

Abstract
Calpains (calcium-dependent cytoplasmic cysteine proteinases) are implicated in processes such as cytoskeleton remodeling and signal transduction. The 2.3-Å crystal structure of full-length heterodimeric [80-kDa (dI-dIV) + 30-kDa (dV+dVI)] human m-calpain crystallized in the absence of calcium reveals an oval disc-like shape, with the papain-like catalytic domain dII and the two calmodulin-like domains dIV+dVI occupying opposite poles, and the tumor necrosis factor α-like β-sandwich domain dIII and the N-terminal segments dI+dV located between. Compared with papain, the two subdomains dIIa+dIIb of the catalytic unit are rotated against one another by 50°, disrupting the active site and the substrate binding site, explaining the inactivity of calpains in the absence of calcium. Calcium binding to an extremely negatively charged loop of domain dIII (an electrostatic switch) could release the adjacent barrel-like subdomain dIIb to move toward the helical subdomain dIIa, allowing formation of a functional catalytic center. This switch loop could also mediate membrane binding, thereby explaining calpains' strongly reduced calcium requirements in vivo. The activity status at the catalytic center might be further modulated by calcium binding to the calmodulin domains via the N-terminal linkers.
The calpains (EC 3.4.22.17; Clan CA, family C02) are a family of calcium-dependent cytosolic cysteine proteinases. They seem to catalyze limited proteolysis of proteins involved in cytoskeletal remodeling and signal transduction but are also implicated in other physiological and pathophysiological processes, such as cell cycle regulation, apoptosis, muscular dystrophies, cataractogenesis, and Alzheimer's or Parkinson's diseases (1–5). In mammals, the calpain family comprises several “tissue-specific” isoforms (n-calpains) besides two “ubiquitous” isoenzymes (μ- and m-calpains). In lower organisms such as insects, nematodes, fungi, and yeast, a number of “atypical” calpain homologues have been found.
The ubiquitous μ- and m-calpains (calpains I and II), by far the best characterized calpains, are heterodimers comprising distinct but quite homologous 80-kDa “large” L-subunits and a common 30-kDa “small” S-subunit. On the basis of amino acid homologies, the L- and S-subunits have been described as consisting of four domains, dI to dIV, and of two domains, dV and dVI, respectively, with domain dII somewhat resembling papain and the calmodulin-like domains dIV and dVI containing EF-hands (6, 7). On exposure to calcium at concentrations of 5–50 μM (μ-calpain) and 200–1,000 μM (m-calpain), both calpains are activated and partially autolyzed. In vivo, both calpains seem to be active at physiological calcium concentrations of 100–300 nM, however, suggesting that other factors such as phospholipids might play a role in activation in addition.
The crystal structures of rat and porcine domain dVI in the absence and presence of calcium have been determined (8, 9). For a full understanding of the activation mechanism and the functioning of calpains, the structures of full-length calpain must to be known. We (10) and others (11) have communicated crystals of full-length human and partially truncated rat m-calpain, respectively. In the following, we describe the fundamental properties of full-length human m-calpain and discuss the possible mechanisms of calcium activation.
Materials and Methods
Full-length human m-calpain containing an N-terminal GlyArgArgAspArgSer L-chain elongation was overexpressed in a baculovirus expression system and was purified (12). Two different crystal forms, both of space group P21, were grown by vapor diffusion at 15% PEG 10,000, spurious amounts of isopropanol and guanidinium chloride, and 100 mM Hepes/NaOH (pH 7.5) at 20°C as described in (10). These crystals show Bragg spacings to 2.1 Å (form II) and 2.8 Å resolution (form I). Of the better form II, two different slightly unisomorphous (with 1.5 Å differences in b axis) heavy metal derivatives (Gold and PheHg) were found, the former of which (with cell constants a = 51.88 Å, b = 169.84 Å, c = 64.44 Å, β = 95.12°) was used for structure solution and refinement. The derivatives Gold and PheHg were prepared by overnight soaking of native crystals with 5 mM gold triethylphosphine and with a spatula of phenyl mercury, respectively, before transferring them via a glycerol-containing cryoprotectant to the cold nitrogen stream. On both derivative crystals, multiple anomalous diffraction data to 2.3 Å and 2.4 Å were measured at the BW6 beam line at Deutsches Elektronen Synchrotron (Hamburg), using a MAR Research (Hamburg) charge-coupled device detector. Complete multiple anomalous dispersion data sets were collected at the gold edge (λ = 1.0092 Å), gold peak (λ = 1.004 Å), mercury edge (λ = 1.0399 Å), mercury peak (λ = 1.0392 Å), and at the remote wavelength (λ = 1.100 Å), respectively. Reflections (400,680 and 421,730 reflections) (10) were evaluated with denzo (13) and were scaled and merged to 47,236 and 48,363 independent reflections (Rmerge = 4.5% and 4.7%, containing 95% and 93% of all reflections expected). Three mercury and five gold positions were localized in anomalous difference Patterson maps and were refined with mlphare/ccp4 (14). In addition, a complete data set to 3.0 Å was collected from a native crystal of form I (10). The 223,726 reflections were merged to 25,010 unique reflections (Rmerge = 5.6%, containing 97% of all expected reflections).
