

Abstract
Hearing in mammals relies on the highly synchronous synaptic transfer between cochlear inner hair cells (IHCs) and the auditory nerve. We studied the presynaptic function of single mouse IHCs by monitoring membrane capacitance changes and voltage-gated Ca2+ currents. Exocytosis initially occurred at a high rate but then slowed down within a few milliseconds, despite nearly constant Ca2+ influx. We interpret the observed secretory depression as depletion of a readily releasable pool (RRP) of about 280 vesicles. These vesicles are probably docked close to Ca2+ channels at the ribbon-type active zones of the IHCs. Continued depolarization evoked slower exocytosis occurring at a nearly constant rate for at least 1 s and depending on “long-distance” Ca2+ signaling. Refilling of the RRP after depletion followed a biphasic time course and was faster than endocytosis. RRP depletion is discussed as a mechanism for fast auditory adaptation.
The inner hair cell (IHC) is the primary sensory cell of the cochlea exciting the auditory nerve in response to incoming sound (reviewed in ref. 1). Deflections of the IHC's stereocilia, caused by movements of the tectorial membrane and hair cells relative to each other, gate stereociliar mechanosensitive transducer channels. The transduction current depolarizes the cell, thereby opening basolateral voltage-gated Ca2+ and K+ channels. The resulting Ca2+ influx at the ribbon-type active zones triggers exocytosis of synaptic vesicles, which probably release glutamate (2) onto glutamate receptors (3, 4) of the postsynaptic auditory nerve fibers. There is little information about the presynaptic function of IHCs, because the small diameter of auditory nerve fibers in mammals hinders postsynaptic recordings. Assumptions about transmitter release have, therefore, mainly been based on auditory nerve fiber spiking rate data (5) or on Furukawa's classical recordings of postsynaptic potentials from goldfish (6).
To study the presynaptic function of mouse IHCs independently of postsynaptic recordings, we detected the exocytic fusion and endocytic retrieval of synaptic vesicle membrane as changes of the membrane capacitance (Cm; ref. 7). The specificity of Cm measurements for reporting exocytosis of neurotransmitters has recently been confirmed in another ribbon-type presynapse, that of the goldfish retinal bipolar nerve terminal, by simultaneously recording transmitter release (8). In these neurons, as well as in neuroendocrine cells, several kinetic components of exocytosis have been observed and attributed to release of functionally different pools of vesicles (9–12). We compare the presynaptic properties of IHCs to the findings in other neurosecretory preparations and discuss them in the context of auditory adaptation and recovery from adaptation.
Materials and Methods
Whole-Cell Recordings.
IHCs from the apical coil of freshly dissected organs of Corti from hearing mice (Naval Medical Research Institute, postnatal days 14–40) were patch-clamped at their basolateral face at room temperature (20–25°C) or near body temperature (35°C). Pipette solutions contained (in mM): 145 Cs gluconate or Cs glutamate, 8 NaCl, 10 CsOH-Hepes, 1 MgCl2, 2 MgATP, 0.3 GTP, and Ca2+ chelators or Ca2+ indicator [EGTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (BAPTA), or FURA-2; Molecular Probes] as specified in the figure legends. For perforated-patch experiments, amphotericin B (250 μg/ml) was added. Changes in intracellular Ca2+ concentration ([Ca2+]i) measured by FURA-2 fluorometry are expressed as the fluorescence ratio at 360- and 390-nm excitation. Ca2+ currents showed a tendency to decrease with age, possibly because of a higher susceptibility to damage by the dissection of the older organs of Corti. To compensate for the possible dissection-associated loss of Ca2+ channels, an elevated extracellular Ca2+ concentration ([Ca2+]e; 10 mM CaCl2) was used for all experiments except for those of Fig. 1b (2 mM CaCl2). The extracellular solution further contained (in mM) 105 NaCl, 35 tetraethylammonium chloride (Pfaltz & Bauer), 2.8 KCl, 1 MgCl2, 10 NaOH-Hepes, and 10 d-glucose (pH 7.2). Solution changes were achieved by bath exchange. IHCs were stimulated electrically rather than mechanically, because Cm measurements require voltage clamp. Unless stated otherwise, a resting period of ≈30 s was kept between depolarizations to allow recovery of exocytosis. EPC-9 amplifiers (HEKA Electronics, Lambrecht/Pfalz, Germany) controlled by pulse software (HEKA Electronics) were used for measurements. All voltages were corrected for liquid junction potentials (−10 mV). Ca2+ current amplitudes were measured during the first 5 ms of the depolarization and are presented without leak correction. Cells with a holding current exceeding −40 pA at −80 mV were excluded from analysis. Means are expressed ± SEM.
