

Abstract
Ethidium (E) is a powerful probe of DNA dynamics and DNA-mediated electron transfer (ET). Molecular dynamical processes, such as solvation and orientation, are important on the time scale of ET. Here, we report studies of the femtosecond and picosecond time-resolved dynamics of E, E with 2′deoxyguanosine triphosphate (GTP) in water, and E with 7-deaza-2′-deoxyguanosine triphosphate (ZTP) in water; E undergoes ET with ZTP but not GTP. These studies elucidate the critical role of relative orientational motions of the donor–acceptor complex on ET processes in solution. For ET from ZTP to E, such motions are in fact the rate-determining step. Our results indicate that these complexes reorient before ET. The time scale for the solvation of E in water is 1 ps, and the orientational relaxation time of E is 70 ps. The impact of orientational and solvation effects on ET between E and mononucleotides must be considered in the application of E as a probe of DNA ET.
Because of its unique properties, ethidium (E) bromide has been used widely for various biochemical and biophysical applications (e.g., refs. 1–10). The cationic dye E intercalates in helical polynucleotides and can be used as a nucleic acid stain. The intercalated probe has different nanosecond fluorescence properties from those of E in water, and one can distinguish between intercalated and nonintercalated E molecules (1–5). Time-resolved studies of E intercalated in DNA on the picosecond time scale provide a probe of torsional dynamics and DNA flexibility, which are important structural features of DNA (7–10).
E has also been employed as a probe of DNA-mediated electron transfer (ET) reactions (11–16). Recently, it has been shown, in systems employing E covalently tethered to DNA duplexes, that on photoexcitation this intercalator acts as an electron acceptor with respect to a modified guanine base, 7-deazaguanine (11), or as a donor with respect to a rhodium intercalator (12). In these studies, measurements of fluorescence quenching suggested fast reaction kinetics (≥1010 s−1) and a sensitivity of quenching to stacking.
Despite the general utility of E as a DNA probe, its photophysical properties on the ultrafast time scale have not been examined extensively. Sommer et al. (17) measured the transient fluorescence spectra with picosecond time resolution and observed a time-dependent spectral shift on a subnanosecond time scale in glycerol. This shift was attributed to an intramolecular relaxation process involving phenyl-group rotation. The fact that they could not resolve the dynamics of this process when water was used as a solvent has been attributed to the lower viscosity, which offers no significant hindrance to phenyl-group rotation.
Studies of the femtosecond dynamics of E in water and ET processes of E
with mononucleotides lay the foundation for studies of ultrafast ET
dynamics in DNA and time-resolved studies of biological dynamics. Here,
we report our studies of the femtosecond dynamics of E and of ET
between E and mononucleotides in water. Experimentally, we used three
different types of measurements, transient absorption (TA),
time-resolved fluorescence, and linear dichroism (LD)/anisotropy, all
with fs time resolution. We also carried out quantum mechanical
calculations to obtain relevant structures and their molecular
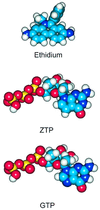
interactions (Scheme
1; ZTP =
7-deaza-2′-deoxyguanosine triphospate).
METHODOLOGY
Experimental.
Fig. 1 gives a schematic of the setup used in the femtosecond TA and fluorescence up-conversion measurements. A Ti:sapphire laser (Spectra-Physics) generated femtosecond pulses (80 fs; ≈800 nm; 2 mJ at 1 kHz). The 2-mJ pulse was split equally to pump two optical parametric amplifiers (OPAs). The signal output from one OPA was then mixed with the residual 800-nm pulse to generate the pump pulse at 480–520 nm. The probe pulse at 570–700 nm was generated by doubling the signal from another OPA. The probe pulse at ≈400 nm was generated by doubling the 800-nm pulse.
Figure 1.

Arrangements for femtosecond TA, LD, and the
fluorescence up-conversion (Inset) used in the
experimental apparatus. OPA, optical parametric amplifier; F, filters;
L, lenses; P, polarizers; PD, photodiodes; PMT, photomultiplier tube;
λ1, excitation beam for up-conversion; λ2,
probe beam for up-conversion;  , half wave plate; BBO, beta
barium borate up-conversion crystal; S, sample cell.
