

Abstract
The present study describes a novel phenomenon in pancreatic acinar cells undergoing regulated exocytosis. When acinar cell preparations were challenged with the secretagogue carbamylcholine, a subpopulation of zymogen granules became coated with filamentous actin. These zymogen granules were always in proximity of the acinar cell apical membrane (the site of exocytosis) but did not appear to have fused yet. They were distinct from regular zymogen granules not only because of their association with filamentous actin, but also because the majority of them lacked the zymogen granule marker rab3D, a small GTPase implicated in regulated exocytosis. The apparent loss of rab3D, presumed to result from the release of rab3D from the granule membranes, could be prevented by agents that modulate the actomyosin system as well as by GTP[γS]. These data suggest that zymogen granules engaging in exocytosis become coated with actin before fusion and that this actin coating is tightly coupled to the release of rab3D. We propose that rab3D is involved in the regulation of actin polymerization around secretory granules and that actin coating might facilitate the movement of granules across the subapical actin network and toward their fusion site.
Regulated exocytosis is the process by which a physiologic stimulus leads to the fusion of storage vesicles with the plasma membrane and the subsequent release of secretory products into the extracellular space (1). The exocytotic event is mediated by an amalgam of docking and fusion proteins. These include the SNAP (soluble N-ethylmaleimide-sensitive fusion protein attachment protein) and SNARE (SNAP receptor) proteins. According to the widely accepted SNARE hypothesis formulated by Rothman and colleagues (2, 3), membrane fusion results from the interaction of vesicle or v-SNAREs (VAMPs for vesicle associated membrane proteins) with target or t-SNARE complexes (syntaxin and SNAP-25) in the plasma membrane. Accessory proteins such as the rab3 low-molecular-weight GTPases, synaptotagmin, and syncollin are thought to regulate this process (3–6).
There has been much speculation about putative roles the actin cytoskeleton may play in regulated exocytosis. In polarized epithelial cells such as pancreatic acinar cells, the cytoplasmic face of the apical membrane is closely associated with a dense network of actin filaments and actin-binding proteins (α-actinin, nonmuscle myosin II, tropomyosin, villin) spanning intercellular junctions and protruding into microvilli (7, 8). This so-called terminal web is interposed between the secretory vesicles and the apical membrane, where vesicle fusion takes place. On the basis of this organization, it has been suggested that the actin cytoskeleton can act as a physical barrier limiting access of secretory vesicles to their target membrane (9, 10). Other postulated roles include that of an actomyosin motor, mediating vesicle transport (11, 12) or generating contractile forces to facilitate the expulsion of secretory material (13). So far, it is unknown how the actin-mediated steps along the exocytotic pathway integrate with the docking and fusion steps regulated by the SNARE machinery. Here we show that the secretory granules of pancreatic acinar cells become “coated” with filamentous actin before membrane fusion and that this actin assembly is accompanied by the loss of the zymogen granule marker rab3D, a low-molecular-weight GTP-binding protein implicated in exocytosis.
Methods
Secretion Assays.
Regulated exocytotic activity was quantified by measuring the release of amylase from pancreatic acini in response to carbamylcholine. The acini were prepared by enzymatic and mechanical dispersion of pancreata from adult male rats (Sprague–Dawley) that had been fasted for 24 h. Details of these techniques have been described elsewhere (14, 15). Stimulation with carbamylcholine was carried out at 37°C and lasted for 1 h. In some experiments, jasplakinolide (3 μM; Molecular Probes), cytochalasin D (10 μM; Sigma), latrunculin B (1 μM; Alexis, San Diego, CA), or 2,3-butanedione monoxime (50 mM; Sigma) was administered, as indicated in Results and Discussion and the figure legends. The drugs were incubated with the acini 30 min before carbamylcholine administration and remained present throughout the 1-h stimulation.
Histochemistry and Immunohistochemistry.
