

Abstract
Magnesiowüstite [(Mg,Fe)O] is the second most abundant mineral of Earth's lower mantle. Understanding its stability under lower mantle conditions is crucial for interpreting the physical and chemical properties of the whole Earth. Previous studies in an externally heated diamond anvil cell suggested that magnesiowüstites decompose into two components, Fe-rich and Mg-rich magnesiowüstites at 86 GPa and 1,000 K. Here we report an in situ study of two magnesiowüstites [(Mg0.39,Fe0.61)O and (Mg0.25,Fe0.75)O] at pressures and temperatures that overlap with mantle conditions, using a laser-heated diamond anvil cell combined with synchrotron x-ray diffraction. Our results show that addition of Mg in wüstite (FeO) can stabilize the rock-salt structure to much higher pressures and temperatures. In contrast to the previous studies, our results indicate that Mg-rich magnesiowüstite is stable in the rock-salt structure in the lower mantle. The physical and chemical properties of magnesiowüstite should change gradually and continuously in the lower mantle, suggesting that it does not make a significant contribution to seismic-wave heterogeneity of the lower mantle. Stable Mg-rich magnesiowüstite in lowermost mantle can destabilize FeO in the core–mantle boundary region and remove FeO from the outer core.
At lower mantle conditions, olivine and orthopyroxene transform to magnesiowüstite [(Mg,Fe)O] and silicate perovskite [(Mg,Fe)SiO3] (1, 2), which together are likely the most important mineral assemblage of Earth's interior. The stability of the magnesiowüstite and silicate perovskite plays a crucial role in understanding the geophysical and geochemical properties of Earth. At ambient conditions, the end members of the MgO–FeO (periclase–wüstite) system form a solid solution and have the same rock-salt (B1) structure. Periclase remains in the B1 structure to at least 227 GPa (3, 4). Wüstite transforms to a rhombohedral structure at pressures above 18 GPa at 300 K (5) and then to the NiAs or anti-NiAs structure (6–8). The topological difference between the pressure–temperature (P–T) phase diagrams of periclase and wüstite indicates that regions of two-phase equilibria should exist. A thermodynamically calculated P–T-composition phase diagram for the system suggests that an increase in pressure in the system would result in a gradual exsolution of an almost pure FeO and an Fe-depleted (Mg,Fe)O (9). Recent studies of three magnesiowüstites [(Mg0.5,Fe0.5)O, (Mg0.6,Fe0.4)O, and (Mg0.8,Fe0.2)O] in an externally heated diamond anvil cell (DAC) up to 86 GPa and 1,000 K suggested that magnesiowüstite decomposes into Mg-rich and Fe-rich magnesiowüstites (10, 11). The decomposition of magnesiowüstite was proposed to occur at the P–T conditions of the lower mantle (10, 11) and to contribute significantly to the seismic-wave heterogeneity of the lower mantle (12, 13). On the other hand, no evidence for a phase transformation in (Mg0.6,Fe0.4)O was found in shock-wave experiments to 201 GPa (14). Here we report the in situ study of structure and stability of magnesiowüstites at P–T conditions of the lower mantle.
Experimental Methods
Polycrystalline magnesiowüstites [(Mg0.39,Fe0.61)O and (Mg0.25,Fe0.75)O] in the cubic B1 structure were obtained from E. K. Graham (Pennsylvania State University, University Park) (15). Electron microprobe analyses showed that the starting materials contained 61.1 (±0.5) and 75.4 (±0.2) mol % FeO (each averaged from at least seven analyses), respectively. Beveled diamonds with an inner culet of 150 μm and an outer culet of 300 μm or flat diamonds with a culet of 250 μm were used. A rhenium or stainless steel gasket was preindented to a thickness of 30 μm and then a hole of 220-μm diameter was drilled in it. An amorphous boron and epoxy mixture (4:1 by weight) was filled and compressed in the hole. Subsequently, another hole of 100 μm was drilled and used as the sample chamber. A sandwich configuration, consisting of dried NaCl as the thermal insulator and pressure medium on both sides of the sample, was used (16–18). The amorphous boron provided higher strength to create a deeper sample chamber, giving stronger x-ray diffraction from a thicker sample and better laser-heating spots attributable to thicker thermal insulating layers.‖ Moreover, use of amorphous boron as an inner gasket also avoided unwanted x-ray diffraction peaks from Re or the stainless steel gasket.