, half wave plate; BBO, beta
barium borate up-conversion crystal; S, sample cell.
The pump pulse was passed through an optical arrangement that was
controlled by a computer for time-delay scans. A chopper in the beam of
the pump pulse was used to optimize the signal-to-noise ratio. A
 wave plate (combined with a polarizer) reduced the
pump-pulse energy to ≈2 μJ. The probe pulse was attenuated to <0.1
μJ by neutral density filters. When the polarization of the probe
pulse was set at 54.7° (magic angle) relative to the polarization of
the pump pulse, the TA signals contained no contribution from the
reorientational motion of the molecules.
wave plate (combined with a polarizer) reduced the
pump-pulse energy to ≈2 μJ. The probe pulse was attenuated to <0.1
μJ by neutral density filters. When the polarization of the probe
pulse was set at 54.7° (magic angle) relative to the polarization of
the pump pulse, the TA signals contained no contribution from the
reorientational motion of the molecules.
Our approach allows us to determine both the reorientational motion and the TA simultaneously. The pump pulse was vertically polarized, and the probe pulse was polarized 45° relative to the pump polarization. The two pulses were passed through the sample cell with a small angle between the two propagating beams. A polarizer placed after the sample cell was used to separate the probe pulse into two components, one parallel and one perpendicular to the pump polarization. The parallel (I∥) and perpendicular (I⊥) signals were detected by two photodiodes. The intensity of the probe pulse was also detected by another photodiode serving as the reference signal (Ir). The three detected signals were sent into a boxcar integrator and then to a computer for data processing. The TA and LD signals are defined (where A = absorbance) as TA = (A∥ + 2A⊥)/3 = [log(Ir/I∥) + 2log(Ir/I⊥)]/3 and LD = A∥ − A⊥ = log(I⊥/I∥). The TA signal detected in this way is equivalent to the one measured at magic angle.
The fluorescence up-conversion setup is shown in Fig. 1 Inset. The fluorescence was initiated by the pump pulse in the sample cell, collected, and focused into a beta barium borate (BBO) crystal (thickness of 1 mm) by two parabolic mirrors. Then, a probe pulse (≈800 nm; 200 μJ) was sent into the BBO crystal for mixing with the fluorescence signal. The up-converted signal was detected by a photomultiplier after passage through a double-grating monochromator to reduce the background signal. The pump polarization was adjusted to be parallel or perpendicular to the fluorescence polarization set by the BBO crystal. Finally, the signal from the photomultiplier was processed via the boxcar integrator and the computer. The measurements were made in a 5-mm quartz cell, which was stirred during the data-collection process. The data have been analyzed by least-squares fitting; for single-exponential decays, we estimated the error to be ≈10%, and for biexponential decays, the errors are somewhat larger.
Sample Preparation.
E bromide was obtained from Aldrich, and ZTP and 2′-deoxyguanosine triphosphate (GTP) were purchased from Amersham Pharmacia. All materials were used as received without further purification. Concentrated stock solutions of E were prepared freshly before each experiment and calibrated by using UV-visible absorption spectroscopy [ɛ480 (E in H2O): 5,680 cm−1⋅M−1; ref. 1] performed on an HP8452 diode array spectrometer (Hewlett–Packard). Concentrated stock solutions of nucleoside triphosphates were obtained by evaporating commercial solutions of known concentrations and redissolving in minimal amounts of water. Samples for spectroscopic experiments then were prepared by adding appropriate amounts of each species to a buffered solution containing 5 mM sodium phosphate and 50 mM sodium chloride, pH 7. The presence of salts in the nucleoside triphosphate solutions resulted in a small increase in ionic strength (≈5 mM) for these samples; however, the measurements included in this study did not seem to be sensitive to the presence of this small amount of added salt.
Computational.
All quantum mechanical calculations have been carried out with the computer program gaussian 98w (18). The structures were optimized with the AM1 Hamiltonian, and the excited-state energies and transition dipole moments were calculated with the standard method of zindo.
RESULTS AND DISCUSSION
Ultrafast Dynamics of E in Water.