For histochemical and immunohistochemical procedures, pancreatic tissue fragments of approximately 1 mm3 were submitted to the same drug treatments as the acini and subsequently fixed in 3% paraformaldehyde. Tissue fragments were used rather than acini to allow for cryosectioning. Routine secretion assays were performed on the tissue fragments to confirm the drug responses measured in the acini. Antibody and phalloidin staining was carried out on 8-μm-thick cryosections. Further details of the fixation and staining techniques have been described elsewhere (16). Mouse monoclonal anti-GP-2 antibody (17) was a generous gift from Anson W. Lowe at Stanford University, Division of Gastroenterology. Rabbit polyclonal anti-rab3D antibody was raised against recombinant rab3D in our laboratory. Secondary goat anti-mouse and goat anti-rabbit antibodies, as well as phalloidin, were fluorescently labeled with either Alexa 488 or Alexa 596 (Molecular Probes). Staining of the acinar cell surface was achieved by incubating fixed fragments of previously stimulated tissue for 2 h in PBS containing 100 μg/ml Texas Red-labeled wheat-germ agglutinin (Molecular Probes), followed by thorough rinsing. The tissue fragments subsequently were cryosectioned and decorated with Alexa-488 phalloidin.
Actin Coating of Secretory Granules in Tissue Extracts.
Pancreata of adult male rats were homogenized in 5 vol of ice-cold homogenization buffer containing 0.3 M sucrose, 25 mM Pipes, 200 μM CaCl2, 200 μM MgCl2, and 2 mM EGTA (pH 6.8) supplemented with 0.02% wt/vol soy bean trypsin inhibitor. The concentration of free Ca2+ in this buffer was estimated at 6 × 10−8 M by using the algorithm by Stockbridge (18) and the Mac freeware computer program free ca finder by Eric Ortega (available at http://electrophys2.ucsf.edu/PatchClamp/Information/calciumprogs.html). In some experiments, CaCl2 was omitted from the buffer to yield a Ca2+-free medium. Homogenates were centrifuged for 10 min at 300 × g and 4°C to generate postnuclear supernatant fractions. These fractions were divided in portions of 500 μl and pipetted into 1.5-ml Eppendorf tubes. ATP stock solution in a volume of 5 μl was added to each tube so that a final concentration of 2 mM was obtained. In some experiments, guanosine 5′-[γ-thio]triphosphate (GTP[γS]) was added as well and in a similar small volume, yielding a final concentration of 100 μM. The samples were incubated for 20 min at 37°C with continuous bidirectional shaking. Immediately after the incubation, each tube received 500 μl of a 2× fixative solution composed of 0.3 M sucrose, 10 mM phosphate buffer, and 6% paraformaldehyde (pH 7.4). The samples were fixed for 10 min and then centrifuged for 10 min at 1,000 × g to generate a crude particulate fraction enriched in secretory granules. The supernatant was removed and the particulate was resuspended in 1 ml of a 50 mM Tris buffer (pH 7.4). Drops of 100 μl of the resuspended granule fraction were pipetted onto gelatin-coated glass slides and granules were allowed to settle for 2 h. Granules then were labeled with fluorescent phalloidin and antibodies by using the same procedures as applied to tissue cryosections (see above).
Digital Imaging Microscopy.
Immunofluorescence was viewed with a Zeiss Axiophot 2 microscope by using a ×100 oil-immersion objective. Images were acquired with a Photometrics Quantix charge-coupled device camera cooled to −35°C. To enhance detail, out-of-focus haze was removed from the digitized images by using the single-image point-spread function of the hazebuster deconvolution software (VayTek, Fairfield, IA).
Results and Discussion
The present study was undertaken to further our understanding of the role of the actin cytoskeleton in pancreatic exocrine secretion and to investigate the relationship between the actin-mediated steps leading to exocytosis and rab3D, a putative regulator of the SNARE-mediated steps along the exocytotic pathway. For this purpose, we first examined the effects of actomyosin modulators on the regulated secretion of amylase from intact acinar cells. As shown in Table 1, the membrane permeable, actin-stabilizing agent jasplakinolide greatly reduced the release of amylase from pancreatic acini challenged with the cholinergic secretagogue carbamylcholine. This finding indicates that remodeling of the actin cytoskeleton is a prerequisite for regulated exocytosis to occur and is in agreement with the hypothesis that the subapical actin network acts as a regulated barrier for secretion. Jasplakinolide also reduced the release of amylase from nonstimulated acini (Table 1). It thus appears that basal secretory activity is actin-mediated as well. In contrast to the inhibitory effect of jasplakinolide, the secretagogue-induced release of amylase was unaffected by the actin-depolymerizing agents cytochalasin D and latrunculin B (Table 1), suggesting that actin is not required for the process of exocytotic membrane fusion itself. However, the myosin-ATPase inhibitor 2,3-butanedione monoxime (BDM) arrested carbamylcholine-induced amylase secretion completely (Table 1), thus raising the possibility that the actomyosin system still might be involved in the fusion process. But because BDM was able to block secretion even in the presence of cytochalasin D or latrunculin B (Table 1), i.e., when the actomyosin system is uncoupled, its mode of action appeared to be more complex than inhibition of myosin ATPase. Indeed, as will be shown below, we found that BDM prevented the depolymerization of actin filaments by cytochalasin D and latrunculin B, suggesting that the effects of BDM led to a stabilization of actin filaments. Taken together, these data indicated that although secretory activity can persist in the absence of the actin terminal web, under physiological conditions when the actin cytoskeleton is present, it needs to be remodeled for exocytosis to take place.