We have used a double-sided Nd:YLF (neodymium: yttrium lithium fluoride) laser heating system, operating in multimode (TEM00+TEM01), to heat the sample from both sides of a DAC at the 13-IDD GeoSoilEnviro-Consortium for Advanced Radiation Sources (GSECARS) sector of the Advanced Photon Source, Argonne National Laboratory (18). The laser beam diameter was ≈25 μm. Graybody temperatures were determined by fitting the thermal radiation spectrum between 670 nm and 830 nm to the Planck radiation function. The temperature uncertainty (1σ) averaged from multiple temperature measurements and temperatures from both sides of the sample across the laser-heated spots was ≈50–150 K. The sample was laser-heated for more than 10 min at each P–T point and was laser-heated for ≈30 min at the highest temperature of each heating cycle to overcome potential kinetic effects on possible phase transformation. The total duration of the laser heating in each heating cycle was ≈2–4 h. A focused monochromatic beam (wavelength = 0.3311 or 0.4246 Å) with a beam size of 7 μm (vertical) × 10 μm (horizontal) was used as the x-ray source for angle-dispersive x-ray diffraction experiments. The diffracted x-rays were collected by an image plate (MAR345) or by a charge-coupled device (Bruker-2k). Pressures were calculated from the room temperature equation of state of NaCl (16, 17) before laser heating. No thermal pressure corrections were made to the pressures at high temperatures in laser-heated DAC experiments.
Results and Discussion
(Mg0.39,Fe0.61)O and (Mg0.25,Fe0.75)O were examined in a laser-heated DAC at pressures up to 102 GPa and 86 GPa and temperatures up to 2,550 (±140) K and 2,580 (±130) K, respectively, and x-ray diffraction patterns were collected before, during, and after laser heating at each pressure (Figs. 1, 2, and 3, respectively) (19). The diffraction patterns were processed with the FIT2D (20), PEAKFIT 4.0, and General Structure Analysis System (gsas) (21, 22) programs. Le Bail and Rietveld crystal structure refinements of the diffraction patterns were performed by using the gsas program. No phase transformation was observed in (Mg0.39,Fe0.61)O, and it was stable in the B1 structure up to 102 GPa and 2,550 (±140) K (Figs. 1 and 3). In the (Mg0.25,Fe0.75)O experiments, a displacive phase transformation from B1 to a rhombohedral structure (R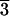 m) was observed at about 60 GPa and 300 K. Because the displacive transition is very sensitive to the hydrostaticity of the sample (5), we used the diffraction patterns after laser heating for phase identification at 300 K. A second phase transformation occurred above 79 GPa as revealed from the observed extra peaks near (003), (101), (104), and (110) peaks of the rhombohedral phase (Fig. 2). The crystal structure of the new high-pressure phase should be closely related to the distorted B1 structure, because its d-spacings are close to the d-spacing of the B1 phase (Fig. 2) (23, 24). The displacive phase transition has also been observed in FeO (5), MnO (24), and CoO (23) under high pressures. MnO and CoO displayed multiple phase transitions under high pressures (23, 24). Nevertheless, the distorted B1 phases transform to the B1 structure upon laser heating, and the B1 structure transforms back to the distorted phases after temperature quench (Fig. 2). The quenched samples from the laser-heated DAC were recovered, polished, and analyzed with a scanning electron microprobe (JEOL 8800L), and the results show that the quenched samples remain chemically homogeneous. Additional details regarding the refinements of the crystal structures and the chemical analyses of the quenched samples are provided in Tables 1 and 2 and Figs. 5–8, which are published as supporting information on the PNAS web site, www.pnas.org.
m) was observed at about 60 GPa and 300 K. Because the displacive transition is very sensitive to the hydrostaticity of the sample (5), we used the diffraction patterns after laser heating for phase identification at 300 K. A second phase transformation occurred above 79 GPa as revealed from the observed extra peaks near (003), (101), (104), and (110) peaks of the rhombohedral phase (Fig. 2). The crystal structure of the new high-pressure phase should be closely related to the distorted B1 structure, because its d-spacings are close to the d-spacing of the B1 phase (Fig. 2) (23, 24). The displacive phase transition has also been observed in FeO (5), MnO (24), and CoO (23) under high pressures. MnO and CoO displayed multiple phase transitions under high pressures (23, 24). Nevertheless, the distorted B1 phases transform to the B1 structure upon laser heating, and the B1 structure transforms back to the distorted phases after temperature quench (Fig. 2). The quenched samples from the laser-heated DAC were recovered, polished, and analyzed with a scanning electron microprobe (JEOL 8800L), and the results show that the quenched samples remain chemically homogeneous. Additional details regarding the refinements of the crystal structures and the chemical analyses of the quenched samples are provided in Tables 1 and 2 and Figs. 5–8, which are published as supporting information on the PNAS web site, www.pnas.org.