After the excitation of E into the S1 state with a femtosecond laser pulse at 480 nm, a biexponential decay of the fluorescence at 570 nm was observed. E shows a fast- and a slow-decay component; the latter is effectively a constant background on the time scale of a few picoseconds. The fast decay occurs with a 1-ps time constant (Fig. 2). The amplitude of this 1-ps component decreases as the detection wavelength increases. To obtain additional information on the ultrafast dynamics, we also carried out TA experiments at the same excitation wavelength. Because our probe wavelengths were chosen to be in the spectral region of the emission, the observed TA signal between 580 and 670 nm is composed of two contributions, excited-state absorption [A(t, λ) (Sn ← S1)] and stimulated emission [F(t, λ) (S1 → S0)]. Although the former gives positive absorbance, the latter leads to a negative contribution, because the probe pulse gains intensity from the emission (Fig. 2).
Figure 2.

The up-converted fluorescence and TA of E in water (short time scans). The excitation pulse at 480 nm was used. (Left) Signals detected at the wavelength indicated. (Right) TA at the indicated probe wavelength. (Insets) Signals of pure water measured under the same experimental conditions.
At 585 nm, we observe a rise with a time constant of 1 ps in the (positive) TA signal, indicating that A(t, λ) is dominant. With increasing probe wavelengths, the sign changes, mainly because of an increased contribution from the stimulated emission. At 665 nm, the observed TA signal is essentially negative, except for a fast positive spike around zero delay, which indicates that F(t, λ) is the dominant contribution. It is important to note that both the time-resolved fluorescence and the observed TA/stimulated-emission signals show the same characteristic time constant of ≈1 ps. The fact that there is a fast initial decay (1 ps) of the fluorescence at shorter wavelengths and a corresponding rise of the emission at longer wavelengths indicates that the overall fluorescence spectrum is shifting on a picosecond time scale toward longer wavelengths. This time-dependent Stokes shift is typical for molecules with dipoles in polar solvents and represents the dynamics of solvation, i.e., the relaxation of the solvent water molecules around the E dipole as a result of changes in its charge distribution on excitation.
The solvation time scale measured here is similar to the one observed by Jiminez et al. (19), who used the fluorescence up-conversion technique for a coumarin salt in water. They observed a strongly biexponential decay in the Stokes-shift correlation function. An ultrafast response of 55 fs was attributed to a librational motion, whereas a second component of 880 fs was related to a diffusional motion of water molecules. The latter time constant agrees well with our 1-ps time component. From our zindo calculations, we estimate the difference between the electric dipole moment of the S0 and the S1 state of E to be ≈4 debye, which represents a large change in the charge distribution for solvation to be effective.
It is worth noting that an additional time constant of ≈7 ps had to be used to fit the TA signal at 665 nm. However, the amplitude of this component is only ≈10%. It is possible that in addition to solvation, intramolecular relaxation processes occur after the excitation. It has been suggested that the phenyl-group rotation takes place on a picosecond time scale in water (17). We have also measured the decay of the E fluorescence at longer times (not shown) and found agreement with previously published results (2, 20); except for the ultrafast dynamics, which occur on the 1- to 10-ps time scale, the fluorescence decay of the S1 state in water takes place with a time constant of 1.6–1.8 ns. Likewise, the TA signal at longer times can be fit with a single decay time constant (≈1.6 ns) at all wavelengths.
Time-Resolved Fluorescence Anisotropy and LD.
We investigated the orientational relaxation in the excited state of E by measuring the fluorescence anisotropy [r(t)]. The fluorescence anisotropy was obtained from the intensity of the parallel [I∥(t)] and perpendicular [I⊥(t)] up-conversion signals by using the following formula: r(t) = [I∥(t) − I⊥(t)]/[I∥(t) + 2I⊥(t)]. We first measured the fluorescence anisotropy of E in water, as shown in Fig. 3a. The fluorescence anisotropy shows a decay with a time constant of 70 ps. Because the fluorescence signal at the magic-angle (54.7°) condition shows a single exponential decay (1.6 ns) without any component on the order of 70 ps, the latter gives the orientational relaxation time of the excited state E* in water. To obtain further evidence, we investigated the viscosity dependence of the fluorescence anisotropy decay. Fig. 3 a and b shows the decays of E* in water and ethanol; we also measured the decay in methanol and butanol (data not shown). As expected, the orientational relaxation time increases as the solvent viscosity increases.