Table 1.
Effects of actin- and myosin-modulating drugs on regulated amylase secretion by pancreatic acini
| Drug | Nonstimulated | Stimulated | ||
|---|---|---|---|---|
| Control | 6.6 ± 3.8 (11) | 19.0 ± 5.9 (11) | ||
| Jasplakinolide (3 μM) | 3.0 ± 0.7 (4) | P < 0.05 | 11.1 ± 1.4 (4) | P < 0.05 |
| Cytochalasin D (10 μM) | 7.8 ± 4.0 (8) | NS | 21.7 ± 7.3 (10) | NS |
| Latrunculin B (1 μM) | 7.0 ± 2.7 (6) | NS | 17.7 ± 7.5 (8) | NS |
| BDM (50 mM) | 7.4 ± 3.6 (6) | NS | 5.9 ± 3.3 (6) | P < 0.001 |
| BDM + cytochalasin D | ND | 3.8 ± 1.2 (4) | P < 0.001 | |
| BDM + latrunculin B | ND | 6.1 ± 1.8 (2) | P < 0.01 | |
The effects of various agents that modulate microfilaments and myosin were tested on pancreatic acini that were either nonstimulated or stimulated with carbamylcholine (10 μM). Values, expressed as mean ± SEM, represent the amount of amylase secreted as a percentage of total amylase (sum of amylase released and amylase remaining in acini). Numbers in parentheses indicate how many experiments were performed. Statistical significance of the various drug treatments was calculated with reference to the controls by using Student's t test. ND, not determined; NS, not significant.
To identify secretagogue-induced rearrangements of actin filaments, we used digital-imaging microscopy techniques on fixed cryosections labeled with fluorescent phalloidin to detect filamentous actin. As illustrated in Fig. 1, sections of pancreatic tissue fragments challenged with carbamylcholine displayed typical phalloidin-positive circular structures that were associated with or in the proximity of the actin terminal web. These structures, which we termed actin-coated granules (ACGs), had a diameter of 0.8 ± 0.2 μm (mean ± SD, determined on 77 ACGs acquired during 8 independent experiments), a value that corresponded exactly to the published size range of zymogen granules in adult rat pancreas (19). The acinar lumen in Fig. 1b was chosen for illustration because it displays the exceptional number of five ACGs in the same focal plane. The occurrence of ACGs, however, was much less frequent than what might be suggested by this figure. In the approximately 1,000 microscopic fields we examined, ACGs were present in not more than 5–10% of all acini, and most (>90%) of them displayed only one ACG. The rareness of the ACG phenomenon is reminiscent of the low frequency at which exocytotic events are observed at the ultrastructural level and suggests that ACGs are very rapidly formed and transient structures. This also explains why, up to now, ACGs have remained unnoticed in the exocrine pancreas, despite the fact that several studies have addressed the role of the actin terminal web in regulated exocytosis (9, 20–22).
Figure 1.

Detection and characterization of actin-coated granules in pancreatic acinar cells. (a) High-magnification view of a pancreatic acinar lumen in nonstimulated tissue. Alexa-488 phalloidin decorates the subapical actin terminal web surrounding the acinar lumen (asterisk). (b) Similar high-magnification view of an acinar lumen in tissue challenged with carbamylcholine (10 μM, 1 h). The lumen (asterisk) is enlarged from secretory activity, and Alexa-488 phalloidin now outlines circular structures (arrows) that are similar in size to zymogen granules. Note that these actin-coated granules are always in proximity of the terminal web and that they are absent in nonstimulated tissue (shown in a). This example was chosen for illustration because it displays the exceptional number of five actin-coated granules around a single lumen. In general, actin-coated granules were much less frequent (see text for quantification). (c–e) Example of an actin-coated granule that is labeled with the zymogen granule marker GP-2 in secretagogue-stimulated tissue. Immunofluorescence for GP-2 is shown in c, and phalloidin fluorescence in the same microscopic field is shown in d. The arrow indicates the position of an actin-coated granule in d and the corresponding GP-2 immunoreactivity in c. A merge of the GP-2 (green) and phalloidin (red) images is shown in e. (f– h) Evidence that the membrane of actin-coated granules is not continuous with the apical membrane. (f) Detail of an acinar lumen (asterisk) outlined by Texas Red-labeled wheat-germ agglutinin which was incubated before cryosectioning with fixed fragments of secretagogue-stimulated tissue. (g) The corresponding phalloidin fluorescence. An actin-coated granule is indicated by the arrow. (h) Merge of the wheat-germ agglutinin and phalloidin fluorescence images. Note that the actin-coated granule is not labeled with wheat-germ agglutinin. [Bars = 5 μm (a and b) and 2 μm in (d and g).]