In contrast to conventional phase combination, the combination of the PheHg phases with the amplitudes from gold [sharp (15)] resulted in a 2.4 Å well contoured electron density map for gold, which allowed tracing of almost 90% of the polypeptide chain (neglecting the first 84 residues of the S-chain) on Silicon Graphics (Mountain View, CA) workstations using turbo frodo (16). This partial model was crystallographically refined, and an improved phase-combined Fourier map allowed completion of the protein model. Calculation of the electron density maps and crystallographic refinement were performed with refmac/ccp4 (14) and x-plor (17). Finally, water molecules were inserted, and individual temperature factors were refined. In the final model of an R factor of 20.6% (Rfree 26.6%) for 38,544 reflections, the polypeptide chains are fully defined from Ala2L to Leu700L and from Thr85S to Ser268S. The final model consists of 7,097 protein nonhydrogen atoms, 5 gold ions, and 352 water molecules. The final rms deviations from target bond and angle values are 0.007 Å and 1.179°. Eighty-five and fifteen percent of all main chain angles fall into the allowed and additionally allowed regions of the Ramachandran plot.
Results and Discussion
The structure analysis shows that the m-calpain molecule forms a flat oval disc. The upper and the lower poles (in the reference orientation used in Fig. 1) are occupied by the two catalytic subdomains dIIa and dIIb and the calmodulin-like domain pair dIV-dVI, respectively. Domain dIII and the N-terminal domains dI and dV connect the two calmodulin-like domains with both catalytic subdomains. The overall similarity of m-calpain in our two crystal forms suggests that the structure of calcium-free m-calpain shown in Fig. 1 is independent of crystal packing.
Figure 1.

Ribbon structure of human m-calpain in the absence of calcium, shown in reference orientation. The 80-kDa L-chain starts in the molecular center (green, dI), folds into the surface of the dIIa subdomain (gold, I→II linker), forms the papain-like left-side part of the catalytic domain dII (gold, dIIa) and the right-side barrel-like subdomain dIIb (red), descends through the open II→III loop (red), builds domain dIII (blue), runs down (magenta, III→IV), and forms the right-side calmodulin-like domain dIV (yellow). The 30-kDa S-chain becomes visible from Thr95S onwards (magenta, dV) before forming the left-side calmodulin domain dVI (orange). The catalytic residues Cys105L, His262L, and Asn286L together with Trp106L, Pro287L, and Trp288L (top) are shown with all non-hydrogen atoms. The figure was made with setor (34).
The 80-kDa L-chain starts with an “anchoring helix” in a semicircular cavity of dVI and runs straight to dIIa, clamping both domains together. At Gly19L [mature human m-calpain numbering (Fig. 2), with suffices L and S indicating residues of the large and small subunit], the L-chain contacts dIIa, where it folds in one α-helix and in multiple turns to form the outer polar surface. From Thr93L onwards, the L-chain folds into the catalytic domain, which is topologically related to papain. However, the domain is broken into two separate subdomains, which differ significantly from the equivalent halves in papain (18), both in length and conformation (Fig. 3). The helical subdomain dIIa shares with papain the N-terminal loop, leading to the catalytic Cys105L, and helices α2II, α4II, and α6II, but includes additional helices and a number of novel strands, including a regular β-hairpin (Figs. 2 and 3). The chain segments and residues that play important roles in substrate binding and catalysis (Gln99L, Cys105L, Gly197L-Gly198L) are arranged as in papain.
Figure 2.
