

Abstract
Fluorescence-based assay technologies play an increasing role in high-throughput screening. They can be classified into different categories: fluorescence polarization, time-resolved fluorescence, fluorescence resonance energy transfer, and fluorescence correlation spectroscopy. In this work we present an alternative analytical technique for high-throughput screening, which we call confocal fluorescence coincidence analysis. Confocal fluorescence coincidence analysis extracts fluorescence fluctuations that occur coincidently in two different spectral ranges from a tiny observation volume of below 1 fl. This procedure makes it possible to monitor whether an association between molecular fragments that are labeled with different fluorophores is established or broken. Therefore, it provides access to the characterization of a variety of cleavage and ligation reactions in biochemistry. Confocal fluorescence coincidence analysis is a very sensitive and ultrafast technique with readout times of 100 ms and below. This feature is demonstrated by means of a homogeneous assay for restriction endonuclease EcoRI. The presented achievements break ground for throughput rates as high as 106 samples per day with using only small amounts of sample substance and therefore constitute a solid base for screening applications in drug discovery and evolutionary biotechnology.
Confocal fluorescence methodologies are based on the detection of laser-induced fluorescence of dye-tagged single molecules in a very small focal volume element of approximately 1 fl. Among confocal methodologies, fluorescence correlation spectroscopy (FCS) (1–3) is a well established method, and for the last few years it has also been used in high-throughput screening (HTS) (4–6). A further development was dual-color fluorescence cross-correlation spectroscopy (4, 7, 8) (dual-color FCS). Because it combines two different fluorophores, dual-color FCS improves on conventional FCS in terms of analysis speed, specificity, and sensitivity. For these reasons, we recently adapted it to HTS of biomolecular samples (rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy RAPID FCS) (9). The new method presented in this article resembles dual-color FCS with regard to dual-color excitation and confocal single-photon detection. Besides some modifications concerning the laser source and a controlled external enhancement of concentration fluctuation, we are introducing a new data evaluation procedure for a specific detection of double-fluorescent molecules. This confocal fluorescence coincidence analysis (CFCA) has clear advantages over RAPID FCS concerning analysis speed and suitability for screening. As in RAPID FCS analysis, the use of two different fluorescent labels with separated emission spectra generally allows monitoring of any reaction in which a chemical bond or physical association between those fluorophores is established or broken. To test CFCA and to compare it with previous RAPID FCS measurements, we chose a well characterized homogeneous assay for restriction endonuclease EcoRI (8) and determined the relation between analysis time and accuracy of detection of enzyme activity.
MATERIALS AND METHODS
Homogeneous EcoRI Assay.
HPLC-purified 66-nt oligonucleotides Cy5ATGGCTAATGACCGAGAATAGGGATCCGAATTCAATATTGGTACCTACGGGCTTTGCGCTCGTATC and RhGGATACGAGCGCAAAGCCCGTAGGTACCAATATTGAATTCGGATCCCTATTCTCGGTCATTAGCCAT (MWG-Biotech, Ebersberg, Germany) that were 5′-labeled with fluorophores Cy-5 (Absmax/Emmax: 649/670 nm) and Rhodamine Green (499/521 nm) were hybridized to reveal a double-stranded DNA (dsDNA) molecule containing the EcoRI recognition site (underlined) framed by two spectrally distinguishable fluorophores. Endonucleolytic assays were carried out for 1 hr at 37°C with 10 nM labeled DNA substrate and 0.25 unit/μl EcoRI (Stratagene) in 150 mM KOAc/37.5 mM Tris acetate, pH 7.6/15 mM MgOAc/0.75 mM 2-mercaptoethanol/515 μg/ml BSA/0.05% Triton X-100/0.5% glycerol.
CFCA Setup.
The optical arrangement is our design and is based on the setup of dual-color FCS (7) (Fig. 1). Main modifications are a multiline laser arrangement to provide two different excitation wavelengths with the same resonator, an on-line data evaluation for sensitive and specific determination of coincidently occurring fluorescence fluctuations at two wavelengths, and a piezo-driven rapid movement of the sample.
Figure 1.

