

Abstract
A cancer treatment is described in which i.m. injection of plasmid DNA (pDNA) encoding murine interferon α (mIFN-α) leads to potent antitumor effects on primary and metastatic tumors in mice. Mice bearing s.c. B16F10 melanoma, Cloudman melanoma, or glioma 261 tumors were injected i.m. with mIFN-α pDNA. In all three tumor models, a significant reduction in tumor volume and enhancement of survival was found after IFN pDNA therapy. The mIFN-α pDNA could be injected as infrequently as once every other week and still produce a significant antitumor effect, and, in a metastatic tumor model, the therapy markedly reduced the number of lung tumor metastases. Depletion of immune cell subsets indicated that CD8+ T cells were required for the antitumor response. These studies demonstrate that primary and metastatic tumors can be treated systemically by i.m. injection of a plasmid encoding a cytokine gene.
Plasmid or viral vectors encoding a therapeutic gene, often an immunomodulatory or suicide gene, currently are undergoing evaluation in a number of cancer gene therapy trials (1, 2). One type of protocol is ex vivo gene therapy in which tumor cells are removed from the patient, transfected in vitro with the plasmid or viral vector, selected, amplified, and reinjected into the patient. Such patient-specific therapy is cumbersome and time-consuming. Another type of protocol is in vivo gene therapy in which a plasmid or viral vector is injected directly into the tumor or locally around the tumor site and allows the patient to be treated in a more timely fashion; however, many late-stage cancer patients have metastatic lesions in internal organs that are too numerous to treat and difficult to access.
In this report, we describe a gene therapy treatment for cancer that does not require ex vivo methods or local injection. Rather, we have found that i.m. injection of a plasmid DNA (pDNA) encoding a cytokine gene can have a significant antitumor effect. In these studies, mice bearing one of three different types of s.c. tumors had reduced tumor growth and increased survival after i.m. injection of a pDNA encoding murine interferon α (mIFN-α). Metastatic melanoma lung tumor nodules also were dramatically reduced after the mIFN-α pDNA therapy. T cell depletion studies suggested that the mIFN-α therapy induced a T helper 1 type of immune response that was required for the antitumor effect. The results of these studies suggest a novel therapeutic approach for the treatment of patients with advanced disease.
MATERIALS AND METHODS
Plasmids.
The mIFN-α cDNA was obtained by amplifying the coding sequence from the plasmid Rous sarcoma virus α1 (3), which was a generous gift from Paula Pitha-Rowe (Johns Hopkins University, Baltimore). The mIFN-α plasmid used in these studies, VR4111, was constructed by cloning the mIFN-α cDNA into the backbone vector VR1055 (4). VR1055 is derived from pUC19, with the β-lactamase (ampicillin-resistance) gene replaced by the aminoglycoside acetyltransferase (kanamycin-resistance) gene from pET9a (Novagen). VR1055 directs eukaryotic gene expression from a cassette containing the human cytomegalovirus (CMV) immediate early I gene promoter/enhancer, 5′ untranslated sequence, and the CMV intron A sequence. After these regulatory elements is a cloning polylinker for insertion of protein-coding sequences and a transcriptional terminator region derived from the rabbit β-globin gene.
The human interferon ω (hIFN-ω) gene was obtained by amplifying the coding sequence from human genomic DNA prepared from a fresh blood sample. The hIFN-ω plasmid, VR4151, was constructed by cloning the hIFN-ω gene into the VR1055 cloning vector.
pDNA Purification.
pDNA was prepared by bacterial fermentation (5) and purified by standard double CsCl-ethidium bromide gradient ultracentrifugation followed by ethanol precipitation and dialysis. All plasmid preparations were free of detectable RNA, and endotoxin levels were less than 0.06 endotoxin units/μg of plasmid DNA. The spectrophotometric A260/A280 ratios were between 1.75 and 2.0.
Cell Lines.
All culture medium was obtained from Life Technologies (Gaithersburg, MD), and all serum was obtained from HyClone. Murine B16F10 cells were a generous gift from F. Suzuki at the University of Texas (Galveston) and grown in RPMI medium 1640 and 5% fetal bovine serum (FBS). Murine Cloudman S91 cells (American Type Culture Collection, Manassas, VA) were grown in Ham’s F-10 medium with 25 mM Hepes/0.1 mM nonessential amino acids/1 mM sodium pyruvate/0.05 mM 2-mercaptoethanol/2.5% FBS/12.5% horse serum. Human melanoma UM449 cells were kindly provided by Gary Nabel at the University of Michigan (Ann Arbor) and were grown in RPMI medium 1640 with 10% FBS.