Several observations suggest that the ACGs are not endosomes. First, endosomes in the apical area of pancreatic acinar cells are much smaller than ACGs (23). In fact, the only structures present in the apical region that are large enough to match the size of the ACGs are zymogen granules. Second, markers for endocytic compartments (rab4, rab5, rab7, rab9, and clathrin antibodies, fluorescent dextran, and horseradish peroxidase) failed to label ACG-like structures (unpublished observations). Moreover, double-label experiments revealed that the ACGs were immunopositive for the zymogen granule membrane marker GP-2 (24) (Fig. 1 c– e). Because ACGs appeared only in stimulated tissue, and because they always localized close to the apical membrane, it seems reasonable to propose that they represent zymogen granules recruited for exocytosis. To assess whether zymogen granules transform into ACGs before or after fusion, we determined whether or not the ACG membranes are continuous with the apical plasma membrane. Fig. 1 f–h demonstrates that fluorescent wheat-germ agglutinin (WGA) readily labeled the apical membrane of acinar cells in fixed fragments of previously stimulated pancreas, whereas WGA failed to outline the ACG shown. Absence of WGA binding was observed in 35 of 36 (97%) ACGs acquired during three independent experiments. Only one ACG (3%) showed WGA binding. Because both the plasma membrane and zymogen granule membranes of pancreatic acinar cells possess WGA-binding sites (25), it follows that the vast majority of the ACGs are intact zymogen granules that have not yet fused with the apical membrane. This, in turn, implies that the coating of zymogen granules with filamentous actin is not simply the result of resealing of the terminal web beneath fused granules. It suggests, rather, that ACG formation is a regulated sequence in the course of events leading to exocytosis. It would have been interesting to determine whether jasplakinolide, which blocks regulated amylase secretion, also prevents the formation of ACGs. Unfortunately, we found that phalloidin staining was totally abolished in jasplakinolide-treated tissue, which can be explained by the fact that jasplakinolide is a competitive inhibitor of phalloidin binding (26).
It has been shown previously in our laboratory that regulated exocytosis in pancreatic acinar cells is accompanied by the redistribution of a rab3-like protein [subsequently identified as rab3D (27)] from zymogen granules to the Golgi complex (28). Importantly, rab3D is never detected at the apical membrane, not even around the typical omega-figures, which are the remnants of fused zymogen granules, suggesting that rab3D is released from the zymogen granule membrane before fusion (28). This and recent speculations in the literature of interactions between rab proteins (in particular, rab3) and the cytoskeleton (29–31) prompted us to study the ACGs in relation to rab3D distribution. Strikingly, we found that most ACGs were devoid of rab3D immunoreactivity (example shown in Fig. 2 a– c). Thus, in 26 ACGs acquired in five independent experiments, 20 (77%) clearly lacked rab3D immunofluorescence, whereas 6 (23%) were immunopositive for rab3D. This finding suggests that rab3D release and actin coating are closely correlated. Furthermore, because about a quarter of the ACGs were still rab3D-positive, it appears that zymogen granules preparing for exocytosis first acquire an actin coat and subsequently release rab3D. This implies that the granules that are positive for both actin and rab3D represent an intermediate form between F-actin-free, rab3D-positive granules and F-actin-positive, rab3D-free granules. It follows that actin coating and rab3D release are temporally linked events.
Figure 2.