Figure 1.
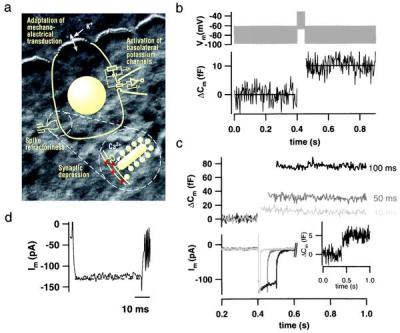
Exocytosis recorded by Cm measurements from single IHCs. (a) Simplified representation of an IHC, the measuring configuration, and an afferent IHC synapse (Inset) overlaid onto a Nomarski image of the explanted mouse organ of Corti (stereocilia of the row of IHCs are shown at the top of the image; cell bodies are covered by supporting cells). Previously suggested mechanisms of adaptation are indicated according to their localizations. Synaptic depression could be due to Ca2+ current inactivation, vesicle depletion, or postsynaptic glutamate receptor desensitization. (b) IHCs kept at 35°C in 2 mM [Ca2+]e were stimulated by a 50-ms depolarizing sinusoidal voltage (1 kHz; 40 mV peak to peak added to a dc potential of −50 mV; Upper). Four Cm traces recorded from three cells in the whole-cell configuration (0.1 mM EGTA added to the pipette solution) were averaged and smoothed by box averaging (n = 3; Lower; horizontal lines represent Cm averages before and after stimulation). Cm estimation is not valid during the depolarization because of nonlinear conductance changes. (c) High time resolution plot of ΔCm and membrane currents measured in response to depolarizations (to −15 mV) of different durations in the perforated-patch configuration. ΔCm traces were smoothed by box averaging (n = 3), and current traces were smoothed by binomial smoothing (n = 5). Outward currents were (incompletely) inhibited by intracellular Cs+ and extracellular tetraethylammonium (35 mM). (Inset) Average of four ΔCm traces in response to 4-ms depolarizations (to −15 mV). (d) Representative membrane current response to a 50-ms depolarization to −15 mV in the presence of intracellular (13 mM) and extracellular (35 mM) tetraethylammonium. Very little decline (inactivation) of Ca2+ current was observed under these conditions.
Capacitance Measurements.
We measured Cm by using the Lindau–Neher technique (ref. 13; implemented in the software “lock-in” module of pulse) combined with compensation of pipette and resting cell capacitances by the EPC-9 compensation circuitries. A 1-kHz, 70-mV peak-to-peak sinusoid was applied at a dc holding potential of −80 mV. After depolarization, slow tail currents were observed (see Fig. 1C Lower). Their reversal potential was estimated as x-axis crossing of a line fitted to the hyperpolarized portion of the current–voltage relationship measured by a voltage ramp (−100 to +60 mV within 20 ms) 20 ms after 1 s of depolarization in separate sweeps (data not shown). Setting the reversal potential used for software lock-in Cm estimation to the measured value (values ranging from −45 to −65 mV) removed the rapid Cm transients observed when an inappropriate reversal potential was entered into the Cm calculation. The mean Cm of IHCs was 8.2 ± 0.1 pF (random sample of 32 cells); the mean access resistances amounted to 9.9 ± 1.0 MΩ (value after 200 s of recording; random sample of 18 recordings) for standard whole-cell and to 17.7 ± 1.8 MΩ (value after 200 s of recording; random sample of 14 cells) for perforated-patch recordings. ΔCm was estimated as the difference of the mean Cm over 400 ms after the end of the depolarization (the initial 30 ms were skipped) and the mean prepulse capacitance (400 ms) for Figs. 3 a and b and 4 c and d. For Figs. 2c and 5, ΔCm was estimated as the difference of the mean Cm over 40 ms after the depolarization (again skipping the initial 30 ms) and the mean prepulse capacitance (40 ms before the depolarization).
Figure 3.