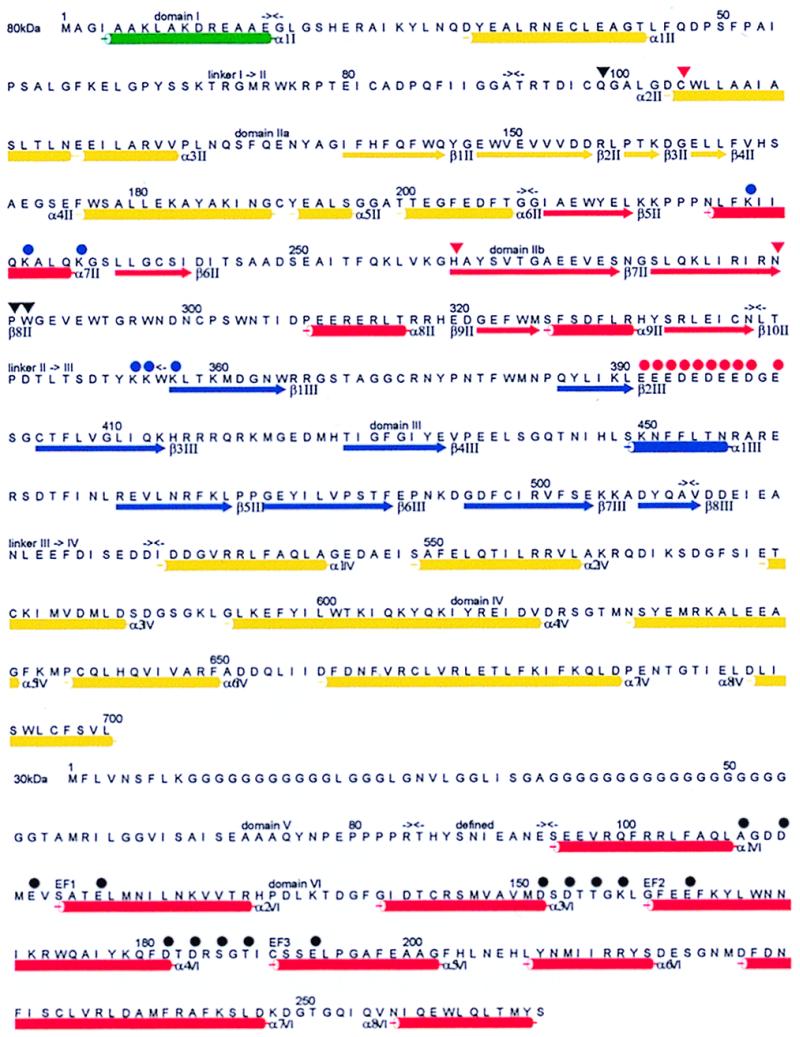
Sequence of the 80-kDa L- and the 30-kDa S-chain of human m-calpain [accession nos. P17655 and P04632 (6, 7)]. β-strands and α-helices are indicated by arrows and cylinders colored as in Fig. 1, and the topologically defined domain boundaries are given by meeting black arrows. Active-site residues and other notable residues (dIIa and dIIb) are marked by red and black triangles, acidic residues of the switch loop (dIII) and opposing alkaline residues (dIIb and II->III) by red and blue circles, and the residues of domain dVI known to be involved in normal calcium binding (8) by black circles, respectively. The figure was made with alscript (35).
Figure 3.