Figure 3.

(a and b) Fluorescence anisotropy (Right) and LD (Left) of E in water (a) and ethanol (b). (a and b) The fluorescence anisotropy E was measured with excitation at 500 nm and emission detected at 645 nm. (Left) The measured parallel (decay) and perpendicular (rise) signals. (c and d) The LD and TA of E in water (λexc = 500 nm; λprobe = 585 nm and 645 nm).
For a quantitative analysis of the viscosity dependence of the orientational relaxation time, we used the Stokes–Einstein–Debye model (see ref. 21). In this model, the rotational diffusion time τr is related linearly to the effective volume (V) of the molecule and the viscosity (η) of the solvent at a given temperature (T): τr = Vη/(kT), in which k is the Boltzmann constant. The viscosities of the alcohols were taken from the literature (22). We found a linear relationship between τr and the solvent viscosity for the alcohols studied and obtained the volume (from the slope of τr vs. η) to be 570 Å3. The volume of the E cation was estimated from ab initio density calculations to be 360 Å3 by using a minimum basis set (STO-3G). With this volume, we obtained the following experimental (and calculated) orientational relaxation times: MeOH (η = 0.597 centipoise), 71 ps (52 ps); EtOH (η = 1.200 centipoise), 146 ps (106 ps); and BuOH (η = 2.948 centipoise), 398 ps (259 ps). It is known that the effective volume involves solvent shell(s), and the effectiveness depends on the shape of the molecule and whether the boundary condition is “slip” or “stick” (see ref. 23, references therein, and ref. 24).
To investigate the orientational relaxation further, we also measured the LD. The wavelength dependence of the TA and LD are shown in Fig. 3 c and d. The LD signals are positive at shorter wavelengths (585 nm) but become negative at longer wavelengths (645 nm), as described above for the TA signal. The LD signal decays with time, again consistent with the results of the anisotropy at different wavelengths, mainly with the 70-ps component. The minor component (≈10%) of 5 ps, as discussed above, can be related to the rotation of the phenyl group, recalling the ≈7-ps component in the TA signal at 665 nm (Fig. 2). Our zindo calculations (see below) indicate that the direction of the transition dipole moments for probing depends on the twisting angle of the phenyl group.
The huge differences in the amplitudes of the TA and LD signals (Fig. 3) are entirely consistent with the fact that the observed signal is composed of both the absorption and the stimulated-emission components, as discussed above. Because the gain observed by the stimulated emission involves the same transition moment (i.e., that of the S1 ← S0 transition), its anisotropy at t = 0 must be 0.4, because r(t) = 0.4⋅P2(cosϑμ1,μ2) ⋅ exp[−t/τr] (ref. 23 and references therein). In contrast, the absorption of S1 to Sn involves a different transition moment, and the initial anisotropy at t = 0 could be much smaller. Indeed, this behavior is evident in our observed transients. However, the decay of the signal is still 70 ps, consistent with the aforementioned results and indicating the probing of the orientational changes of E* in the solvent.
The zindo calculations predict the possible transitions from the S1 state of E to higher states and their transition dipole moments with respect to the initial transition dipole moment of the excitation; two transitions (S4 ← S1 and S7 ← S1) with substantial oscillator strengths and approximately the appropriate energy have been found that could account for the TA signal probed between 585 and 665 nm. The angles between the dipole moments of the two transitions and the initial excitation dipole moment are about 25° and 49°, respectively. The corresponding anisotropies are 0.25 and 0.05, respectively (i.e., much lower than 0.4). The small values of the anisotropies and the presence of multiple transitions account for the observed LD and TA behavior as a function of wavelength.