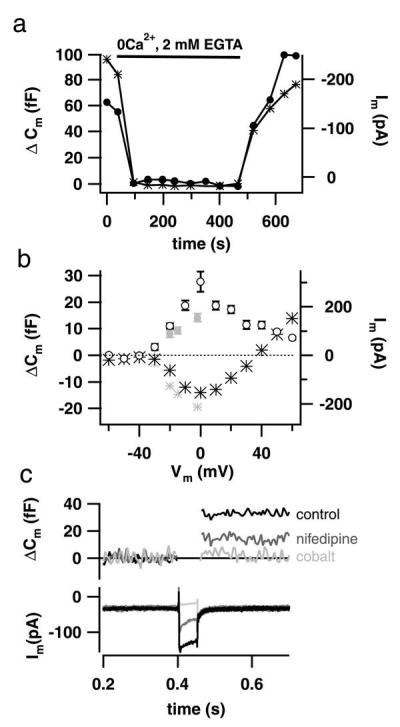
Cm changes depend strongly on voltage-gated Ca2+ entry. (a) Omission of extracellular Ca2+ (nominally Ca2+-free extracellular solution/2 mM EGTA/3 mM MgCl2) reversibly abolished both voltage-gated Ca2+ entry (asterisks, right axis reversed) and Cm increments (filled circles) stimulated by 100-ms depolarizations to −15 mV. (b) The voltage dependence of ΔCm caused by depolarizations of 200 ms (open circles, two cells) and 25 ms (small filled circles, seven cells) was similar to that of the Ca2+ current (large asterisks, 200 ms; small asterisks, 25 ms). The current–voltage relation is shifted in a positive direction, most likely because of surface charge screening. (c) Nifedipine (3 μM; dark gray) blocked about 50% of both the Ca2+ current (Lower) and the ΔCm (Upper) in this representative example, whereas 5 mM Co2+ (light gray, in the presence of Ca2+) led to full suppression of both Ca2+ current and ΔCm. Cells were depolarized to −15 mV for 50 ms. ΔCm and current traces were smoothed by box averaging (n = 5) and binomial smoothing (n = 5), respectively. All experiments (a–c) were performed in perforated-patch configuration.
Figure 4.
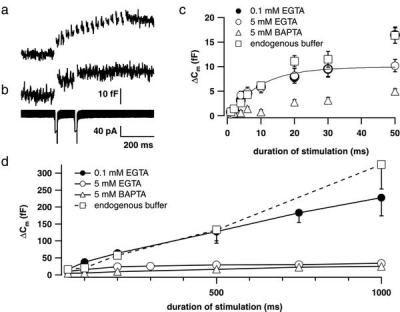
Kinetics of exocytosis in IHCs. (a) Recordings (n = 10) of ΔCm responses to trains of 15 depolarizations to −15 mV for 10 ms (20-ms interval) were averaged (scaling as in b). The first depolarization caused the largest response. (b) A paired-pulse paradigm (two 15-ms depolarizations to −15 mV; 100-ms interpulse interval) resulted in a larger first than second ΔCm (Upper). Secretory depression occurred, although Ca2+ currents were nearly identical (Lower). Data represent an average of four responses. (a and b) Experiments were performed in the perforated patch-configuration. (c) ΔCm triggered by depolarizations of different durations to −15 mV plotted against pulse duration up to 50 ms to emphasize the fast secretory component. Experiments were performed in perforated-patch (squares; n = 25) or whole-cell configuration, and then Ca2+ chelators were introduced to the IHC's cytosol as specified. Mixtures of equal amounts of Ca2+-loaded and Ca2+-free chelators were used to avoid possible depriming effects of very low [Ca2+]i levels. Stimulation was started 60 s after establishing the whole-cell configuration. Filled circles, low buffering capacity (0.1 mM Ca2+-free EGTA/0.1 Ca2+-EGTA; n = 33 cells); open circles, high buffering capacity, slower Ca2+-binding (5 mM Ca2+-free EGTA/5 mM Ca2+-EGTA; n = 26 cells); triangles, high buffering capacity, faster Ca2+-binding (5 mM Ca2+-free BAPTA/5 mM Ca2+-BAPTA; n = 16 cells); solid line, exponential fit to the first 50 ms of the “high EGTA” data. (d) Same data set as in c. Slow Cm rise during continued depolarization.
Figure 2.
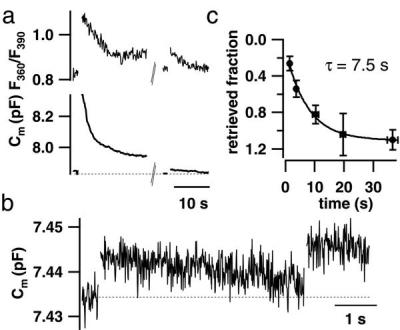
Endocytosis in IHCs. (a) Low time resolution plot of [Ca2+]i (Upper) and Cm (Lower) of a cell. Both Cm and [Ca2+]i increased after a 500-ms pulse, and, 30 s later, smaller responses were elicited by a 50-ms depolarization. After the depolarizations, Cm and [Ca2+]i declined in parallel (whole-cell configuration; 0.1 mM FURA-2 added to the pipette solution). (b) Paired pulses (twice for 15 ms to −15 mV) with variable interval between both stimuli were used to measure the time course of endocytosis in 11 perforated-patch experiments. b shows an example Cm trace with an interval of 5 s (smoothed by box averaging; n = 5). (c) Cm values measured right after the first stimulus and before the second stimulus were used to calculate the endocytosed fraction of the Cm increment elicited by the first depolarization (ΔCm1) at a certain interval. The solid line is an exponential fit to the data.