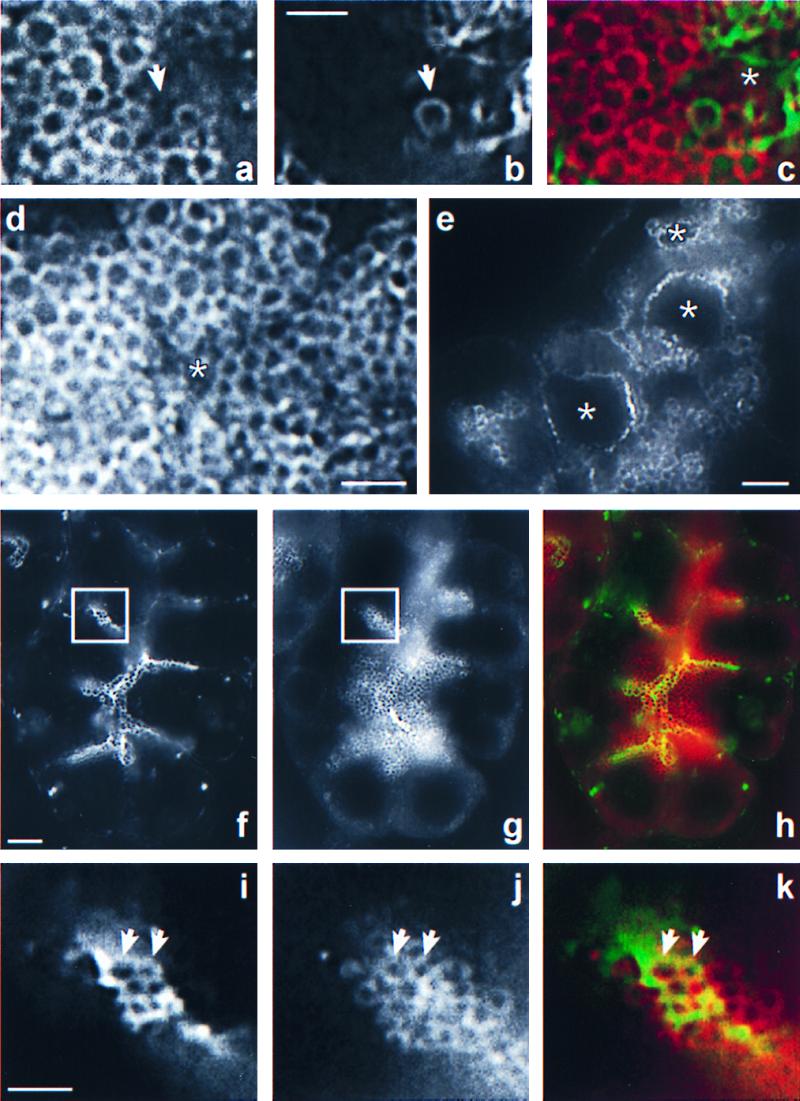
Rab3D release and actin coating of zymogen granules are interdependent events. (a– c) Double localization of rab3D and filamentous actin in tissue challenged with carbamylcholine (10 μM, 1 h). A close-up of part of an acinar lumen (asterisk) and the underlying zymogen granule field is shown. Rab3D immunofluorescence is depicted in a, and the corresponding phalloidin fluorescence is shown in b. The merge of the rab3D and phalloidin fluorescence images is shown in c. Note that the actin-coated granule (arrow) is not outlined by rab3D immunoreactivity, in contrast to the other zymogen granules. (d) Rab3D immunofluorescence in an acinus of tissue that was stimulated with carbamylcholine, showing absence of rab3D immunoreactivity on the apical membrane surrounding the lumen (asterisk). (e) Rab3D immunofluorescence in an acinus stimulated with carbamylcholine in the presence of cytochalasin D. The dramatically enlarged lumen (asterisks) is now outlined by strong rab3D staining. (f–h) Double localization of rab3D and filamentous actin in an acinus stimulated with carbamylcholine in the presence of latrunculin B and 2,3-butanedione monoxime. Phalloidin fluorescence is shown in (f). Note the presence of numerous actin-coated granules packed in clusters. Also note that the acinar lumen has not expanded. The corresponding rab3D immunofluorescence is depicted in g, and the merge between both fluorescence images is shown in h. Boxes in f and g outline an area that is shown at higher magnification in i (phalloidin), j (rab3D), and k (merge). Note that the actin-coated granules (two are indicated by arrows) are rab3D-immunoreactive. [Bars = 2 μm (b, d, and i) and 5 μm (e and f).]