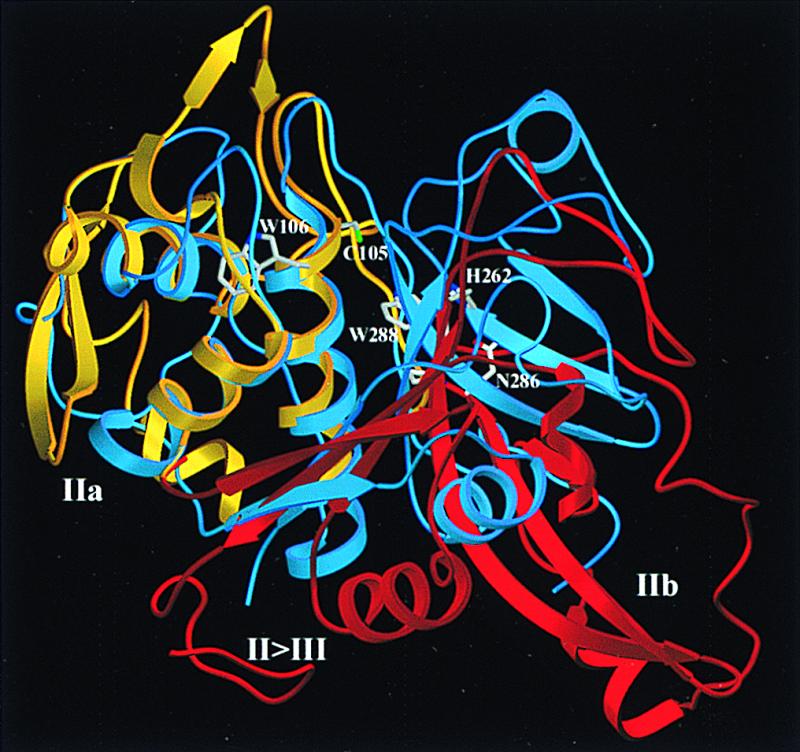
Superposition of the m-calpain catalytic domain and papain. The papain-like part of the catalytic domain (gold, dIIa) and the barrel-like subdomain dIIb (red) are superimposed with papain (18) (blue) after optimal fit of the left-side papain half to the helical subdomain dIIa. The active site residues Cys105L, His262L, and Asn286L, and Pro287L, Trp288L, and Trp106L are shown in full structure. This “standard view” of papain-like cysteine proteinases (18) is obtained from Fig. 1 by a 90° rotation around a horizontal axis. The figure was made with setor (34).
At the Gly209L-Gly210L “hinge,” the L-chain passes over to the barrel-like subdomain dIIb, forming the typical six-stranded β-pleated sheet (strands β5II to β10II) rolled into an open barrel carrying helices α9II, α7II, and α8II (Figs. 2 and 3). Although this barrel has a similar shape as in papain, the β7tβ8 and in particular the β9tβ10 loop are quite extended. Noteworthy, the central Asn286L-Pro-Trp288L segment following β8II (with Asn286L representing the third catalytic residue) exhibits a different conformation than the characteristic Asn-Ser-Trp loop in papain (18). Similar to papain, the sixth barrel strand β10II traverses over to IIa, before turning through an open loop and entering dIII.
Domain dIII, essentially made of two opposing β-sheets, each comprising four antiparallel strands, exhibits the tertiary fold of a compact β-sandwich (Fig. 4), with a topology that [only distantly related to C2 domains of phospolipases C and protein kinases C (19)] resembles the tumor necrosis factor α monomer (20) and several virus coat proteins. Noteworthy are the basic His415L-His427L loop situated in the calpain center and the solvent-exposed negatively charged β2tβ3 loop, which carries 10 acidic residues within the 11-residue Glu392L-Glu402L segment (Figs. 2 and 4). This latter loop, including all side chains, is extended but fully defined in our structure. It is spatially adjacent to helix α7II and the open loop of dIIb, and certainly interacts electrostatically with the many positive charges of both dIIb segments.
Figure 4.
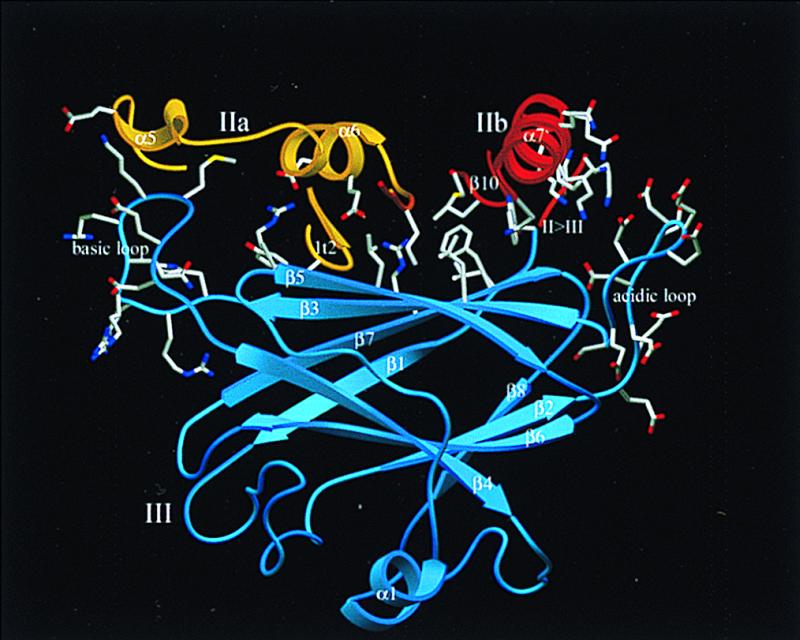
Ribbon plot of domain dIII (blue) and contacting segments from subdomains dIIa (gold) and dIIb (red). The side chains of the right-side acidic loop and of the left-side basic loop, and some polar and hydrophobic residues forming the polar (center, left) and hydrophobic (center, right) interface between both catalytic subdomains and domain dIII are given. Reference orientation is as in Fig. 1. The figure was made with setor (34).