CFCA setup. Fluorescence excitation was achieved by epi-illumination of a water-immersion objective (UPLAPO 60×/1.2 W; Olympus, Tokyo) with radiation of wavelengths 476/483 nm and 647 nm from a krypton ion laser (INNOVA 90-K; Coherent, Palo Alto, CA) operating in multiline mode. The dichroic beamsplitter A (AHF Analysentechnik, Tübingen, Germany) reflected at <502 nm and 585–655 nm and transmitted at 502–585 and >655 nm. Additional laser lines at 531 nm and 568 nm were blocked with a custom-made excitation/notch filter (>OD 5; AHF Analysentechnik). Appropriate relative laser excitation power for both wavelengths was obtained by the combination of an absorption glass filter (BG 40; Andover Corporation, Salem, NH) and an attenuation filter (OD 0.6; Spindler & Hoyer, Göttingen, Germany). The sample holder was connected to a high-speed two-dimension piezo actor (Piezosystem Jena), which in turn was mounted onto a high-precision x–y mechanical scan table (Märzhäuser). Fluorescence photons were separated at dichroic beamsplitter B (585DCLP02; Omega Optical, Brattleboro, VT) and, after filtering in the red (667EFLP; Omega) and the green channel (535RDF45; Omega), were imaged to two avalanche photo diodes (APD) (SPCM-AQ 131-FS; EG & G, Quebec). Digital pulses of APDs were recorded either by a dual input multiscaler PC card (MCD-2; FAST ComTec, München, Germany) for analysis of time traces at high temporal resolution, or by an on-board processor PC card (Adwin-9LD; Jäger Meβtechnik, Lorsch, Germany), which was programmed to perform on-line data processing. Alternatively, for comparison with dual-color FCS, the APD pulses were crosscorrelated by a PC correlator card (ALV-5000 multiple-τ; ALV, Langen, Germany).
One-Beam Two-Color Excitation.
Using one laser in multiline mode rather than two distinct laser beams facilitates the adjustment procedure and improves the long-term stability of the confocal setup decisively. To characterize the chosen optical setup, dual-color fluorescence cross-correlation curves of the test assay were measured. The results are shown in Fig. 2. The experimental data curves were fitted by using a three-dimensional model for a single diffusing species (10):
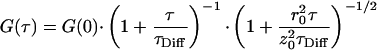 |
1 |
G(0) denotes the correlation amplitude at τ = 0, which is proportional to the concentration of double-labeled molecules in the observed rotary elliptical volume element with half-axis z0 parallel to the laser beam axis and radius r0. The parameter τDiff is inversely related to the diffusion coefficient D, according to the relation τDiff = r02/4D. The fitting procedure was carried out by using the software package microcal origin 4.10 (Microcal Software, Northampton, MA). Table 1 summarizes the fit results. The ratio of the cross-correlation amplitudes G(0) resulting from an uncleaved dsDNA substrate (‘negative’ sample) and from an identical but EcoRI-cleaved dsDNA substrate (‘positive’ sample), G(0)neg/G(0)pos, represented a measure of the capability of the chosen optical setup to detect the considered cleaving reaction. In the present case, this ratio was nearly 4. All measurements described in the remainder of this article were performed by using the excitation power 1.3 mW at 476/483 nm and 0.6 mW at 647 nm, which corresponded to average count rates of approximately 5 × 105 counts per second in both detection channels.
Figure 2.

Dual-color FCS curves measured with the described confocal setup (correlation times 60 s) and results of the fitting procedure (see Eq. 1). The parameters obtained from the fitting routine are given in Table 1. The two pairs of curves correspond to two different laser excitation powers (measured at the objective aperture). Each pair describes the dual-color FCS curve measured from a sample of 10 nM double-labeled dsDNA molecules (upper curves) and the curve measured from an equivalent sample after EcoRI endonuclease treatment (lower curves), respectively. [Smaller G(0) values at lower excitation power resulted from insufficient saturation of the sample solution.] The experimental cross-correlation curves and the results of the fitting procedure indicated that the optical setup used was well suited to perform a dual-color confocal fluorescence coincidence analysis, as it is presented in this work.
Table 1.
Parameters resulting from the fit of the experimental fluorescence cross-correlation spectroscopy data shown in Fig. 2
| Excitation power, mW | λ, nm | dsDNA sample | G(0) | τDiff, ms | G(0)negG(0)pos |
|---|---|---|---|---|---|
| 1.3 and | 476/483 | Uncleaved “negative” | 0.0194 | 1.35 | 3.7 |
| 0.6 | 647 | Cleaved “positive” | 0.00522 | 0.791 | |
| 0.25 and | 476/483 | Uncleaved “negative” | 0.0109 | 0.998 | 3.8 |
| 0.15 | 647 | Cleaved “positive” | 0.00286 | 0.894 |
The fit function is given in Eq. (1). The ratio z0/r0 was determined from autocorrelation calibration measurements by using a solution of free dye and was fixed to 5. The ratio of the cross-correlation amplitudes G(0) resulting from untreated (“negative”) and EcoRI-treated (“positive”) dsDNA substrates, G(0)neg/G(0)pos served as a measure of the capability of the chosen optical setup to detect the cleaving reaction and was the essential parameter in the context of this work. For both sets of applied laser excitation energies this ratio was nearly the same.