Figure 5.
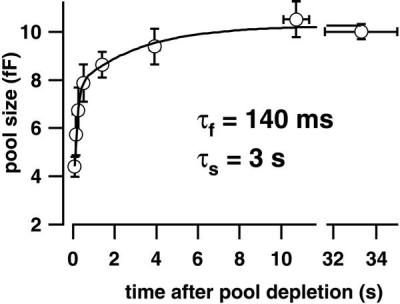
Rapid recovery from RRP depletion. Paired pulses (twice for 15 ms to −15 mV) were used to measure the time course of pool recovery in 11 perforated-patch experiments (see Fig. 2b for an example trace). The second response (ΔCm2), measured at variable times after the first pool-depleting response (ΔCm1), indicated the refilling state of the RRP. The summary plot shows ratios ΔCm2/ΔCm1 multiplied by the steady state ΔCm2 (10.2 fF), which was similar to our RRP size estimate (see Fig. 4C). Solid line, double exponential fit to the data (see Results).
Results
Exocytosis and Endocytosis of IHCs Monitored by Cm Measurements.
Fig. 1a schematically shows a patch-clamped IHC, overlaid onto a Nomarski image of the organ of Corti. In addition, Fig. 1a lists some of the mechanisms that have been suggested to contribute to fast adaptation of hearing (see Discussion). Fig. 1b shows an average ΔCm response to a 50-ms sinusoidal stimulation that mimics physiological conditions (35°C; 2 mM [Ca2+]e; stimulus comparable to strong IHC depolarizations achieved by mechanoelectrical transduction; ref. 1). The resulting Cm increment of about 10 fF most likely reflects exocytosis of synaptic vesicles (about 280, as determined with a conversion factor of 37 aF per vesicle; ref. 14). Fig. 1c shows ΔCm and Ca2+ currents in response to step depolarizations of different lengths. ΔCm could be detected after very short stimuli and increased with longer stimulation. The inward current showed some reduction with time, in part reflecting activation of some unblocked K+ channels and perhaps a very slow Ca2+ current inactivation for longer pulses. When tetraethylammonium, in addition to its extracellular presence, was introduced intracellularly (13 mM), even less decline of the inward current was observed (Fig. 1d).
After termination of both long and short Ca2+ current injections, the elevated [Ca2+]i and Cm declined slowly (Figs. 2 a and b), the latter most likely reflecting endocytosis. Because of its slow time course, endocytosis was unlikely to complicate measurements of exocytosis, which were performed shortly after the end of the depolarization (see Materials and Methods). Membrane added by brief stimuli (Fig. 2b) was retrieved after a single exponential time course (τ = 7.5 s; Fig. 2c).
The interpretation of the observed Cm changes as Ca2+-triggered exocytosis followed by endocytic membrane retrieval is strongly supported by their dependence on Ca2+ influx (Fig. 3). Omission of extracellular Ca2+ completely and reversibly abolished the ΔCm (Fig. 3a). The voltage dependence of ΔCm mirrored that of the Ca2+ influx (Fig. 3b). Both ΔCm and Ca2+ influx reached their maximum around 0 mV in 10 mM [Ca2+]e. The remaining ΔCm at positive potentials most likely represents exocytosis triggered by Ca2+ tail currents. The dihydropyridine l-type Ca2+ channel blocker nifedipine (3 μM; Fig. 3c) partially inhibited ΔCm and Ca2+ influx, proving that l-type Ca2+ channels are involved in stimulus-secretion coupling of IHCs. ΔCm and Ca2+ influx were abolished by addition of the inorganic Ca2+ channel blocker cobalt (5 mM; Fig. 3c).
Kinetics of IHC Exocytosis in Differing Cytosolic Ca2+ Buffering Conditions.