After leaving dIII, the chain runs alongside the calmodulin-like dIV in an extended conformation, presenting several acidic residues toward the solvent. At Ile530L, the L-chain enters dIV, which has a similar fold and structure as the isolated dVI domains from rat and porcine calpains (8, 9) (Fig. 5). Both domains have been shown to resemble other calcium-binding EF-hand proteins (21). dIV comprises eight α-helices connected through characteristic linkers, together forming five well known EF-hand supersecondary structural elements (8, 9).
Figure 5.

Ribbon plot of the calmodulin-like domains dIV (yellow) and dVI (orange) superimposed with the calcium-containing rat dVI-dVI homodimer (8, 9) (white and green). Shown are also the 2 × 3 calcium ions (pink spheres I to III) and the acidic residues found in the homodimer at medium calcium levels and involved in calcium binding, respectively (8). Reference orientation is as in Fig. 1. The figure was made with setor (34).
The N-terminal part of the calpain S-chain, rich in glycine residues, is not represented by clear electron density, but might be arranged on the front side (Fig. 1) of m-calpain. The visible part of dV, from Thr85S onwards, runs to the molecular periphery, where it enters dVI (Figs. 1 and 5). This domain, likewise equipped with five EF-hand motifs (8, 9), together with dIV forms a quasi-symmetrical heterodimer, with helices α6, α7, and α8 and the α7tα8 linkers involved in symmetrical interdimer contacts. Astonishingly, the overall fit of our dIV-dVI-m-calpain heterodimer to the calcium-bound rat dVI-dVI homodimer succeeds slightly better than that to the rat apo-homodimer (8). A comparison of our calcium-free calpain structure with the calcium-bound homodimer (8) reveals neither large overall structural deviations nor considerable differences in hydrophobic surface exposure. Thus, the calmodulin-like domains of calpains would appear to play a structural rather than a regulatory role (22). Noteworthy, however, is the unfolding of the linkers involved in the three calcium-binding EF-hand motifs (8), which change their local geometry on calcium binding, mainly to allow the involved acidic residue side chains to coordinate the bound calcium ions (Fig. 5). Domains dIV and dVI might nevertheless help to direct m-calpain to certain target proteins (23). It is possible that calcium binding is not favorably compatible with the interheterodimer contacts and might facilitate heterodimer separation, in agreement with data indicating dissociation of human and rabbit calpains (24, 25).
As mentioned above, the catalytic domain is broken in two halves. For superposition of both calpain subdomains with both papain halves, the barrel-like dIIb would have to be rotated and translated by 50° and 12 Å, respectively (Fig. 3). Thus, the active center of calcium-free m-calpain is disrupted, so that Cys105L Sγ is 10 Å apart from the catalytic His262L Nδ1. The hydrogen bond between His262L Nɛ2 and Asn286L Nδ2, however, is not, as in papain, shielded against the bulk solvent by the indole moiety of Trp288L, which, because of a main chain kink at the intermediate Pro287L, is differently placed (Fig. 3). In addition to the active-site residues, the whole substrate binding cleft is disrupted and open, incompatible with productive binding of peptide substrates.
The overall homology with papain strongly suggests that in active m-calpain both subdomains dIIa and dIIb fuse into a papain-like catalytic domain. A simple movement of dIIb toward dIIa leads to considerable sterical hindrance in the domain center, mainly caused by the Pro287L-Trp288L loop. In active calpain, this peptide must certainly be flipped over, so that the His262L imidazole side chain would approach Cys105L, and so that its hydrogen bond to Asn286L would become shielded from bulk water by the Trp288L indole moiety. Furthermore, at the reverse side (Fig. 3), the acidic residues of Asp96L and Glu172L (dIIa) would come into close contact with Glu320L and Asp321L (dIIb), so that the charge repulsion would request charge compensation (by calcium?). Upon such a “fusion,” an active-site cleft with relatively low rims would be formed, which could accommodate peptide substrates in a papain-like manner (18). The P2 side chain would extend into a shallow, essentially hydrophobic groove based on Ala263L, suggesting a papain-like specificity.