Figure 1.

Representative angle-dispersive x-ray diffraction patterns of (Mg0.39,Fe0.61)O at ≈98 GPa in a laser-heated DAC. A monochromatic beam (wavelength = 0.3311 Å) was used as the x-ray source and the diffracted x-rays were collected by an image plate (MAR345). The diffraction patterns were integrated with the FIT2D program (20), and the backgrounds were subtracted with PEAKFIT 4.0. (Mg0.39,Fe0.61)O is stable in the B1 structure up to 2,390 (±130) K and remains in the B1 structure after laser heating at 97 GPa and 300 K. Peak identifications are mw, (Mg0.39,Fe0.61)O in the B1 structure; and B2, NaCl in the cesium chloride structure (B2) (17).
Figure 2.

Representative angle-dispersive x-ray diffraction patterns of (Mg0.25,Fe0.75)O at ≈79 GPa in a laser-heated DAC. A monochromatic beam (wavelength = 0.3311 Å) was used as the x-ray source. The backgrounds were subtracted with PEAKFIT 4.0. Le Bail and Rietveld crystal structure refinements, performed with the GSAS program (21, 22), confirmed that (Mg0.25,Fe0.75)O is in the rhombohedral structure (R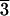 m) at 79 GPa and 300 K. The rhombohedral phase transforms to the B1 structure upon laser heating, and the B1 phase changes back to the rhombohedral phase after temperature quench. A higher pressure phase of (Mg0.25,Fe0.75)O may exist above 79 GPa and 300 K as shown from the observed extra peaks near (003), (101), (104), and (110) peaks of the rhombohedral phase. Peak identifications are mw, (Mg0.25,Fe0.75)O in the B1 structure; R, (Mg0.25,Fe0.75)O in the rhombohedral structure (R
m) at 79 GPa and 300 K. The rhombohedral phase transforms to the B1 structure upon laser heating, and the B1 phase changes back to the rhombohedral phase after temperature quench. A higher pressure phase of (Mg0.25,Fe0.75)O may exist above 79 GPa and 300 K as shown from the observed extra peaks near (003), (101), (104), and (110) peaks of the rhombohedral phase. Peak identifications are mw, (Mg0.25,Fe0.75)O in the B1 structure; R, (Mg0.25,Fe0.75)O in the rhombohedral structure (R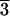 m); and B2, NaCl in the B2 structure; *, extra diffraction peaks from a higher pressure phase of (Mg0.25,Fe0.75)O.
m); and B2, NaCl in the B2 structure; *, extra diffraction peaks from a higher pressure phase of (Mg0.25,Fe0.75)O.
Figure 3.

Phases observed in (Mg0.39,Fe0.61)O and (Mg0.25,Fe0.75)O under high P–T. The temperature uncertainty (1σ) averaged from multiple temperature measurements and temperatures from both sides of the sample across the laser-heated spots was ≈50–150 K. Solid lines represent the phase diagram of FeO (6). FeO transforms to a rhombohedral structure at pressures above 18 GPa at 300 K (5) and then to the NiAs or anti-NiAs structure (6–8). Gray line shows the approximate mantle geotherm (19). □ and ○, (Mg0.39,Fe0.61)O and (Mg0.25,Fe0.75)O in the B1 structure, respectively; ×, (Mg0.25,Fe0.75)O in the rhombohedral structure.
Comparison to estimated geotherms of the deep Earth (19) shows that our high P–T experiments overlap with the P–T conditions of the lower mantle. Thus, (Mg0.39,Fe0.61)O and (Mg0.25,Fe0.75)O are stable in the B1 structure in the lower mantle (Fig. 3). The effect of adding MgO in FeO on the B1–rhombohedral phase transformation is manifested in the pressure-composition phase diagram (Fig. 4) (3–6, 25–27, ∗∗). As shown, addition of MgO strongly stabilizes the B1 structure to much higher pressures. The B1 structure has a wide P–T stability field in the Mg-rich magnesiowüstite (ferropericlase). The addition of MgO should also decrease the magnetoelastic coupling in wüstite, because the displacive B1–rhombohedral phase transition is related to the magnetic transition from the cubic paramagnetic phase to the rhombohedral antiferromagnetic phase (28–30).
Figure 4.