Murine glioma 261 tumor fragments were obtained from the Division of Cancer Treatment Tumor Repository (National Cancer Institute, Frederick Cancer Research and Development Center, Frederick, MD). The glioma 261 tumor fragments were grown by serial transplantation in the flanks of C57BL/6 mice (6). A cell line was established from minced tumor fragments and grown in Iscove’s tissue culture medium with 10% FBS.
In Vitro Transfections with IFN Plasmids.
UM449 cells were plated at a concentration of 2 × 105 cells per well in a six-well plate and incubated for 24 h. Medium was removed from the cells, which were washed with PBS followed by the addition of the pDNA and cationic lipid DMRIE/DOPE complex (1:1, 1 μg of each, 1 ml/well) in Optimem medium (Life Technologies). After incubation of 4–5 h at 37°C, 1 ml Optimem with 30% fetal calf serum (FCS) was added to each well, followed by the addition of 1 ml Optimem with 10% FCS the next day. Tissue culture supernatants were collected 48 h after the start of the in vitro transfection.
Cell Proliferation Assay.
In vitro transfection of UM449 cells with the mIFN-α and control pDNA was performed as described previously, and the supernatants were tested in a cell proliferation assay of murine tumor cell lines using the Boehringer Mannheim Cell Proliferation Kit II (XTT). Murine tumor cells were plated in 96-well plates at a concentration of 5 × 103 cells/ml for B16F10 cells and 5 × 104 cells/ml for the Cloudman S91 and glioma 261 cells. After 24 h, tissue culture supernatants from UM449 cells transfected in vitro with either mIFN-α or control pDNA were added to the wells. mIFN-α protein (ICN) was used as a positive control. The XTT labeling reagent was added 24–48 h later, and the OD at 490 nm was determined 6–24 h later. The percent reduction in cell proliferation was determined by the formula:
Antiviral Assay.
In vitro transfections were performed as described previously, and supernatants were collected from cells transfected with either the mIFN-α or control pDNA. An encephalomyocarditis (EMC) plaque-reduction assay was used to determine the antiviral activity of the supernatants (IIT Research Institute, Chicago). Briefly, tissue culture supernatants were added to L929 cells in 96-well plates (2.5 × 104 cells per well) and incubated for 24 h. The cells were washed and murine EMC virus was added at a multiplicity of infection of 0.04 and incubated for 24 h. The cells were fixed with 5% formalin and stained with 1% crystal violet. As a positive control, cells were incubated with mIFN-α/β reference standard (National Institutes of Health, Bethesda, MD) before the addition of EMC virus.
Murine Tumor Models.
C57BL/6, DBA/2, nude (nu/nu), and beige-nude (bg/nu/xid) female mice between the ages of 6 and 8 weeks were obtained from Harlan–Sprague–Dawley. All animal experiments were conducted in accordance with Vical’s Institutional Animal Care and Use Committee as well as the standards set forth in the National Research Council guidelines concerning animal care and use.
To establish s.c. B16F10 melanoma tumors, C57BL/6, nude, or nude/beige mice were injected s.c. with 104 B16F10 cells. The Cloudman melanoma model was established by s.c. injection of 105 Cloudman S91 cells in DBA/2 mice, and the glioma 261 model was established by s.c. injection of 5 × 105 glioma 261 cells in C57BL/6 mice. Tumor dimensions were determined by measuring with calipers (length × width × height), and the values were inserted into the formula: tumor volume (mm3) = 0.52 (length × width × height) (7).
To establish lung metastases of B16F10 melanoma, C57BL/6 mice were injected i.v. with 2 × 104 B16F10. On day 25 after tumor cell injection, the mice were sacrificed, and lungs were removed and fixed in 10% buffered formalin (Fisher Scientific) followed by counting of tumor nodules.
Intramuscular Injections.
Fifty micrograms of plasmid DNA in 50 μl of saline was injected into the rectus femoris muscle of each hind leg for a total DNA dose of 100 μg. The muscle injections were performed by using a sterile, 300-μl tuberculin syringe fitted with a 28G ½ needle (Becton Dickinson) modified with a plastic collar cut from a 200-μl micropipette tip. The collar length was adjusted to limit the needle from penetrating further than 2 mm into the rectus femoris muscle.
Determination of Serum Levels of IFN-ω.
To determine the serum levels of hIFN-ω, C57BL/6 mice received a single i.m. injection of 100 μg of hIFN-ω pDNA. Serum was collected every 2–3 days for 2 weeks after injection (four mice per day) and analyzed with a hIFN-ω ELISA kit (Alexis, San Diego), which was sensitive to 2 pg/ml.
Antibody Depletion of Immune Cell Subsets.