It seems to be justified to assume that both calpain subdomains possess the inherent tendency to fuse, forming a functional catalytic domain, and that this fusion is hindered in the absence of calcium. How can calcium binding trigger this fusion? It is generally accepted that m-calpain exhibits (depending on calcium concentration) three to four calcium binding sites in each calmodulin-like domain (8, 9, 25). Because of participation of Asp154S in the EF2-hand calcium binding site (Fig. 5), calcium binding to dVI would disrupt the Lys7L-Asp154S salt bridge between the L-chain anchoring helix and dVI, facilitating liberation of this helix and its possible unfolding, a prerequisite for any (autocatalytic) cleavage at Ala9L-Lys10L and Gly19L-Ser20L (26). Release of the N-terminal helix might mechanically relieve tension between dVI and dIIa, facilitating the latter's approach to dIIb and formation of a functional catalytic domain, in agreement with the observed lower calcium requirement of autolyzed m- and μ-calpain (22). The lost anchoring capability of the autolyzed m-calpain species would explain the easier dissociation of autolyzed calpain.
Calcium binding to both calmodulin-like domains could, furthermore, be signaled to the catalytic domain through the noncovalent contacts via dIII. If the conformational changes in the m-calpain heterodimer on calcium addition should be equally small as those observed in the crystals of the rat dVI homodimer (8) (Fig. 5), however, it is difficult to understand how calcium-induced changes in the calmodulin-like domains alone could push both catalytic subdomains together. We believe that our m-calpain structure provides good evidence that dIII itself plays a major role in calcium regulation of the calpain activity.
Based on sequence alignment and calcium binding studies, a “sixth” calpain EF-hand motif flanking His319L-Phe331L was predicted for calpains (27). This hypothetical “EF6-hand” was located to the region between dII and dIII but is now identified as an integral constituent of dIIb, where it forms the central loop leading to β9II and is flanked by α8II and α9II (see Figs. 1–3). Upon subdomain fusion, the contained acidic residues Glu320L and Asp321L would come into the neighborhood of acidic residues from dIIa. Binding of calcium here would facilitate or even trigger such a fusion. This negatively charged cluster created in the catalytic domain center does not resemble any typical proteinaceous calcium binding site, however.
A much better calcium binding candidate seems to be the extended, extremely acidic β2tβ3 loop of dIII, whose 10 negatively charged side chains all extend away from one another because of strong electrostatic repulsion. This acidic loop directly contacts the amphipathic α7II-helix and the open loop of dIIb, which provide the basic side chains of Lys residues 226L, 230L, and 234L, and Lys residues 354L, 355L, and 357L (Fig. 4). A few of these acidic and basic residues form direct interdomain salt bridges, and the negative and the positive electrostatic potentials mutually attract one another. It is tempting to speculate that positively charged particles such as calcium ions would bind to this acidic loop under (partial) charge compensation. The large number of clustered, negatively charged residues suggests that not only one but two or even three calcium ions might be bound to this loop in close proximity. This could create a more compact fold of this loop but also reduce its strongly negative potential. Lowering the electrostatic interaction would allow dIIb to turn over to dIIa, leading to the inherent fusion of the catalytic domain. The hydrophobic interface between dIIb-strands β5II and β10II and dIII-strands β3III, β5III, and β7III would easily allow such a rolling motion of dIIb whereas dIIa, clamped through many polar contacts to dIII via the novel hairpin loop and α5II and α6II, would remain rigidly oriented with respect to dIII (Fig. 4).
Such an “electrostatic switch mechanism” could account for different calcium activations: m-calpains possessing the largest number of acidic residues in the switch loop (Fig. 4) might require the highest calcium concentration for full charge compensation, because of charge repulsion of clustered calcium ions; for μ-calpains, with “only” eight negative charges distributed over a longer segment, this charge repulsion might be slightly weaker; the atypical calpains (1, 28), with a shorter and less acidic loop, might not need calcium compensation. A similar order is seen for the opposing basic residues of dIIb: Lys226L is found only in m-calpains, and Lys230L is present in all vertebrate calpains, whereas residue 234L always exists as Arg or Lys (1); similarly, Lys357L occurs only in m-calpains, and Lys355L is present in all m- and μ-calpains, whereas almost all calpains possess a basic residue at position 354L. Stronger switch loop-dIIb interactions would require higher calcium concentrations for release of dIIb. Of note, p94, which lacks basic residues at positions 354L, 355L, and 357L, will lack a strong switch-dIIb attraction, in agreement with proteolytic activity even in the presence of EGTA (1, 28). It is conceivable that release of dIIb exposes a novel surface site for interaction with calpastatin (29). We note that an electrostatic switch mechanism has been postulated for the calcium-binding C2A domain of the synaptic vesicle exocytosis-involved synaptotagmin I, for which binding of three closely packed calcium ions to two adjacent acidic loops attached to a similar sandwich structure has been shown by NMR (30).