Data Evaluation.
The aim of CFCA data evaluation is to identify fluctuations that arise coincidently in the two different spectral emission ranges, thereby quantifying the number of double-labeled fluorescent molecules. There is a variety of possible algorithms that may solve this task. As a first approach, we tested here the normalized product sum algorithm:
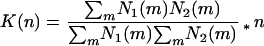 |
2 |
K(n) represents the coincidence value as a measure of the frequency of coincident events in the two detection channels. N1(m) and N2(m) are the number of counts at the different emission ranges in time channel m, and n is the total number of time channels in the trace. The analysis times are determined by the time channel width and the number of channels n. This data recording procedure minimizes the readout parameter to one value without any further fitting routine, as it is required with FCS analysis.
Piezo-Driven Sample Movement.
The sample holder was connected to a high-speed two-dimensional piezo actor (Piezosystem Jena, Germany), which in turn was mounted onto a high-precision x–y mechanical scan table (Märzhäuser, Wetzlar, Germany). In this work, the sample was moved with frequencies up to 250 Hz. The enforced sample movement relative to the focal volume was done (i) to avoid the accumulation of photobleached fluorophores in a zone around the focal volume, thereby increasing the effective fluorophore concentration, and (ii) to enhance the number of fluorescent particles traversing the focal volume in a given time. Concomitantly, the residence time values of the molecules in the focal element, τres, should have been shifted to shorter τ values. This shortening was verified by cross-correlation analysis (results not shown here): Increased oscillation frequencies lead to correlation curves shifted to lower correlation times τ, which corresponded to shorter mean residence times.
RESULTS AND DISCUSSION
To characterize CFCA, we examined essential parameters as time channel width, frequency of oscillation, and analysis time per sample systematically (Figs. 3 and 4). K resulting from the normalized product sum algorithm (cf. Eq. 2) served as a measure of coincidence. For each parameter combination, a set of 300 measurements of a model sample (10 nM double-labeled dsDNA) was evaluated. To characterize the resulting distributions of K, Gaussian curve fits were applied (cf. also Fig. 5). The extracted standard deviation σ was standardized by a division by the distance Δxc between the actual center point (xc) and the center point of an ideal sample with no coincidence at all (xc,0 = 1.0). This relative standard deviation σ/Δxc was then considered as a measure of the detection error. Generally, the detection error strongly depended on oscillation frequency and analysis time. In contrast, changing the time channel width had only a minor influence; only at the highest oscillation frequencies applied, which corresponded to shortest molecular residence times in the observation volume, did the range of optimal time channel widths become quite narrow. Particularly noteworthy was the large decrease of the detection error in a sample oscillating at 3 Hz compared with a resting sample. We assume this phenomenon to be caused mainly by suppression of photobleaching effects.
Figure 3.

Relative standard deviation σ/Δxc of Gaussian curve fits applied to distributions of K as a function of the time channel width at different frequencies of sample oscillations: K was evaluated from multiscaler time traces with analysis times per sample of 500 ms. At every frequency, there was a range of minimum σ/Δxc in the central part of the covered time channel width. At time channel widths that were in the order of magnitude of the triplet state lifetime (<5 μs) or that were larger than the average residence time τres (which was a function of oscillation frequency), the relative standard deviation increased significantly. The effect of increased frequency was even more pronounced. As the frequency increased, σ/Δxc diminished drastically. Simultaneously, the plateau of time channel width at minimum relative standard deviation narrowed down to the range of 10–30 μs at 216 Hz.
Figure 4.

Relative standard deviation σ/Δxc of Gaussian curve fits applied to distributions of K vs. sample oscillation frequency at analysis times ranging from 50 to 500 ms. The abscissa indicates the frequency in y-direction, which was superimposed by a constant frequency of 3 Hz in x-direction (except for the oscillation-free case). Coincidence analysis was performed by using the on-board processor PC card with a time channel width of 12.5 μs. The curves showed similar courses with a large reduction in relative standard deviation when the oscillation of the sample was started. Toward higher frequencies, a further moderate decrease could be noticed. At shortened analysis times the curves were shifted in parallel toward increasing σ/Δxc.