We typically observed high secretory rates during the first 10 ms of stimulation followed by slower exocytic activity. Thus, the first stimulus within a train of 10-ms depolarizations evoked the largest ΔCm (Fig. 4a). As shown in Fig. 4b, a pair of 15-ms, equally strong depolarizations evoked paired-pulse depression. Note that the two Ca2+ currents were virtually identical, proving that depression of exocytic activity occurred despite unabated Ca2+ influx. The biphasic pattern of secretion was most obvious when we probed ΔCm in response to depolarizations of different durations in whole-cell experiments (0.1 mM EGTA; Fig. 4 c and d, filled circles). The initial, rapid Cm rise (Fig. 4c) was followed by a slower, up to 1 s nearly linear Cm rise (Fig. 4d, slope between 30 ms and 1 s; 223 fF⋅s−1 or 6,030 vesicles⋅s−1). Separation of the rapid and sustained secretory components was facilitated by intracellular application of high concentrations of EGTA (5 mM; Fig. 4 c and d, open circles), which left the rapid component unchanged (Fig. 4c) but strongly inhibited the slower Cm rise (Fig. 4d, slope: 23.8 fF⋅s−1 or 640 vesicles⋅s−1). Therefore, exponential fitting to the Cm rise during the initial 50 ms of stimulation in the intracellular presence of 5 mM EGTA was used to estimate amplitude (10 fF or 280 vesicles) and time constant (10 ms) of the fast secretory component, yielding a maximum secretory rate of 28,000 vesicles⋅s−1. We interpret this component as exocytosis of a limited readily releasable pool (RRP) of vesicles. BAPTA (5 mM), a chelator with faster binding kinetics, also effectively suppressed the release of the RRP (Fig. 4 c and d, triangles).
The effects of exogenously added chelators highlight the impact that cellular Ca2+ buffers (15, 16) can have on stimulus-secretion coupling depending on their Ca2+-binding properties and concentration. In the sole presence of endogenous Ca2+ buffer (perforated-patch configuration; no washout of cellular Ca2+ binding molecules into the pipette; no exogenously added chelator; Fig. 4 c and d, squares), size and release kinetics of the RRP were comparable to the whole-cell data (loss of mobile endogenous Ca2+ buffer) with exogenously added EGTA. However, they were very different from the results obtained with millimolar BAPTA. The endogenous buffer interfered far less with the sustained component (slope: 322 fF⋅s−1 or 8,700 vesicles⋅s−1) than 5 mM EGTA (slope: 640 vesicles⋅s−1).
Rapid Biphasic Recovery of the RRP from Depletion.
To study the kinetics of vesicle supply after termination of voltage-gated Ca2+ entry, we measured the recovery of the RRP from depletion by using pairs of short stimuli (15 ms) separated by varying intervals. Experiments were performed in the perforated-patch configuration to minimize secretory rundown (for a typical measurement, see Fig. 2b). The recovery time course was biphasic (Fig. 5); double exponential fitting yielded time constants of 140 ms and 3 s with amplitudes of 60% and 26% of the final RRP size and an initial level of depression of 86%. At the shortest interval measured (85 ms), the pool size amounted already to about 40% of the initial RRP. Thus, IHCs have a rapid mode of pool recovery with a maximal rate of vesicle supply of about 1,200 vesicles⋅s−1 (number of rapidly recovered vesicles⋅τ−1).
Discussion
Our study provides a quantitative description of exocytosis and endocytosis in cochlear IHCs. Exocytosis depended on voltage-gated Ca2+ influx, which was at least partially mediated by l-type Ca2+ channels. The time course of endocytosis in IHCs (τ = 7.5 s) compares to time constants of 12 s in frog vestibular hair cells (17) and of 1–2 s in retinal bipolar nerve terminals of goldfish (18), two other ribbon-type synapses.
The RRP of IHCs.
Exocytosis of IHCs initially occurred at very high rates but slowed after a few milliseconds of stimulation. We favor the interpretation that the secretory depression was caused by the depletion of a pool of about 280 release-ready vesicles, because the Ca2+ current of mouse cochlear hair cells did not significantly inactivate. Similarly, the Ca2+ current of chick cochlear hair cells has been reported to show little if any inactivation (19).
The observed differential effects of slow (EGTA) versus fast (BAPTA) Ca2+ chelators on the release of the RRP are expected for the case of tight spatial coupling between release sites and Ca2+ channels (20). Therefore, we suggest that the release-ready vesicles are docked in close vicinity of the Ca2+ channels, which are clustered at the active zones (21) and most likely form part of the ultrastructurally observed presynaptic intramembrane particles (22). Rapid release of a similar pool was observed in the bipolar neurons of the goldfish retina (9). Their pool size compared well to the number of vesicles found by electron microscopy to be docked to the presynaptic membrane at the base of the ribbons (22 vesicles per active zone; ref. 23). Thus far, no electron microscopic counts of active zones and/or docked vesicles are available for mouse IHCs. Indirect estimation, relying on counts of the numbers of IHCs and approaching myelinated afferent nerve fibers within a stretch of organ of Corti, yields about 25 active zones per IHC for the apical part of the cochlea (M. C. Liberman, personal communication). Assuming that each active zone contributes equally to transmitter release, one would expect about 14 release-ready vesicles per active zone. The resulting high initial exocytic rates (about 1,400 vesicles per second and synapse) would be sufficient to drive the auditory nerve fibers toward the high spiking rates observed at the onset of sound (5).