The anti-CD4 (clone GK1.5, rat IgG) and anti-CD8 (clone 2.43, rat IgG) hybridomas were obtained from the American Type Culture Collection. The anti-CD8 hybridoma was grown as ascites in nude mice, and the mAb was purified from ascites by using ion exchange chromatography (Harlan Bioproducts for Science, San Diego). The anti-CD4 hybridoma was grown in vitro with DMEM/10% FBS/low IgG, and the mAb was purified from tissue culture supernatant by ammonium sulfate precipitation to 30%. The protein pellet was resolubilized and dialyzed extensively in Dulbecco’s Ca2+/Mg2+-free PBS (Zymed).
For depletion of CD4+ and CD8+ T cells, mice were injected i.p. with 500 μg of either the anti-CD4 or anti-CD8 mAb. The mAbs were administered 1 day before each i.m. pDNA injection (twice per week for 3 weeks). Control tumor-bearing mice were injected i.p. with 500 μg of normal rat IgG (Sigma). To ensure complete depletion, sentinel mice were injected according to the same regimen and, once per week, spleens were collected, dissociated, and assessed for the presence of CD4+ and CD8+ T cells. Spleen cells were stained with fluorescein isothiocyanate-conjugated anti-CD4 and phycoerythrin-conjugated anti-CD8 mAbs (PharMingen) and analyzed by flow cytometry (Cytometry Research Services, San Diego). The depletion of CD4+ and CD8+ T cells was consistently greater than 98% as determined by flow cytometry.
Statistical Analyses.
Tumor volume was analyzed by using the Mann–Whitney U nonparametric statistical test to identify groups having significantly different tumor sizes. Mouse survival was analyzed by using a Kaplan–Meier survival plot followed by a Logrank (Mantel–Cox) test to identify significant differences in survival between groups. Differences were considered statistically significant when the P value was <0.05.
RESULTS
Biological Activity of mIFN-α pDNA.
To ensure that the mIFN-α pDNA used in these studies encoded biologically active mIFN-α, both cell proliferation and antiviral assays were conducted on supernatants from cultured cells that were transfected in vitro with the mIFN-α pDNA. Cell proliferation assays were performed to measure the inhibition of growth of several murine tumor cell lines after addition of supernatants from UM449 cells transfected with either the mIFN-α pDNA or the control pDNA (backbone plasmid without mIFN-α). The supernatants from the mIFN-α pDNA-transfected cells inhibited the proliferation of murine B16F10 melanoma, Cloudman melanoma, and glioma 261 cell lines by 40, 42, and 17%, respectively. An antiviral assay also was performed that evaluated the ability of the supernatants from the mIFN-α pDNA-transfected cells to protect murine L929 cells from infection by EMC virus. The antiviral assay identified 30,000 units/ml of mIFN-α in tissue culture supernatants after transfection with mIFN-α pDNA.
mIFN-α pDNA Treatment Inhibits Primary Tumor Growth.
The mIFN-α pDNA was evaluated for antitumor efficacy by i.m. injection of the plasmid into mice bearing s.c. tumors. Tumor-bearing mice were injected with 100 μg of mIFN-α pDNA or control pDNA, twice per week for 3 weeks, beginning on day 4 after tumor cell implant. C57BL/6 mice bearing s.c. B16F10 melanoma tumors and injected i.m. with mIFN-α pDNA had an 89% reduction in tumor volume by day 28 after tumor cell implant compared with control pDNA-treated mice (P = 0.0003) (Fig. 1a). Survival of the B16F10 tumor-bearing mice injected i.m. with mIFN-α pDNA was enhanced significantly, with 90% of the mIFN-α pDNA-treated mice surviving to day 39 of the study compared with only 10% of the control pDNA-treated mice surviving (P = 0.003) (Fig. 1b). B16F10 tumor-bearing mice injected i.m. with saline had the same rate of tumor growth as the mice treated with the control pDNA (data not shown).
Figure 1.

Antitumor efficacy of mIFN-α pDNA delivered by i.m. injection in three murine tumor models. Tumor growth and survival are shown for C57BL/6 mice bearing B16F10 melanoma (a and b) or glioma 261 (c and d) and DBA/2 mice bearing Cloudman melanoma (e and f). Mice were injected s.c. with 104 B16F10 melanoma cells, 5 × 105 glioma 261 cells, or 105 Cloudman melanoma cells on the flank (n = 8–10 mice per group). Beginning on day 4 after tumor cell implant, mice were injected i.m. with 100 μg of either the mIFN-α pDNA or the control pDNA twice a week for 3 weeks, for a total of six injections. A significant reduction in tumor volume (P ≤ 0.02) and a significant increase in survival (P ≤ 0.01) were found for all three tumor types treated with the mIFN-α pDNA compared with the tumor-bearing mice treated with the control pDNA. In a the dip in the tumor-growth curve of the control pDNA-treated mice from day 23 to 25 reflects euthanasia of one mouse bearing a tumor >6,000 mm3 on day 23.