Figure 5.

Histogram plots of 300 measurements of the coincidence quantity K at sample oscillation frequencies 216 Hz (y) and 3 Hz (x) (superimposed) in samples containing pure substrate (10 nM double-labeled dsDNA, black bars) and cleaved products (endonuclease-treated 10 nM double-labeled dsDNA, gray bars), respectively. The slices (A–D) are related to different analysis times. The distributions were fitted with Gaussian functions. As a measure of detection error, the relative standard deviation σ/Δxc and the overlap in percentage between corresponding fit functions were calculated.
The described properties of CFCA show the great importance of the presented new technique for fast and reliable yes-or-no decisions in screening applications. The procedure of measuring the number of coincident events in two distinct time traces combined with piezo-driven sample oscillations leads to a considerable reduction of the tolerance of spot checks in investigated biochemical systems. By rapidly assaying a large number of positive and negative samples, we examined the number of false negative and false positive determinations in relation to the analysis time (Fig. 5). At analysis times of 200 ms, the overlap between substrate and product distributions amounted to less than 0.2%. At 100 ms, the overlap was 1.4%, which still corresponded to a tolerable number of false negative samples. Even at 50 ms, the intersection of the distributions amounted to only 8%. The presented results constitute a considerable advance in the effort to shorten the analysis times for screening biomolecular samples. Up to now, RAPID FCS has achieved the shortest analysis times in this context (9). Compared with RAPID FCS, the results presented here lead to a further increase in analysis speed of up to 10 times.
CONCLUSIONS
The limited throughput rate is still the main bottleneck of screening approaches: In drug discovery, large libraries of compounds of natural or combinatorial origin are screened with regard to their effect on biomolecular interactions with medical significance (5, 6). Likewise, optimization of enzymes by means of evolutionary biotechnology requires screening of huge libraries of mutants to select very rare variants having increased or more specific catalytic activities (4). CFCA-based screening is especially suited to overcoming the remaining difficulties in this context. Its short analysis times make it possible to judge reliably up to one million samples per day, in combination with the well-known advantages of confocal fluorescence methodologies such as highest sensitivity, lowest consumption of reagents, and a very general applicability. Furthermore, the data evaluation algorithm not only evades fitting procedures ensuing the measurement (as in FCS), but also offers the possibility of adapting the analysis time to the actual experimental results on-line. Because of the method’s high performance, it is very well suited for miniaturization. For the future, this possibility of size reduction strongly suggests a comprehensive integration of CFCA-based screening into nanotechnology.
Acknowledgments
This work was supported by the German Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (Grant FK 0310739 A) and by Evotec BioSystems, Hamburg. T.W. thanks the Deutsche Forschungsgemeinschaft for a grant.
ABBREVIATIONS
- CFCA
confocal fluorescence coincidence analysis
- dsDNA
double-stranded DNA
- FCS
fluorescence correlation spectroscopy
- dual-color FCS
dual-color fluorescence cross-correlation spectroscopy
- HTS
high-throughput screening
- APD
avalanche photo diodes
- RAPID FCS
rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy
References
- 1.Magde D, Elson E L, Webb W W. Biopolymers. 1974;13:29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 2.Rigler R, Mets U, Widengren J, Kask P. Eur Biophys J. 1993;22:169–175. [Google Scholar]
- 3.Maiti S, Haupts U, Webb W W. Proc Natl Acad Sci USA. 1997;94:11753–11757. doi: 10.1073/pnas.94.22.11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auer M, Moore K J, Meyer-Almes F-J, Guenther R, Pope A J, Stoeckli K A. Drug Discovery Today. 1998;3:457–465. [Google Scholar]
- 6.Rogers M V. Drug Discovery Today. 1997;2:156–160. [Google Scholar]
- 7.Schwille P, Meyer-Almes F-J, Rigler R. Biophys J. 1997;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kettling U, Koltermann A, Schwille P, Eigen M. Proc Natl Acad Sci USA. 1998;95:1416–1420. doi: 10.1073/pnas.95.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koltermann A, Kettling U, Bieschke J, Winkler T, Eigen M. Proc Natl Acad Sci USA. 1998;95:1421–1426. doi: 10.1073/pnas.95.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rigler R, Widengren J. Bioscience. 1990;3:180–183. [Google Scholar]