The calcium levels observed in target cells in vivo are much lower than required for activation of solubilized m/μ-calpains in vitro, pointing to the additional involvement of cellular components such as membranes or calpain “activators” (31). Occupation of the calcium coordination spheres of dIII might remain incomplete in the isolated calpain and attract additional ligands, as provided by the negatively charged phosphatidyl head groups of acidic phospholipids. Indeed, acidic phospholipids such as phosphorylated phosphatidyl inositols greatly reduce the calcium concentration necessary for activity, with the calcium requirement shifted to lower concentrations with increasing degree of phosphorylation (32). In the cell, the calcium ions might target and link the m-calpain molecules to membranes. Upon membrane binding, several basic residues clustered at the back of dIIb (Fig. 3) could come into close contact with the cognate membrane surface, making favorable electrostatic interactions. We note that the p94 insertion sites IS1 and IS2, suggested to locate this tissue-specific calpain to the nucleus, are situated on the same molecular side as the switch loop, offering additional membrane binding sites.
Our structure explains the inactivity of m-calpain in the absence of calcium, and the postulated switch model can be checked by well defined experiments in the near future. Several questions, including the role of the S-chain, the calcium requirement of chimeras (33), the interaction geometry of calpastatin (29), or the real interaction site of “specific” calpain inhibitors such as PD150606 (9, 34), remain open and are the subject for future studies.
Acknowledgments
We thank Dr. M. Stubbs for valuable suggestions. The financial support by the Sonderforschungsbereichs 469 (W.B.), by the Biotech project B104-CT98-0418 and the Training-and-Mobility-of-Researcher project ERB FMRX-CT98-0193 of the European Union (W.B.), by the Human Frontier Science Program RG-203/98 (W.B. and H.S.), by the Fonds der Chemischen Industrie (W.B.), and by the Ministry of Education, Science, Sports and Culture of Japan (K.S.) are kindly acknowledged.
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1DKV).
References
- 1.Sorimachi H, Ishiura S, Suzuki K. Biochem J. 1997;328:721–732. doi: 10.1042/bj3280721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carafoli E, Molinari M. Biochem Biophys Res Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- 3.Richard I, Roudaut C, Saenz A, Pogue R, Grimbergen J E, Anderson L V, Beley C, Cobo A M, de Diego C, Eymard B, et al. Am J Hum Genet. 1999;64:1524–1540. doi: 10.1086/302426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goll D E, Thompson V F, Taylor R G, Zalewska T. BioEssays. 1992;14:549–556. doi: 10.1002/bies.950140810. [DOI] [PubMed] [Google Scholar]
- 5.Johnson G V W, Guttmann R P. BioEssays. 1997;19:1011–1018. doi: 10.1002/bies.950191111. [DOI] [PubMed] [Google Scholar]
- 6.Imajoh S, Aoki K, Ohno S, Emori Y, Kawasaki H, Sugihara H, Suzuki K. Biochemistry. 1988;27:8122–8128. doi: 10.1021/bi00421a022. [DOI] [PubMed] [Google Scholar]
- 7.Ohno S, Emori Y, Suzuki K. Nucleic Acids Res. 1986;14:5559. [PMC free article] [PubMed] [Google Scholar]
- 8.Blanchard H, Yunge L, Cygler M, Kay C M, Arthur J S C, Davies P L, Elce J S. Nat Struct Biol. 1997;4:532–538. doi: 10.1038/nsb0797-532. [DOI] [PubMed] [Google Scholar]
- 9.Lin G D, Chattopadhyay D, Maki M, Wang K K, Carson M, Jin L, Yuen P W, Takano E, Hatanaka M, DeLucas L J, et al. Nat Struct Biol. 1997;4:539–547. doi: 10.1038/nsb0797-539. [DOI] [PubMed] [Google Scholar]