To determine whether the mIFN-α pDNA therapy could be applied to other murine tumor models, the therapy was evaluated in C57BL/6 mice implanted with s.c. glioma 261. Mice bearing s.c. glioma and injected i.m. with mIFN-α pDNA twice per week for 3 weeks had a 71% reduction in tumor volume by day 35 compared with mice treated with the control pDNA (P = 0.01) (Fig. 1c). By day 53 of the study, 70% of the glioma-bearing mice treated with mIFN-α pDNA were alive compared with only 10% of the control pDNA-treated mice (P = 0.01) (Fig. 1d).
To ensure that the mIFN-α pDNA therapy was not unique to C57BL/6 mice, the treatment was evaluated in DBA/2 mice implanted with s.c. Cloudman melanoma. Mice bearing Cloudman melanoma and treated i.m. with mIFN-α pDNA had a 61% reduction in tumor volume by day 26 compared with the control pDNA-treated tumors (P = 0.02) (Fig. 1e). By day 42 after tumor cell injection, 70% of the mIFN-α pDNA-treated mice were alive compared with only 20% of the control pDNA-treated mice (P = 0.01) (Fig. 1f). In summary, although none of the mice became tumor-free, significant reductions in tumor growth and increases in survival were found after mIFN-α pDNA therapy in three different mouse tumor models. In addition, no IFN-related side effects were observed in any of the studies.
mIFN-α pDNA Dose Response in the B16F10 Melanoma Model.
A study was conducted to evaluate the optimal dose and regimen of administration of mIFN-α pDNA required to obtain antitumor efficacy. C57BL/6 mice bearing s.c. B16F10 tumors were injected i.m. with either 100 or 50 μg of mIFN-α pDNA or control pDNA over a 6-week period. Mice received pDNA injections twice per week, once per week, or once every other week. All injections began 4 days after the initial B16F10 tumor cell implant. Mice that received i.m. injections of 100 μg of mIFN-α pDNA for any of the treatment regimens had a significant reduction in tumor volume by day 19 of the study (P < 0.005) and a significant increase in survival compared with the controls (P < 0.007) (Fig. 2 a and b). As few as three injections of 100 μg of mIFN-α pDNA administered once every other week had a significant antitumor effect. In contrast, the 50-μg dose of mIFN-α pDNA revealed a dose response based on the frequency of injection. Mice treated once or twice per week with 50 μg of mIFN-α pDNA had a significant reduction in tumor volume by day 19 of the study (P < 0.03) and a significant increase in survival (P < 0.003) compared with the controls, whereas mice treated once every other week with 50 μg of mIFN-α pDNA had no significant antitumor response (Fig. 2 c and d).
Figure 2.

Dose response of the mIFN-α pDNA treatment in the B16F10 melanoma model. Tumor growth and survival are shown for C57BL/6 mice bearing s.c. B16F10 melanoma tumors and treated with either 100 μg (a and b) or 50 μg (c and d) of mIFN-α pDNA or control pDNA. Beginning on day 4 after s.c. injection with 104 B16F10 cells, mice were injected i.m. with either 100 or 50 μg of the mIFN-α pDNA or the control plasmid, twice per week, once per week, or once every other week for 6 weeks (n = 10 mice per group). Treatment of the mice with 100 μg of the mIFN-α pDNA for any of the regimens resulted in a significant reduction in tumor growth by day 19 (P < 0.005) and a significant increase in survival compared with the controls (P < 0.007). The mice receiving 50 μg of the mIFN-α pDNA once per week or twice per week also had a significant reduction in tumor growth by day 19 (P < 0.03) and a significant increase in survival (P < 0.003) compared with the controls. No significant effects on tumor growth or survival were found for the mice injected every other week with 50 μg of mIFN-α pDNA.
Therapy with mIFN-α pDNA Inhibits the Growth of Tumor Metastases.
Because metastatic tumors often pose a difficult therapeutic challenge, the mIFN-α pDNA therapy was evaluated for effects on tumor metastases. A metastatic tumor model was used in which C57BL/6 mice were injected i.v. with B16F10 melanoma cells to establish lung metastases. On day 4 after the tumor cell injection, the mice received i.m. injections of 100 μg of either mIFN-α pDNA or control pDNA twice weekly for 3 weeks. On day 25 after tumor cell injection, the mice were sacrificed and lung tumor nodules were counted. Although 90% of the mice treated with mIFN-α pDNA had less than 15 lung tumor nodules, only 30% of the mice treated with control pDNA had a similar low number of tumor nodules (Fig. 3). In fact, the majority of the control pDNA-treated mice (70%) had tumor nodules that were too consolidated and numerous to count, whereas none of the mIFN-α pDNA-treated mice had such extensive tumor growth. The number of lung metastases for the control pDNA-treated group was similar to the number of lung metastases for saline-treated mice (data not shown).