As a final test, we examined the effect of deuterium [H2O vs. 2H2O (D2O)] on the observed anisotropy and fluorescence lifetime. With a slight deviation (≈10°) from the magic-angle condition, as expected, we observed the 70-ps component with an amplitude of about 30%. In D2O, we observed a substantial increase in the fluorescence lifetime (by a factor of ≈4–5), in agreement with the report by Olmsted and Kearns (20), but with no change in the 70-ps decay. This result is consistent with the orientational relaxation picture discussed above. The change in the fluorescence lifetime when changing H2O to D2O is known to involve excited-state proton transfer from E* to water (20) and is common in many systems.
From the studies described, we conclude that there are two key time scales for E in water besides the usual fluorescence decay of 1.6 ns. These are the solvation process, which occurs in 1 ps, and the orientational relaxation time of E, which was determined to be 70 ps.
ET Between E and Mononucleotides.
The molecular structures are displayed in Scheme 1. Note that the base in ZTP differs from the natural guanine base only by one atom: the nitrogen in the 7-position of GTP is replaced by a C—H group in ZTP. This small structural change reduces the oxidation potential by 0.3 V. Electrochemical studies (11) determined that the free energy change ΔG (for ET) is ≈−0.2 eV (E*/ZTP) and +0.1 eV (E*/GTP). Hence, quenching is apparent with ZTP but not GTP. Energy transfer does not seem to contribute to quenching, given the absence of spectral overlap between donor and acceptor and the lack of concentration dependence on the rates. Likewise, proton transfer does not account for significant quenching, as evidenced by the lack of a D2O effect. Because ZTP and GTP are different only by one atom, other nonradiative processes could be excluded.
Fig. 4 shows the effect of GTP and ZTP on the TA probed at 400 nm (at the magic angle of 54.7°). At this probe wavelength, we observe the lifetime decay of E (1.6 ns). Although the lifetime increased in the presence of GTP, the lifetime is shortened dramatically in the presence of ZTP. The decay of the excited-state population becomes biexponential, and the amplitude of the new short time component (70 ps) depends strongly on the ZTP concentration. However, the decay time (70 ps) is essentially unchanged (within the accuracy of ≈10 ps).
Figure 4.

The TA (at the magic angle) of E in water (b) and with 5 mM GTP (a), 2 mM ZTP (c), and 5 mM ZTP (d); excited at 500 nm and probed at 400 nm; see Methodology for details about the medium used, water for E, and buffer for ZTP and GTP with E.
This new decay caused by the presence of ZTP and the change of its amplitude (not time constant) with concentration elucidates the dynamical model describing the ET process. From the transients in Fig. 4, it is clear that the nanosecond component is present even when ZTP is quenching the excited state. Accordingly, the ensemble is a two-component system in equilibrium, one made of complexes of E⋅ZTP and the second of free E. The complexed species, on fs excitation, reorient in order for ET to take place. Thus, the picture is
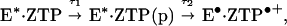 |
where the first step involves reorientation to reach the preferred (p) geometry for ET; note that E* is actually the excited cation, i.e., E+*.
The time scales of the two elementary steps can be separated with knowledge of the population and reorientation transient decays. For this reason, we studied the LD of E in the presence of ZTP and GTP (Fig. 5) and also the TA anisotropy [r(t)]. For the LD, we observe a 70-ps component for the E*⋅ZTP system, and a 220-ps (60%) component for E*⋅GTP; a 30% component shows a 70-ps decay, and the remaining 10% is the 5-ps component discussed above. The anisotropy gives a decay time of ≈400 ps or more for E*⋅ZTP in contrast to a 250-ps (i.e., 220-ps, corrected for the nanosecond lifetime) decay for E*⋅GTP. The fact that the anisotropy decay of E*⋅ZTP (quenching in 70 ps) is longer than that of E*⋅GTP (not quenching) indicates the correlation of reorientation with quenching, i.e., that fast-orienting E*⋅ZTP complexes have a higher probability of quenching, leaving only the more slowly orienting molecules to be observed. The τ2-type process must drain the population of E*⋅ZTP. Hence, in this case, the rate-determining step in the process is the step in which the system is brought into the preferred structure. τ2 is much shorter than τ1. Only in the presence of the τ2-type process does the reorientation dynamics appear in the TA, even at 54.7°. The concentration of the intermediate E*⋅ZTP(p) is very small because of the small ratio between τ2 and τ1.