Pressure–composition phase diagram of magnesiowüstite. Open symbols, +, and × represent magnesiowüstite in the B1 structure; solid symbols and ticks indicate the phase-transition pressure from B1 to rhombohedral structure. The dashed curve represents a simple polynomial fit to all of the phase-transition pressures observed in magnesiowüstites. Two shock-wave studies on MgO and (Mg0.6,Fe0.4)O are under high P–T conditions (3, 14), and other studies are under high pressure and 300 K. ■, Shu et al. (5); solid ticks, Shu et al.**; ▴, Mao et al. (27); ♦, Richet et al. (25); ● and □, this study; +, Richet et al. (25); ⋄, Vissiliou and Ahrens (14); ×, Fei et al. (26); ○, Duffy et al. (4); and ▵, Vissiliou and Ahrens (3).
High P–T experiments on the Mg–Fe partitioning between magnesiowüstite and silicate perovskite indicate that the FeO content in magnesiowüstite decreases with increasing P–T and with the addition of Al2O3 (31–33), suggesting that Mg-rich oxide (ferropericlase) exists in the deep lower mantle. Fig. 4 shows that indeed this is the case. Because the rhombohedral phase of wüstite is stable only up to ≈1,000 K (6) and the addition of MgO in FeO should further depress the stability field of the rhombohedral phase (Fig. 4), the B1–rhombohedral phase transformation in the ferropericlase is unlikely to occur under lower mantle conditions. Furthermore, the B1–NiAs phase transformation (6) in the ferropericlase is also not expected to occur in the lower mantle, because the addition of MgO in FeO also depresses the stability of the NiAs structure. In accord with these findings, high P–T experiments on the assemblage of magnesiowüstite and perovskite also showed no evidence of a phase transformation in the Mg-rich magnesiowüstite up to 120 GPa and 2,300 K (32, 33).
In contrast to recent studies on three magnesiowüstites [(Mg0.5,Fe0.5)O, (Mg0.6,Fe0.4)O, and (Mg0.8,Fe0.2)O] in an externally heated DAC up to 86 GPa and 1,000 K by Dubrovinsky et al. (10, 11), our in situ high P–T experiments, at conditions of the lower mantle show that Mg-rich magnesiowüstite is stable in the B1 structure in the lower mantle. Magnesiowüstite in the lower mantle is likely to exchange elements with silicate perovskite as a function of P and T (31–33), which may result in a gradual, continuous change in physical and chemical properties of the lower mantle. Our results suggest that recently documented seismic-wave heterogeneity of the lower mantle (12, 13) must be explained by phenomena other than high P–T phase transformation or decomposition of magnesiowüstite as proposed by previous studies (10, 11). Oxygen is considered a possible light element in the outer core and core-mantle boundary (34). FeO has been observed in the reaction product of the silicate perovskite [(Mg,Fe)SiO3] and the liquid iron (34). It is conceivable that the stable Mg-rich magnesiowüstite chemically reacts with FeO in the core–mantle boundary and the outer core, and hence destabilizes FeO in the core–mantle boundary region and removes FeO from the outer core. The reaction may cause silicate material to accumulate at the core–mantle boundary (35).
Supplementary Material
Acknowledgments
We thank GSECARS at the Advanced Photon Source, Argonne National Laboratory, for providing the synchrotron beamtime. We also thank I. Steele, A. Campbell, N. Sata, V. Prakapenka, Y. Ding, C. Prewitt, P. Dera, Y. Fei, and C. Hadidiacos for help and discussions. This research is supported by the Department of Energy and the National Science Foundation. GSECARS is supported by the Department of Energy, the National Science Foundation, and the W. M. Keck Foundation.