The Sustained Secretory Component in IHCs.
Like frog saccular hair cells (17), IHCs had a prominent sustained component of exocytosis with a nearly constant secretory rate up to 1 s. In contrast to hair cells, the sustained release in bipolar nerve terminals slows after about 150 ms, probably because of depletion of a pool of about 6,000 vesicles (18), which compares well to the pool of vesicles being morphologically tethered to the ribbons in these terminals (23). It has been suggested that this difference between hair cells and bipolar terminals may be due to the different shape of the presynaptic ribbons present in these cells (ref. 23; spheroid body in saccular hair cells and platelet ribbon in bipolar cells). This view has to be questioned now, because the IHCs investigated in the present study possess platelet ribbons (24) but show a linear sustained secretory component up to 1 s. The sustained component was sensitive also to the slowly binding Ca2+ chelator EGTA, which is in line with results obtained in bipolar terminals (18) and neuroendocrine cells (25). Effects of EGTA on cellular processes are commonly taken as evidence for a dependence on “long-distance” Ca2+ signaling rather than on local domains of high [Ca2+] (e.g., around Ca2+ channels), because the slowly binding chelator is expected to bind Ca2+ only a few milliseconds after Ca2+ entered the cytosol (20). Ca2+ spreading, therefore, is required for the sustained secretory component in the various preparations. Two mechanisms have been suggested to underlie sustained exocytosis. First, it may result from a rapid vesicle supply to the RRP (9, 10). In ribbon synapses, this vesicle supply may be due to mobilization of vesicles, which were previously attached to the ribbons (23). Second, fusion of release-ready vesicles located at longer distance to Ca2+ channels (12) may contribute to the sustained exocytosis. If the slow Cm rise in IHCs exclusively represented vesicle supply to the active zones of the IHCs (supply of about 9,000 vesicles⋅s−1), the RRP would be turned over about 30 times within 1 s during strong stimulation. Although we observed a rapid resupply of release-ready vesicles in IHCs after RRP depletion, the maximal rate of vesicle supply in IHCs (1,200 vesicles⋅s−1) was much slower than the rate of sustained secretion during stimulation. It is possible that the high [Ca2+]i during the stimulation causes an acceleration of vesicle supply as has been described for neuroendocrine cells (26) as well as for neurons (10, 27). However, it seems unlikely that such acceleration could fully account for the difference between the rate of sustained exocytosis during stimulation (high [Ca2+]i) and that of vesicle supply after stimulation (decaying [Ca2+]i). Therefore, we cannot exclude that at least part of the slow Cm rise represents parallel exocytosis of vesicles residing at larger distance to Ca2+ channels.
Biphasic Recovery of the RRP.
RRP recovery from depletion in IHCs showed a rapid, biphasic time course. Somewhat slower biphasic RRP recovery has recently been described for retinal bipolar cells (10) and the calyx of Held, a calyx-type synapse in the auditory brainstem (28). Both studies argued that the initial phase of pool refilling was Ca2+ independent. Although we currently cannot rule out that, in IHCs, this phase represents Ca2+-accelerated vesicle supply during the elevated poststimulus [Ca2+]i (26), we consider a further possible reason for the biphasic recovery time. IHCs are believed to possess functionally distinct active zones that contact auditory nerve fibers of different spontaneous rates and thresholds (29). It has been suggested that different release properties among the active zones could account for the variable spontaneous rates of the auditory nerve fibers (29). Similarly, the observed biphasic pool refilling could reflect different recovery kinetics at functionally distinct active zones. Indeed, the time needed for recovery from prior stimulation differs among high (215 ms) and low (2 s) spontaneous rate auditory nerve fibers (ref. 30; for comparison, the time constants for RRP recovery were 140 ms and 3 s).
Endocytosis seems too slow (τ = 7.5 s; Fig. 2c) for vesicle recycling to account for pool recovery from depletion in IHCs. However, endocytic vesicles are expected to enter the maturation cascade (31), thereby ensuring supply of release-ready vesicles on longer time scales. Indeed, vesicles containing endocytosed horseradish peroxidase were seen to populate the synaptic bodies of IHCs within few minutes of incubation with the tracer (32).