Figure 3.

Reduction in melanoma lung tumor metastases after treatment of mice with i.m. mIFN-α pDNA. C57BL/6 mice were injected i.v. with 2 × 104 B16F10 cells (n = 10 mice per group). Four days after tumor cell implant, mice were injected i.m. with 100 μg of either the mIFN-α or control pDNA twice a week for 3 weeks. On day 25 after tumor cell implant, mice were sacrificed and the lungs were fixed in formalin followed by counting of lung tumor nodules. (a) Number of lung tumor nodules per mouse in each treatment group. TNTC denotes lungs with nodules that were too numerous to count. Seventy percent of the mice injected with the control pDNA had lung tumor nodules that were too numerous to count, whereas 90% of the mice injected with mIFN-α pDNA had ≤12 lung tumor nodules. Representative formalin-fixed lobe of a lung from a mouse injected with the control pDNA (b) and the mIFN-α pDNA (c).
Serum Levels of IFN After i.m. Injection of IFN pDNA.
Because the i.m. mIFN-α pDNA therapy could be delivered as infrequently as once every other week for a total of three injections and still have potent antitumor effects, we reasoned that a single injection may result in serum levels of IFN. Since no mIFN-α ELISA was commercially available at the time of these studies and our attempts to develop an ELISA resulted in low sensitivity, we measured serum levels of another type I interferon, hIFN-ω, for which a sensitive ELISA was available. C57BL/6 mice received a single i.m. injection of 100 μg of hIFN-ω pDNA, and serum samples were collected every 2–3 days over a 2-week period and analyzed in the ELISA. Peak serum levels were found 6 days after injection (254 pg/ml), and expression continued to day 14 (55 pg/ml), the final time point of the study (Fig. 4). Thus, using a sensitive ELISA, IFN could be detected in the serum after a single i.m. injection of an IFN-encoding pDNA.
Figure 4.

Pharmacokinetics of serum hIFN-ω after a single i.m. injection of hIFN-ω pDNA. C57BL/6 mice received a single i.m. injection of 100 μg of hIFN-ω pDNA. Serum was collected every 2–3 days for 2 weeks after pDNA injection, and samples were analyzed in an ELISA specific for hIFN-ω. Each data point represents the mean hIFN-ω serum level of four mice. Peak serum levels of 254 pg/ml were found on day 6 after a pDNA injection, and expression continued to day 14, the last time point of the study. Mice injected with the control pDNA were negative in the IFN-ω ELISA.
Mechanism of the Antitumor Effect.
Because IFNs are known to have potent immunomodulatory effects, several studies were conducted to evaluate the role of immune cells in the mIFN-α-mediated antitumor response. To investigate the role of NK and T cells, mIFN-α pDNA therapy was evaluated in nude mice (T cell-deficient) and beige-nude mice [natural killer (NK)- and T cell-deficient] bearing s.c. B16F10 melanoma tumors. In these studies, mice received the same regimen of i.m. injections of mIFN-α pDNA (twice per week for 3 weeks) as in the earlier antitumor efficacy studies in the B16F10 melanoma model in C57BL/6 mice. Interestingly, there was neither a significant reduction in tumor volume nor enhancement of survival in the nude and beige-nude mice, suggesting that T cells are required for the antitumor response and NK cells alone could not mediate the antitumor effect (data not shown).
To further explore the role of T cells in the antitumor efficacy of mIFN-α pDNA therapy, C57BL/6 mice bearing s.c. B16F10 tumors were injected with depleting doses of mAbs specific for either CD4+ or CD8+ T cells. The mice were injected with mAbs 1 day before each i.m. injection of pDNA (twice per week for 3 weeks). By day 21 after tumor cell injection, the mIFN-α pDNA therapy had significantly reduced tumor growth (P < 0.003) and enhanced survival (P < 0.009) of both normal mice and mice depleted of CD4+ T cells, suggesting that CD4+ T cells were not required for the response (Fig. 5). In contrast, mice depleted of CD8+ T cells and injected with mIFN-α pDNA had tumor volumes and survival that were not significantly different from mice treated with the control pDNA, indicating a requirement for CD8+ T cells in the antitumor response.
Figure 5.