Does rab3D interact with the actin cytoskeleton? A clue to answer this question is provided by experiments in which we examined the distribution of rab3D in tissue challenged with carbamylcholine while an actin-disrupting agent was present. Interestingly, rab3D, which normally does not relocate to the apical membrane (Fig. 2d), strongly labeled the apical membrane in the presence of cytochalasin D (Fig. 2e) or latrunculin B (not shown). Thus, at least under these conditions, regulated exocytosis does not depend on the dissociation of rab3D from the secretory granule membranes. The rab3D immunoreactivity associated with the apical membrane was amplified by a dramatic enlargement of the apical membrane surface area because of the insertion of zymogen granule membrane. This, as we have shown previously, can be ascribed to the actin depolymerization inhibiting compensatory membrane retrieval after exocytosis (16).
The above data indicate that the release of rab3D from zymogen granule membranes and the coating with filamentous actin are coupled events. Namely, when actin filaments are dispersed, rab3D no longer dissociates from the zymogen granule membrane and remains with the apical plasma membrane once fusion has been completed. This, in turn, suggests that rab3D interacts, either directly or via an effector protein, with the actin cytoskeleton. As a matter of fact, we were able to identify an experimental condition in which this putative interaction appears to be “frozen” so that rab3D and filamentous actin colocalize around ACGs. This condition consists of the stimulation of pancreas fragments with carbamylcholine in the presence of a combination of latrunculin B and BDM. As mentioned earlier, BDM blocked regulated amylase release even when combined with either cytochalasin D or latrunculin B (Table 1). This perplexing observation was explained by our finding that BDM prevented the cytochalasin D- and latrunculin B-induced (not shown and Fig. 2f, respectively) depolymerization of actin filaments monitored by fluorescent phalloidin. It thus appeared that BDM, presumably via its action on myosin, stabilized actin filaments, thereby rendering the actin-depolymerizing agents ineffective. Strikingly, in the presence of latrunculin B but not cytochalasin D, BDM led to the accumulation of numerous ACGs in carbamylcholine-challenged tissue. These ACGs were clustered in patches surrounding the terminal web of acinar cells (Fig. 2f). Double staining for filamentous actin and rab3D revealed that the clustered ACGs were rab3D-positive (Fig. 2 f–k). Quantification in 20 acini taken from four independent experiments revealed that the number of rab3D-positive ACGs in a 0.2-μm focal plane varied between 31 and 128 (mean = 67 ± 26) per acinus. Because treatment with BDM and latrunculin B blocked carbamylcholine-induced amylase secretion while inducing a dramatic accumulation of ACGs, these experiments provide further evidence that ACGs are zymogen granules that have not yet fused with the apical membrane. Why the aggregation of ACGs occurred only in the presence of BDM and latrunculin B and not with BDM and cytochalasin D must await further investigation. However, it is interesting to mention that the actin-depolymerizing drugs act via distinct mechanisms [cytochalasin D caps the barbed end of actin filaments whereas latrunculin B sequesters actin monomers (32)].
So far, our data could not rule out the possibility that ACGs represent zymogen granules that are merely enmeshed by the actin terminal web. We therefore set about to determine whether zymogen granules are able to undergo actin coating in a cell-free system under conditions (GTP[γS], presence of Ca2+) that mimic secretagogue stimulation. Postnuclear supernatants prepared from pancreatic homogenates were incubated for 20 min at 37°C with the addition of Ca2+-EGTA, ATP, and GTP[γS]. A crude zymogen granule fraction was obtained by centrifugation of the fixed postnuclear supernatants for 10 min at 1,000 × g. The zymogen granules were resuspended, settled on gelatin-coated slides, and processed for phalloidin fluorescence and immunofluorescence. In a total of 1,921 zymogen granules, viewed in 16 microscopic fields chosen randomly from 3 independent experiments, 58 (3%) were labeled with fluorescent phalloidin (Fig. 3). ACGs were never observed among a comparable number of zymogen granules taken from three control experiments in which GTP[γS] (Fig. 3) or Ca2+ (not shown) was omitted from the incubation mixture. Interestingly, all 58 ACGs found in cell extracts incubated in the presence of GTP[γS] and Ca2 were immunopositive for rab3D (Fig. 3). Although this observation is in apparent contradiction with the ACGs found in intact cells being mostly devoid of rab3D, it is most readily explained by the nonhydrolyzable nature of GTP[γS]. Namely, the dissociation of rab proteins from their target membranes requires GTP hydrolysis (33), and, thus, GTP[γS] will prevent the release of rab3D from the ACGs.
Figure 3.