Figure 5.

The LD of E excited at 500 nm and probed at 645 nm with 10 mM ZTP (red), 10 mM GTP (blue), and pure water (black). The Inset shows the corresponding TA signals.
Steady-state UV-visible absorption and NMR spectra indicate that E forms complexes with GTP and ZTP in aqueous solution. Previous experiments (11) have shown that only ZTP (and not GTP) can act as a quencher with respect to the excited E. We have used the concentration-dependent shift in the absorption band to determine the association constant (equilibrium constant) in the ground state, estimated to be ≈300 M−1 in both systems, by using the Hildebrand–Benesi formalism (25). In our experiments, we used an E concentration of 100 μM and a GTP/ZTP concentration of 10 mM, corresponding to ≈70% complexed and ≈30% uncomplexed E. These values are in agreement with the amplitudes shown in Fig. 4. Note that at the concentration of E used, no significant dimer formation occurs (26); spectroscopically, we also showed that at higher salt concentrations, the dimer formation becomes observable.
The following questions are important: what is the nature of the orientational motion leading to quenching, and why is the quenching time of E*⋅ZTP similar to the orientation time of E* in water? The E* molecule can reorient on the “substrate” ZTP as a result of collisions with water molecules in a diffusive process; the time scale of 70 ps is much longer than the inertial and solvation time scales. Thus, there will be two types of motions of the E* molecule, one that is “internal” in the complex and another that is a “global” cooperative motion involving the entire complex, because E interacts with ZTP. The latter occurs on a longer time scale relative to the former. Given that the internal motion in the complex structure is controlled by a diffusive interaction with water molecules, it is reasonable to expect the similarity in time scales for E* orientation in water and E*⋅ZTP quenching. However, the absence of a 70-ps decay in the r(t) of E*⋅ZTP suggests that the motion in the complex on the time scale of 70 ps does not significantly alter the direction of the transition dipole in the process toward ET (note that the decay of the correlation function for E* in water is single exponential). We estimate that the “average” angle for trajectories to reach the preferred geometry is close to 45°. The decay of the anisotropy at long times (≥400 ps), therefore, reflects the changes caused by the global motion (of the remaining population) of the complex and/or the slow relative motions of the two partners with dipole directional change. For E*⋅GTP, the ≈250-ps anisotropy decay reflects the global motion of the entire population.
In the ground state, GTP and ZTP form complexes with E of comparable, albeit weak stability, as evidenced by the similarities in their association constants. In the absence of water, the calculated structures for both types of complexes are determined strongly by the electrostatic interaction of the polar carbonyl group on the base and the E cation. In fact, this electrostatic interaction between both moieties leads to an orientation where the C⩵O group is pointed toward the E, providing no overlap between the two π-systems. Because these calculations do not take interactions with water into account, it is possible that E and the nucleotides are already in a stacked geometry but not in an orientation favorable for ET. However, our AM1 calculations (in vacuum) of the binding energies give ≈10 kcal/mol for E⋅ZTP and ≈13 kcal/mol for E⋅GTP, whereas, from the equilibrium constant, we obtained ≈3 kcal/mol for both. Regardless of the actual geometries required, these studies point out the sensitivity of ET in this system to orientation, an issue that is likely to be critical in elucidating mechanisms for ET processes mediated by DNA.
Future studies should include different DNA systems and molecular structure and dynamical calculations in the presence of water.
Acknowledgments
We thank Chris Treadway for preparing the E/mononucleotide samples and Dr. J. Spencer Baskin for valuable discussions and very helpful comments on the manuscript. We also thank the two referees for their useful suggestions. T.F. is grateful for financial support from the Deutsche Forschungsgemeinschaft. This work was supported by the National Science Foundation.