Evaluation of the role of CD4+ and CD8+ T cells in the mIFN-α pDNA antitumor response. Tumor growth (a) and survival (b) are shown for the mice bearing s.c. B16F10 and treated with mIFN-α or control pDNA. Beginning on day 4 after s.c. injection with 104 B16F10 tumor cells, mice were injected i.m. with 100 μg of either the mIFN-α or control pDNA, twice per week for 3 weeks. One day before each pDNA injection, the mice were injected i.p. with 500 μg of either anti-CD4 mAb (clone GK1.5, rat IgG), anti-CD8 mAb (clone 2.43, rat IgG), or normal rat IgG (n = 10 mice per group). Mice injected with the mIFN-α pDNA and treated with normal IgG had a significant reduction in tumor volume (P < 0.003) by day 21 and a significant increase in survival (P < 0.009) compared with the mice injected with control pDNA and treated with normal IgG. Mice injected with mIFN-α pDNA and treated with anti-CD4 mAb still had a significant reduction in tumor volume (P < 0.003) by day 21 and a significant increase in survival (P < 0.009) compared with the controls. In contrast, mice injected with mIFN-α pDNA and depleted of CD8+ T cells had no significant reduction in tumor volume or increase in survival compared with the controls.
DISCUSSION
This report demonstrates that primary and metastatic tumors in mice can be treated by distant i.m. delivery of a plasmid encoding a cytokine gene. In these studies, i.m. injection of a pDNA expressing mIFN-α resulted in a potent antitumor effect in three different mouse tumor models, including a marked reduction in the number of melanoma lung tumor metastases. This method differs from previous in vivo cancer gene therapies (1, 2) in that it does not require local injection or identification of existing tumor nodules, thus facilitating treatment of primary or metastatic tumors at distant sites.
Cancer gene therapy using pDNA encoding a cytokine gene has important advantages over injection of a bolus of the corresponding therapeutic protein. Recombinant IFN-α protein currently is used as adjuvant therapy for metastatic melanoma, and the treatment involves a high-dose, intensive regimen of i.v. injection of 10–20 million units (MU)/m2 5 days per week for 1 month followed by s.c. injection of 10 MU/m2 3 days per week for 11 months (8, 9). This dosing regimen, although producing clinical benefits, approaches the maximally tolerated dose and is very toxic. Frequent injection of the recombinant protein is required because peak serum levels occur by 1–8 h after protein injection via i.v., s.c., or i.m. routes, with negligible serum levels by 24 h (10–12). In mouse melanoma tumor studies demonstrating antitumor effects of mIFN-α protein, frequent injections of the protein similarly were required (13–15). In contrast, we have found that a single i.m. injection of IFN-ω pDNA in mice resulted in relatively stable serum levels over a 2-week period. Moreover, mIFN-α pDNA therapy in mice bearing melanoma tumors was effective even when pDNA injection occurred as infrequently as once every 2 weeks for a total of only three injections. By avoiding the sharp peaks and drops in serum levels that occur after bolus protein injection, pDNA therapy may sustain a lower-level but more stable concentration of IFN in the serum, possibly reducing the severity of side effects and prolonging the therapeutic effect.
IFN-α has been shown to have a multitude of antitumor effects including activation of immune cells, inhibition of cell proliferation, induction of cell differentiation, up-regulation of class I major histocompatibility complex antigens, inhibition of angiogenesis, and establishment of a T helper 1 (Th1)-type response (16–18). In the present study, the mechanism of the antitumor effect of systemic IFN-α appeared to involve primarily the induction of a Th1-type immune response. This was evidenced by the fact that depletion of CD8+ T cells, but not CD4+ T cells, abolished the antitumor effect of systemic mIFN-α therapy in mice bearing melanoma tumors. Although in vitro assays revealed the ability of mIFN-α to have direct antiproliferative effects on murine tumor cell lines, this did not appear to be the predominant effect in vivo.
In the present study, the IFN-α pDNA was injected directly into muscle, which differs considerably from previous studies of IFN-α cancer gene therapy in which pDNA or viral vectors were injected locally, intratumorally, or delivered by ex vivo methods (19–26). A number of other studies also have evaluated the effect of i.m. injection of a cytokine-expressing pDNA; however, none of these studies evaluated the effect of this type of therapy on tumor growth. In these studies, immunological effects were found after i.m. injection of pDNA encoding either interleukin (IL)-2, IL-4, IL-5, or transforming growth factor β (TGF-β) (27, 28). In several mouse models of disease, i.m. injection of a pDNA encoding TGF-B was found to have beneficial effects for the treatment of lupus (29), diabetes (30), and arthritis (31), and i.m. injection of a pDNA encoding IL-10 was found to have therapeutic effects in diabetes (32). Intramuscular injection of mIFN-α pDNA was demonstrated to have antiviral effects in mice infected with murine cytomegalovirus (33). Unlike the latter studies, we have demonstrated that i.m. cytokine pDNA can be effective in mouse tumor models. These results expand upon the earlier i.m. pDNA research by demonstrating that i.m. injection of pDNA encoding a cytokine gene can have potent antitumor effects on both solid and metastatic tumors.