Actin coating of zymogen granules in a cell-free system. Postnuclear supernatants were incubated for 20 min at 37°C in the presence of Ca2+-EGTA, ATP, and GTP[γS], subsequently fixed, centrifuged to generate a crude zymogen granule fraction, and processed for phalloidin and immunofluorescence. (a) Bright-field image of a microscopic field containing approximately 22 zymogen granules. (b) Phalloidin-fluorescent image of the microscopic field shown in a. Note the presence of five actin-coated granules whose location corresponds to that of five zymogen granules shown in a (arrowheads). (c) Rab3D immunofluorescence in the same microscopic field as in a and b, showing that all zymogen granules, including the actin-coated granules, are rab3D-positive. (d) Merge of the images shown in b (green) and c (red). Yellow indicates overlap between phalloidin and rab3D fluorescence. (e) Bright-field microscopic view of zymogen granules that were incubated as in a–d, but without GTP[γS]. (f) Phalloidin-fluorescent image of the microscopic field shown in e. Note the absence of actin-coated granules. (g) Rab3D immunofluorescence in the same microscopic field as shown in e and f. (h) A merge of the images shown in f (green) and g (red).
It has been shown previously that actin antibodies, which do not discriminate between globular and filamentous actin, label the membranes of the entire zymogen granule pool in rat pancreatic acinar cells (8, 34). This, combined with our data showing that only very few zymogen granules are coated with filamentous actin and exclusively in tissue challenged with secretagogue, suggests that zymogen granules normally are covered with globular actin. It therefore is possible that actin coating results from the polymerization of actin already present on the zymogen granules. In the light of a recent study that reported that myosin I is present on zymogen granules in rat pancreas as well (11), it appears that zymogen granules possess a self-contained actomyosin system. These data emphasize the complexity and importance of the role that the actin cytoskeleton plays in regulated exocytosis.
Taken together, our results indicate that a select pool of secretory granules becomes coated with actin filaments in response to secretagogue stimulation. We propose that the functional significance of actin coating is to allow for movement of primed secretory granules through the terminal web while preserving the integrity of the fusion barrier that the subapical actin network forms. Our data also indicate that the formation of ACGs and the release of rab3D from zymogen granules are interdependent events and, furthermore, that the release of rab3D is microfilament dependent. This implies that rab3D, in contrast to rab3A (35), is involved in an early step of regulated exocytosis that involves the actin cytoskeleton and that precedes the docking and fusion steps mediated by SNARE proteins. It is likely that rab3D is implicated in the regulation of the actin cytoskeleton, but further studies will be needed to determine the nature of the interaction between rab3D and the cytoskeleton and the functional significance thereof.
Acknowledgments
This study was funded by National Institutes of Health Grants DK 07017, DK 17389, and PO1 CA46128.
Abbreviations
- ACG
actin-coated granule
- BDM
2,3-butanedione monoxime
- GTP[γS]
guanosine 5′-[γ-thio]triphosphate
- SNAP
soluble N-ethylmaleimide-sensitive fusion protein attachment protein
- SNARE
SNAP receptor
References
- 1.Palade G E. Science. 1975;189:347–356. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- 2.Rothman J E, Söllner T H. Science. 1997;276:1212–1213. doi: 10.1126/science.276.5316.1212. [DOI] [PubMed] [Google Scholar]
- 3.Ferro-Novick S, Jahn R. Nature (London) 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- 4.Fischer von Mollard G, Stahl B, Li C, Südhof T C, Jahn R. Trends Biochem Sci. 1994;19:164–168. doi: 10.1016/0968-0004(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 5.Geppert M, Südhof T C. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 6.Edwardson J M, An S, Jahn R. Cell. 1997;90:325–333. doi: 10.1016/s0092-8674(00)80340-2. [DOI] [PubMed] [Google Scholar]