Relating Vesicle Pool Depletion to Fast Auditory Adaptation.
In response to a constant sound, the auditory nerve spiking rate is maximum at the onset but then declines rapidly, a phenomenon called adaptation (5). Fast types of peripheral auditory adaptation include rapid and short-term adaptation (gerbil, ref. 33; guinea pig, ref. 34). Rapid adaptation (τ = 1–25 ms) occurs faster at high sound intensity, and its time course also depends on the spontaneous rate of the fiber (35). Short-term adaptation follows a somewhat slower time course (τ = 15–60 ms, depending on the animal species). The fast adaptation of the peripheral auditory system to sound is probably important for speech recognition, because it may contribute to peripheral coding of complex stimuli (36).
Synaptic depression at the hair cell afferent synapse has been suggested to underlie fast auditory adaptation (6). Specifically, depletion of presynaptic transmitter stores was assumed, although the evidence was indirect because it relied on postsynaptic recordings from the goldfish sacculus. Our presynaptic measurements now show a slowing of the secretory rate of IHCs, most likely reflecting depletion of the RRP, with a time course (τ = 10 ms) compatible to the time courses of rapid and short-term adaptation. Moreover, we observed a biphasic time course for the RRP recovery from depletion, which was similar to that of the compound action potential recovery from adaptation in the auditory nerve (37). We therefore conclude that presynaptic depletion of release-ready vesicles may contribute to fast auditory adaptation. Inactivation of Ca2+ current was negligible in IHCs (Fig. 1d) and is therefore unlikely to play a major role in fast auditory adaptation.
The abundance of two types of fast adaptation in nerve fiber recordings suggests that presynaptic vesicle depletion is not the only reason for adaptation. Three other mechanisms could potentially contribute to rapid auditory adaptation: (i) decline of the receptor potential caused by opening of basolateral Ca2+-activated potassium channels (1), (ii) desensitization of postsynaptic glutamate receptors (3), and (iii) spike refractoriness in the auditory nerve fiber (38). Adaptation of mechanoelectrical transduction, because of its high-pass-filter characteristics (39), is not expected to reduce acoustical sensitivity, except for ultralow frequency sounds. Fig. 1a lists these mechanisms according to their site of action. The implication of depletion of release-ready vesicles during peripheral auditory adaptation adds one more physiological perspective to the concept of depletion and refilling of presynaptic vesicle pools.
Acknowledgments
We thank Drs. A. Rüsch and D. Oliver for help in setting up the preparation; Drs. R. Klinke, T. Voets, C. J. Kros, A. Gummer, H. v. Gersdorff, and E. Neher for critical feedback on the manuscript; and Mr. M. Pilot for technical assistance. We are grateful to Dr. E. Neher for generous support and helpful discussions. This work was supported by a Deutsche Forschungsgemeinschaft grant to T.M. and a Fortuene grant.
Abbreviations
- IHC
inner hair cell
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate
- [Ca2+]i/e
intracellular/extracellular Ca2+ concentration
- Cm
membrane capacitance
- RRP
readily releasable pool
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kros C J. In: The Cochlea. Dallos P, Popper A N, Fay R R, editors. New York: Springer; 1996. pp. 318–385. [Google Scholar]
- 2.Kataoka Y, Ohmori H. J Physiol (London) 1994;477:403–414. doi: 10.1113/jphysiol.1994.sp020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raman I M, Zhang S, Trussell L O. J Neurosci. 1994;14:4998–5010. doi: 10.1523/JNEUROSCI.14-08-04998.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsubara A, Laake J H, Davanger S, Usami S, Ottersen O P. J Neurosci. 1996;16:4457–4467. doi: 10.1523/JNEUROSCI.16-14-04457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiang N Y-S, Watanabe T, Thomas E C, Clark L F. Discharge Pattern of Single Fibers in the Cat's Auditory Nerve. Cambridge, MA: MIT Press; 1965. [Google Scholar]