ABBREVIATIONS
- E
ethidium
- ET
electron transfer
- LD
linear dichroism
- TA
transient absorption
- ZTP
7-deaza-2′-deoxyguanosine triphosphate
References
- 1.Le Pecq J-B, Paoletti C. J Mol Biol. 1967;27:87–106. doi: 10.1016/0022-2836(67)90353-1. [DOI] [PubMed] [Google Scholar]
- 2.Burns V W F. Arch Biochem Biophys. 1969;133:420–424. doi: 10.1016/0003-9861(69)90471-8. [DOI] [PubMed] [Google Scholar]
- 3.Douthart R J, Burnett F W, Beasley F W, Frank B H. Biochemistry. 1973;12:214–220. doi: 10.1021/bi00726a006. [DOI] [PubMed] [Google Scholar]
- 4.Bresloff J L, Crothers D M. J Mol Biol. 1975;95:103–123. doi: 10.1016/0022-2836(75)90339-3. [DOI] [PubMed] [Google Scholar]
- 5.Waring M J. J Mol Biol. 1965;13:269–282. doi: 10.1016/s0022-2836(65)80096-1. [DOI] [PubMed] [Google Scholar]
- 6.Tuite E, Nordén B. Bioorgan Med Chem. 1995;3:701–711. doi: 10.1016/0968-0896(95)00061-k. [DOI] [PubMed] [Google Scholar]
- 7.Millar D P, Robbins R J, Zewail A H. Proc Natl Acad Sci USA. 1980;77:5593–5597. doi: 10.1073/pnas.77.10.5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millar D P, Robbins R J, Zewail A H. J Chem Phys. 1982;76:2080–2094. [Google Scholar]
- 9.Fujimoto B S, Schurr J M. Nature (London) 1990;344:175–178. doi: 10.1038/344175a0. [DOI] [PubMed] [Google Scholar]
- 10.Delvin P R, Cook D N, Pon N G, Bauer W R, Klein M P, Hearst J E. Science. 1992;255:82–85. doi: 10.1126/science.1553534. [DOI] [PubMed] [Google Scholar]
- 11.Kelley S O, Barton J K. Chem Biol. 1998;5:413–425. doi: 10.1016/s1074-5521(98)90158-2. [DOI] [PubMed] [Google Scholar]
- 12.Kelley S O, Holmlin R E, Stemp E D A, Barton J K. J Am Chem Soc. 1997;119:9861–9870. [Google Scholar]
- 13.Baguley B C, LeBret M. Biochemistry. 1984;23:937–943. doi: 10.1021/bi00300a022. [DOI] [PubMed] [Google Scholar]
- 14.Baguley B C. Biophys Chem. 1990;35:203–212. doi: 10.1016/0301-4622(90)80009-v. [DOI] [PubMed] [Google Scholar]
- 15.Brun A M, Harriman A. J Am Chem Soc. 1992;114:3656–3660. [Google Scholar]
- 16.Fromherz P, Rieger B. J Am Chem Soc. 1986;108:5361–5362. [Google Scholar]
- 17.Sommer J H, Nordlund T M, McGuire M, McLendon G. J Phys Chem. 1986;90:5173–5178. [Google Scholar]
- 18.Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Zakrzewski V G, Montgomery J A, Jr, Stratmann R E, Burant J C, et al. gaussian 98. Pittsburgh: Gaussian; 1998. , Version A.3. [Google Scholar]
- 19.Jimenez R, Fleming G R, Kumar P V, Maroncelli M. Nature (London) 1994;369:471–473. [Google Scholar]
- 20.Olmsted J, III, Kearns D R. Biochemistry. 1977;16:3647–3654. doi: 10.1021/bi00635a022. [DOI] [PubMed] [Google Scholar]
- 21.Millar D P, Shah R, Zewail A H. Chem Phys Lett. 1979;66:435–440. [Google Scholar]
- 22.Weast R C, editor. CRC Handbook of Chemistry and Physics. 67th Ed. Boca Raton, FL: CRC Press; 1986–1987. pp. F37–F44. [Google Scholar]
- 23.Eisenthal K B. Acc Chem Res. 1975;8:118–124. [Google Scholar]
- 24.Hu C M, Zwanzig R. J Chem Phys. 1974;60:4354–4357. [Google Scholar]
- 25.Benesi H A, Hildebrand J H. J Am Chem Soc. 1949;71:2703–2707. [Google Scholar]
- 26.Guenza M, Cuniberti C. Spectrochim Acta, Part A. 1988;44:1359–1364. [Google Scholar]