- 7.Drenckhahn D, Mannherz H G. Eur J Cell Biol. 1983;30:167–176. [PubMed] [Google Scholar]
- 8.Bendayan M. Can J Biochem Cell Biol. 1985;63:680–690. doi: 10.1139/o85-085. [DOI] [PubMed] [Google Scholar]
- 9.Muallem S, Kwiatkowska K, Xu X, Yin H L. J Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vitale M L, Seward E P, Trifaró J-M. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- 11.Poucell-Hatton S, Perkins P S, Deerinck T J, Ellisman M H, Hardison W G M, Pandol S J. Gastroenterology. 1997;113:649–658. doi: 10.1053/gast.1997.v113.pm9247487. [DOI] [PubMed] [Google Scholar]
- 12.Bi G Q, Morris R L, Liao G C, Alderton J M, Scholey J M, Steinhardt R A. J Cell Biol. 1997;138:999–1008. doi: 10.1083/jcb.138.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segawa A, Yamashina S. Cell Struct Funct. 1989;14:531–544. doi: 10.1247/csf.14.531. [DOI] [PubMed] [Google Scholar]
- 14.Bruzzone R, Halban P A, Gjinovci A, Trimble E R. Biochem J. 1985;226:621–624. doi: 10.1042/bj2260621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernfeld P. Methods Enzymol. 1955;1:149–158. [Google Scholar]
- 16.Valentijn K, Gumkowski F D, Jamieson J D. J Cell Sci. 1999;112:81–96. doi: 10.1242/jcs.112.1.81. [DOI] [PubMed] [Google Scholar]
- 17.Lowe A W, Luthen R E, Wong S M E, Grendell J H. Gastroenterology. 1994;107:1819–1827. doi: 10.1016/0016-5085(94)90826-5. [DOI] [PubMed] [Google Scholar]
- 18.Stockbridge N. Comput Biol Med. 1987;17:299–304. doi: 10.1016/0010-4825(87)90019-9. [DOI] [PubMed] [Google Scholar]
- 19.Ermak T H, Rothman S S. Cell Tissue Res. 1981;214:51–66. doi: 10.1007/BF00235144. [DOI] [PubMed] [Google Scholar]
- 20.Schäfer C, Ross S E, Bragado M J, Groblewski G E, Ernst S A, Williams J A. J Biol Chem. 1998;273:24173–24180. doi: 10.1074/jbc.273.37.24173. [DOI] [PubMed] [Google Scholar]
- 21.Bauduin H, Stock C, Vincent D, Grenier J F. J Cell Biol. 1975;66:165–181. doi: 10.1083/jcb.66.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Konski M S, Pandol S J. J Clin Invest. 1990;86:1649–1657. doi: 10.1172/JCI114887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver C, Tolbert C L, Waters J F. J Histochem Cytochem. 1989;37:49–56. doi: 10.1177/37.1.2908883. [DOI] [PubMed] [Google Scholar]
- 24.Rindler M J, Hoops T C. Eur J Cell Biol. 1990;53:154–163. [PubMed] [Google Scholar]
- 25.Willemer S, Kohler H, Naumann R, Kern H F, Adler G. Histochemistry. 1990;93:319–326. doi: 10.1007/BF00266395. [DOI] [PubMed] [Google Scholar]
- 26.Bubb M R, Senderowicz A M J, Sausville E A, Duncan K L K, Korn E D. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 27.Valentijn J A, Sengupta D, Gumkowski F D, Tang L H, Konieczko E M, Jamieson J D. Eur J Cell Biol. 1996;70:33–41. [PubMed] [Google Scholar]
- 28.Jena B P, Gumkowski F D, Konieczko E M, Fischer von Mollard G, Jahn R, Jamieson J D. J Cell Biol. 1994;124:43–53. doi: 10.1083/jcb.124.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber E, Berta G, Tousson A, St. John P, Green M W, Gopalokrishnan U, Jilling T, Sorscher E J, Elton T S, Abrahamson D R, et al. J Cell Biol. 1994;125:583–594. doi: 10.1083/jcb.125.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato M, Sasaki T, Ohya T, Nakanishi H, Nishioka H, Imamura M, Takai Y. J Biol Chem. 1996;271:31775–31778. doi: 10.1074/jbc.271.50.31775. [DOI] [PubMed] [Google Scholar]
- 31.Valentijn J A, Jamieson J D. Biochem Biophys Res Commun. 1998;243:331–336. doi: 10.1006/bbrc.1997.7824. [DOI] [PubMed] [Google Scholar]
- 32.Spector I, Shochet N R, Blasberger D, Kashman Y. Cell Motil Cytoskel. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- 33.Pfeffer S R, Dirac-Svejstrup A B, Soldati T. J Biol Chem. 1995;270:17057–17059. doi: 10.1074/jbc.270.29.17057. [DOI] [PubMed] [Google Scholar]
- 34.Bendayan M. Histochem J. 1983;15:39–58. doi: 10.1007/BF01006070. [DOI] [PubMed] [Google Scholar]
- 35.Geppert M, Goda Y, Stevens C F, Südhof T C. Nature (London) 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]