- 6.Furukawa T, Matsuura S. J Physiol (London) 1978;276:193–209. doi: 10.1113/jphysiol.1978.sp012228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neher E, Marty A. Proc Natl Acad Sci USA. 1982;79:6712–6716. doi: 10.1073/pnas.79.21.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Gersdorff H, Sakaba T, Berglund K, Tachibana M. Neuron. 1998;21:1177–1188. doi: 10.1016/s0896-6273(00)80634-0. [DOI] [PubMed] [Google Scholar]
- 9.Mennerick S, Matthews G. Neuron. 1996;17:1241–1249. doi: 10.1016/s0896-6273(00)80254-8. [DOI] [PubMed] [Google Scholar]
- 10.Gomis A, Burrone J, Lagnado L. J Neurosci. 1999;19:6309–6317. doi: 10.1523/JNEUROSCI.19-15-06309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas P, Wong J G, Almers W. EMBO J. 1993;12:303–306. doi: 10.1002/j.1460-2075.1993.tb05657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voets T, Neher E, Moser T. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- 13.Lindau M, Neher E. Pflügers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- 14.Lenzi D, Runyeon J W, Crum J, Ellisman M H, Roberts W M. J Neurosci. 1999;19:119–132. doi: 10.1523/JNEUROSCI.19-01-00119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts W M. J Neurosci. 1994;14:3246–3262. doi: 10.1523/JNEUROSCI.14-05-03246.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helmchen F, Borst J G, Sakmann B. Biophys J. 1997;72:1458–1471. doi: 10.1016/S0006-3495(97)78792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons T D, Lenzi D, Almers W, Roberts W M. Neuron. 1994;13:875–883. doi: 10.1016/0896-6273(94)90253-4. [DOI] [PubMed] [Google Scholar]
- 18.von Gersdorff H, Matthews G. Nature (London) 1994;367:735–739. doi: 10.1038/367735a0. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs P A, Evans M G, Murrow B W. J Physiol (London) 1990;429:553–568. doi: 10.1113/jphysiol.1990.sp018272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neher E. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 21.Issa N P, Hudspeth A J. Proc Natl Acad Sci USA. 1994;91:7578–7582. doi: 10.1073/pnas.91.16.7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts W M, Jacobs R A, Hudspeth A J. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Gersdorff H, Vardi E, Matthews G, Sterling P. Neuron. 1996;16:1221–1227. doi: 10.1016/s0896-6273(00)80148-8. [DOI] [PubMed] [Google Scholar]
- 24.Sobkowicz H M, Rose J E, Scott G E, Slapnick S M. J Neurosci. 1982;2:942–957. doi: 10.1523/JNEUROSCI.02-07-00942.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moser T, Neher E. J Neurosci. 1997;17:2314–2323. doi: 10.1523/JNEUROSCI.17-07-02314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith C, Moser T, Xu T, Neher E. Neuron. 1998;20:1243–1253. doi: 10.1016/s0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- 27.Stevens C F, Wesseling J F. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 28.Wu L G, Borst J G. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 29.Liberman M C. Science. 1982;216:1239–1241. doi: 10.1126/science.7079757. [DOI] [PubMed] [Google Scholar]
- 30.Relkin E M, Doucet J R. Hear Res. 1991;55:215–222. doi: 10.1016/0378-5955(91)90106-j. [DOI] [PubMed] [Google Scholar]
- 31.Betz W J, Angleson J K. Annu Rev Physiol. 1998;60:347–363. doi: 10.1146/annurev.physiol.60.1.347. [DOI] [PubMed] [Google Scholar]
- 32.Siegel J H, Brownell W E. J Neurocytol. 1986;15:311–328. doi: 10.1007/BF01611434. [DOI] [PubMed] [Google Scholar]
- 33.Westerman L A, Smith R L. Hear Res. 1984;15:249–260. doi: 10.1016/0378-5955(84)90032-7. [DOI] [PubMed] [Google Scholar]
- 34.Yates G K, Robertson D, Johnstone B M. Hear Res. 1985;17:1–12. doi: 10.1016/0378-5955(85)90124-8. [DOI] [PubMed] [Google Scholar]
- 35.Rhode W S, Smith P H. Hear Res. 1985;18:159–168. doi: 10.1016/0378-5955(85)90008-5. [DOI] [PubMed] [Google Scholar]
- 36.Delgutte B. J Acoust Soc Am. 1980;68:843–857. doi: 10.1121/1.384824. [DOI] [PubMed] [Google Scholar]
- 37.Relkin E M, Doucet J R, Sterns A. Hear Res. 1995;83:183–189. doi: 10.1016/0378-5955(95)00004-n. [DOI] [PubMed] [Google Scholar]
- 38.Gaumond R P, Molnar C E, Kim D O. J Neurophysiol. 1982;48:856–873. doi: 10.1152/jn.1982.48.3.856. [DOI] [PubMed] [Google Scholar]
- 39.Corey D P, Hudspeth A J. J Neurosci. 1983;3:942–961. doi: 10.1523/JNEUROSCI.03-05-00942.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]