The gene therapy method described in this report has several important advantages over traditional cancer treatments. First, unlike recombinant cytokine protein therapies, the lower, but sustained, serum levels of a therapeutic protein after i.m. injection of a pDNA encoding the cytokine gene may reduce side effects while still maintaining a therapeutic effect. Second, in contrast to current in vivo cancer gene therapy protocols, the method described here does not require identification of existing tumor nodules, thereby facilitating the treatment of small lesions in internal organs. In summary, the results of this research suggest a novel type of in vivo cancer gene therapy that may be used to treat primary tumors as well as to prevent the development of metastases in advanced cancer patients.
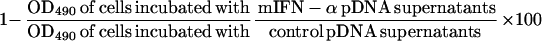
Acknowledgments
We thank W. Nishioka for providing the anti-CD4 antibody, M. Yankauckas for advice on the antibody depletion study, K. Tonsky and the Vivarium staff for excellent animal care, R. Newlin and L. Vahlsing for graphics, L. Sukhu and J. Stupack for tissue culture support, J. Meek and the Vical DNA production staff for preparation of plasmid DNA stocks, and M. Manthorpe for critical review of the manuscript.
ABBREVIATIONS
- IFN
interferon
- pDNA
plasmid DNA
- EMC
encephalomyocarditis
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Ross G, Erickson R, Knorr D, Motulsky A G, Parkman R, Samulski J, Straus S E, Smith B R. Hum Gene Ther. 1996;7:1781–1790. doi: 10.1089/hum.1996.7.14-1781. [DOI] [PubMed] [Google Scholar]
- 2.Roth J A, Cristiano R J. J Natl Cancer Inst. 1997;89:21–39. doi: 10.1093/jnci/89.1.21. [DOI] [PubMed] [Google Scholar]
- 3.Kelley K A, Pitha P M. Nucleic Acids Res. 1985;13:805–823. doi: 10.1093/nar/13.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartikka J, Sawdey M, Cornefert-Jensen F, Margalith M, Barnhart K, Nolasco M, Vahlsing H L, Meek J, Marquet M, Hobart P, et al. Hum Gene Ther. 1996;7:1205–1217. doi: 10.1089/hum.1996.7.10-1205. [DOI] [PubMed] [Google Scholar]
- 5.Horn N A, Meek J A, Budahazi G, Marquet M. Hum Gene Ther. 1995;6:565–573. doi: 10.1089/hum.1995.6.5-565. [DOI] [PubMed] [Google Scholar]
- 6.Saris S C, Solares G R, Wazer D E, Cano G, Kerley S E, Joyce M A, Adelman L S, Harling O K, Madoc-Jones H, Zamenhof R G. Cancer Res. 1992;52:4672–4677. [PubMed] [Google Scholar]
- 7.Tomayko M M, Reynolds C P. Cancer Chemother Pharmacol. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 8.Kirkwood J M, Strawderman M H, Ernstoff M S, Smith T J, Borden E C, Blum R H. J Clin Oncol. 1996;14:7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 9.Sondak V K, Wolfe J A. Curr Opin Oncol. 1997;9:189–204. doi: 10.1097/00001622-199703000-00015. [DOI] [PubMed] [Google Scholar]
- 10.Kirkwood J M, Ernstoff M S, Davis C A, Reiss M, Ferraresi R, Rudnick S A. Ann Intern Med. 1985;103:32–36. doi: 10.7326/0003-4819-103-1-32. [DOI] [PubMed] [Google Scholar]
- 11.Radwanski E, Perentesis G, Jacobs S, Oden E, Affrime M, Symchowicz S, Zampaglione N. J Clin Pharmacol. 1987;27:432–435. doi: 10.1002/j.1552-4604.1987.tb03044.x. [DOI] [PubMed] [Google Scholar]
- 12.Gutterman J U, Fine S, Quesada J, Horning S J, Levine J F, Alexanian R, Bernhardt L, Kramer M, Spiegel H, Colburn W. Ann Intern Med. 1982;96:549–556. doi: 10.7326/0003-4819-96-5-549. [DOI] [PubMed] [Google Scholar]