Abbreviations
- P–T
pressure–temperature
- DAC
diamond anvil cell
- GSECARS
GeoSoilEnviro-Consortium for Advanced Radiation Sources
Footnotes
References
- 1.Liu L-G. Nature. 1975;258:510–512. [Google Scholar]
- 2.Shim S-H, Duffy T S, Shen G. Nature. 2001;411:571–574. doi: 10.1038/35079053. [DOI] [PubMed] [Google Scholar]
- 3.Vissiliou M S, Ahrens T J. Geophys Res Lett. 1981;8:729–732. [Google Scholar]
- 4.Duffy T S, Hemley R J, Mao H K. Phys Rev Lett. 1995;74:1371–1374. doi: 10.1103/PhysRevLett.74.1371. [DOI] [PubMed] [Google Scholar]
- 5.Shu J, Mao H K, Hu J, Fei Y, Hemley R J. N Jb Mineral Abh. 1998;172:309–323. [Google Scholar]
- 6.Fei Y, Mao H K. Science. 1994;266:1678–1680. doi: 10.1126/science.266.5191.1678. [DOI] [PubMed] [Google Scholar]
- 7.Mao H K, Shu J, Fei Y, Hu J, Hemley R J. Phys Earth Planet Inter. 1996;96:135–145. [Google Scholar]
- 8.Mazin I I, Fei Y, Downs R, Cohen R. Am Mineral. 1998;83:451–457. [Google Scholar]
- 9.McCammon C A, Ringwood A E, Jackson I. Geophys J R Astron Soc. 1983;72:577–595. [Google Scholar]
- 10.Dubrovinsky L S, Dubrovinskaia N A, Saxena S K, Annersten H, Hålenius E, Harryson H, Tutti F, Rekhi S, Le Bihan T. Science. 2000;289:430–432. doi: 10.1126/science.289.5478.430. [DOI] [PubMed] [Google Scholar]
- 11.Dubrovinsky L S, Dubrovinskaia N, Annersten H, Hålenius E, Harryson H. Eur J Mineral. 2001;13:857–861. [Google Scholar]
- 12.van der Hilst R D, Widiyantoro S, Engdahl E R. Nature. 1997;386:578–584. [Google Scholar]
- 13.Garnero E. Annu Rev Earth Planet Sci. 2000;28:509–537. [Google Scholar]
- 14.Vissiliou M S, Ahrens T J. Geophys Res Lett. 1982;9:127–130. [Google Scholar]
- 15.Bonczar L J, Graham E K. J Geophys Res. 1982;87:1061–1078. [Google Scholar]
- 16.Birch F. J Geophys Res. 1978;83:1257–1268. [Google Scholar]
- 17.Heinz D L, Jeanloz R. Phys Rev B. 1984;30:6045–6050. [Google Scholar]
- 18.Shen G, Rivers M L, Wang Y, Sutton S R. Rev Sci Instrum. 2001;72:1273–1282. [Google Scholar]
- 19.Brown J M, Shankland T J. Geophys J R Astron Soc. 1981;66:579–596. [Google Scholar]
- 20.Hammersley A P. FIT2D V10.3 Reference Manual. Grenoble, France: European Synchrotron Radiation Facility; 1998. , Version 4.0. [Google Scholar]
- 21.Larson A C, Von Dreele R B. GSAS Manual. Los Alamos, NM: Los Alamos Natl. Lab.; 1988. , Report LAUR 86-748. [Google Scholar]
- 22.Toby B H. J Appl Crystallogr. 2001;34:210–213. [Google Scholar]
- 23.Noguchi Y, Atou T, Kondo T, Yagi T, Syono Y. Jpn J Appl Phys. 1999;38:L7–L9. [Google Scholar]
- 24.Kondo T, Yagi T, Syono Y, Noguchi Y, Atou T, Kilegawa T, Shimomura O. J Appl Phys. 2000;87:4153–4159. [Google Scholar]
- 25.Richet P, Mao H K, Bell P M. J Geophys Res. 1989;94:3037–3045. [Google Scholar]
- 26.Fei Y, Mao H K, Shu J, Hu J. Phys Chem Miner. 1992;18:416–422. [Google Scholar]
- 27.Mao W, Shu J, Hu J, Hemley R J, Mao H K. J Phys Condens Matter. 2002;14:1–6. [Google Scholar]
- 28.Cohen R E, Mazin I I, Isaak D G. Science. 1997;275:654–657. doi: 10.1126/science.275.5300.654. [DOI] [PubMed] [Google Scholar]
- 29.Badro J, Struzhkin V V, Shu J, Hemley R J, Mao H K, Kao C C, Rueff J P, Shen G. Phys Rev Lett. 1999;83:4101–4104. [Google Scholar]
- 30.Struzhkin V V, Mao H K, Hu J Z, Schwoerer-Böhning M, Shu J F, Hemley R J, Sturhahn W, Hu M Y, Alp E E, Eng P, Shen G. Phys Rev Lett. 2001;87:255501. doi: 10.1103/PhysRevLett.87.255501. , 1–4. [DOI] [PubMed] [Google Scholar]
- 31.Mao H K, Shen G, Hemley R J. Science. 1997;278:2098–2100. doi: 10.1126/science.278.5346.2098. [DOI] [PubMed] [Google Scholar]
- 32.Andrault D. J Geophys Res. 2001;106:2079–2087. [Google Scholar]
- 33.Kesson S E, Fitz Gerald J D, O'Neill H, St C, Shelley J M G. Phys Earth Planet Int. 2002;121:85–102. [Google Scholar]
- 34.Knittle E, Jeanloz R. Science. 1991;251:1438–1443. doi: 10.1126/science.251.5000.1438. [DOI] [PubMed] [Google Scholar]
- 35.Buffett B A, Garnero E J, Jeanloz R. Science. 2000;290:1338–1342. doi: 10.1126/science.290.5495.1338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}