biol Chem. 2002;277:3061–3064. doi: 10.1074/jbc.R100059200. [DOI] [PubMed] [Google Scholar]
- Tzivion G, Shen YH, Zhu J. Oncogene. 2001;20:6331–6338. doi: 10.1038/sj.onc.1204777. [DOI] [PubMed] [Google Scholar]
- Urano T, Takahashi S, Suzuki T, Fujimura T, Fujita M, Kumagai J, Horie-Inoue K, Sasano H, Kitamura T, Ouchi Y, Inoue S. Biochem Biophys Res Commun. 2004;319:795–800. doi: 10.1016/j.bbrc.2004.05.056. [DOI] [PubMed] [Google Scholar]
- Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. J Biol Chem. 2003;278:36323–36327. doi: 10.1074/jbc.M304689200. [DOI] [PubMed] [Google Scholar]
- Yatabe Y, Osada H, Tatematsu Y, Mitsudomi T, Takahashi T. Oncogene. 2002;21:8310–8319. doi: 10.1038/sj.onc.1206014. [DOI] [PubMed] [Google Scholar]
- Yu F, Thiesen J, Stratling WH. Nucleic Acids Res. 2000;28:2201–2206. doi: 10.1093/nar/28.10.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Karas M, Zhao H, Yakar S, LeRoith D. J Biol Chem. 2004;279:34353–34360. doi: 10.1074/jbc.M401300200. [DOI] [PubMed] [Google Scholar]