- 13.Bart R S, Porzio N R, Kopf A W, Vilcek J T, Cheng E H, Farcet Y. Cancer Res. 1980;40:614–619. [PubMed] [Google Scholar]
- 14.Iigo M, Sakurai M, Tamura T, Saijo N, Hoshi A. Cancer Res. 1988;48:260–264. [PubMed] [Google Scholar]
- 15.Fleischmann W R, Masoor J, Wu T Y, Fleischmann C M. J Interferon Cytokine Res. 1998;18:17–20. doi: 10.1089/jir.1998.18.17. [DOI] [PubMed] [Google Scholar]
- 16.Gutterman J U. Proc Natl Acad Sci USA. 1994;91:1198–1205. doi: 10.1073/pnas.91.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pestka S, Langer J A. Annu Rev Biochem. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 18.Pfeffer L M, Dinarello C A, Herberman R B, Williams B R G, Borden E C, Bordens R, Walter M R, Nagabhushan T L, Trotta P P, Pestka S. Cancer Res. 1998;58:2489–2499. [PubMed] [Google Scholar]
- 19.Zhang J F, Hu C, Geng Y, Selm J, Klein S B, Orazi A, Taylor M W. Proc Natl Acad Sci USA. 1996;93:4513–4518. doi: 10.1073/pnas.93.9.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang F Z, Hu C, Geng Y, Blatt L M, Taylor M W. Cancer Gene Ther. 1996;3:31–38. [PubMed] [Google Scholar]
- 21.Santodonato L, D’Agostini G, Santini S M, Carlei D, Musiani P, Modesti A, Signorelli P, Belardelli F, Ferrantini M. Gene Ther. 1997;4:1246–1255. doi: 10.1038/sj.gt.3300518. [DOI] [PubMed] [Google Scholar]
- 22.Santodonato L, Ferrantini M, Gabriele L, Proietti E, Venditti M, Musiani P, Modesti A, Modica A, Lupton S D, Belardelli F. Hum Gene Ther. 1996;7:1–10. doi: 10.1089/hum.1996.7.1-1. [DOI] [PubMed] [Google Scholar]
- 23.Kaido T, Bandu M T, Maury C, Ferrantini M, Belardelli F, Gresser I. Int J Cancer. 1995;60:221–229. doi: 10.1002/ijc.2910600216. [DOI] [PubMed] [Google Scholar]
- 24.Ferrantini M, Proietti E, Santodonato L, Gabriele L, Peretti M, Plavec I, Meyer F, Kaido T, Gresser I, Belardelli F. Cancer Res. 1993;53:1107–1112. [PubMed] [Google Scholar]
- 25.Ferrantini M, Giovarelli M, Modesti A, Musiani P, Modica A, Venditti M, Peretti E, Lollini P L, Nanni P, Forni G, Belardelli F. J Immunol. 1994;153:4604–4615. [PubMed] [Google Scholar]
- 26.Belldegrun A, Tso C L, Sakata T, Duckett T, Brunda M J, Barsky S H, Chai J, Kaboo R, Lavey R S, McBride W H, deKernion J B. J Natl Cancer Inst. 1993;85:207–216. doi: 10.1093/jnci/85.3.207. [DOI] [PubMed] [Google Scholar]
- 27.Tokui M, Takei I, Tashiro F, Shimada A, Kasuga A, Ishii M, Ishii T, Takatsu K, Saruta T, Miyazaki J I. Biochem Biophys Res Commun. 1997;233:527–531. doi: 10.1006/bbrc.1997.6485. [DOI] [PubMed] [Google Scholar]
- 28.Raz E, Watanabe A, Baird S M, Eisenberg R A, Parr T B, Lotz M, Kipps T J, Carson D A. Proc Natl Acad Sci USA. 1993;90:4523–4527. doi: 10.1073/pnas.90.10.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raz E, Dudler J, Lotz M, Baird S M, Berry C C, Eisenberg R A, Carson C A. Lupus. 1995;4:286–292. doi: 10.1177/096120339500400409. [DOI] [PubMed] [Google Scholar]
- 30.Piccirillo C A, Chang Y, Prud’homme G J. J Immunol. 1998;161:3950–3956. [PubMed] [Google Scholar]
- 31.Song X-Y, Gu M, Jin W-W, Klinman D M, Wahl S M. J Clin Invest. 1998;101:2615–2621. doi: 10.1172/JCI2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nitta Y, Tashiro F, Tokui M, Shimada A, Takei I, Tabayashi K, Miyazaki J I. Hum Gene Ther. 1998;9:1701–1707. doi: 10.1089/hum.1998.9.12-1701. [DOI] [PubMed] [Google Scholar]
- 33.Yeow W-S, Lawson C M, Beilharz M W. J Immunol. 1998;160:2932–2939. [PubMed] [Google Scholar]