- 10.Masumoto, H., Nakagawa, K., Irie, S., Sorimachi, H., Suzuki, K., Bourenkov, G. P., Bartunik, H., Fernandez-Catalan, C., Bode, W. & Strobl, S. (2000) Acta Crystallogr. D, in press. [DOI] [PubMed]
- 11.Hosfield C M, Ye Q, Arthur J S C, Hegadorn C, Croall D E, Elce J S, Jia Z. Acta Crystallogr D. 1999;55:1481–1486. doi: 10.1107/s0907444999007386. [DOI] [PubMed] [Google Scholar]
- 12.Masumoto H, Yoshizawa T, Sorimachi H, Nishino T, Ishiura S, Suzuki K. J Biochem. 1998;124:957–961. doi: 10.1093/oxfordjournals.jbchem.a022213. [DOI] [PubMed] [Google Scholar]
- 13.Otwinowski Z, Minor W. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 14.Collaborative Computing Project 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 15.de la Fortelle E, Bricogne G. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 16.Roussel A, Cambileau C. turbo frodo. Marseille, France: Centre National de la Recherche Scientifique/Université Aix-Marseille; 1989. , Version Open Gl.1. [Google Scholar]
- 17.Brünger A. x-plor Version 3.1; A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 18.Kamphuis I G, Kalk K H, Swarte B A, Drenth J. J Mol Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 19.Rizo J, Südhoff T C. J Biol Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 20.Jones E Y, Stuart D I, Walker N P C. Nature (London) 1989;338:225–228. doi: 10.1038/338225a0. [DOI] [PubMed] [Google Scholar]
- 21.Kretsinger R H. Nat Struct Biol. 1996;3:12–15. doi: 10.1038/nsb0196-12. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki K, Sorimachi H, Yoshizawa T, Kinbara K, Ishiura S. Biol Chem Hoppe-Seyler. 1995;376:523–529. doi: 10.1515/bchm3.1995.376.9.523. [DOI] [PubMed] [Google Scholar]
- 23.Meador W E, Means A R, Quiocho F A. Science. 1993;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 24.Yoshizawa T, Sorimachi H, Tomioka S, Ishiura S, Suzuki K. FEBS Lett. 1995;358:101–103. doi: 10.1016/0014-5793(94)01401-l. [DOI] [PubMed] [Google Scholar]
- 25.Michetti M, Salamino F, Minafra R, Melloni E, Pontremoli S. Biochem J. 1997;325:721–726. doi: 10.1042/bj3250721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown N, Crawford C. FEBS Lett. 1993;322:65–68. doi: 10.1016/0014-5793(93)81112-d. [DOI] [PubMed] [Google Scholar]
- 27.Andresen K, Tom T D, Strand M. J Biol Chem. 1991;266:15085–15090. [PubMed] [Google Scholar]
- 28.Ono Y, Shimada H, Sorimachi H, Richard I, Saido T C, Beckmann J S, Ishiura S, Suzuki K. J Biol Chem. 1998;273:17073–17078. doi: 10.1074/jbc.273.27.17073. [DOI] [PubMed] [Google Scholar]
- 29.Takano E, Ma H, Yang H Q, Maki M, Hatanaka M. FEBS Lett. 1995;362:93–97. doi: 10.1016/0014-5793(95)00219-y. [DOI] [PubMed] [Google Scholar]
- 30.Shao X, Fernandez I, Südhoff T C, Rizo J. Biochemistry. 1998;37:16106–16115. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- 31.Saido T C, Shibata M, Takenawa T, Murofushi H, Suzuki K. J Biol Chem. 1992;267:24585–24590. [PubMed] [Google Scholar]
- 32.Vilei E M, Calderara S, Anagli J, Berardi S, Hitomi K, Maki M, Carafoli E. J Biol Chem. 1997;272:25802–25808. doi: 10.1074/jbc.272.41.25802. [DOI] [PubMed] [Google Scholar]
- 33.Wang K K W, Wang K K, Nath R, Posner A, Raser K J, Buroker-Kilgore M, Hajimohammadreza I, Probert A W, Jr, Marcoux F W, Ye Q, Takano E, et al. Proc Natl Acad Sci USA. 1996;93:6687–6692. doi: 10.1073/pnas.93.13.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans S V. J Mol Graph. 1990;11:134–138. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 35.Barton G J. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]